

Abstract
Clinical manifestations of tetanus and botulism result from an intricate series of interactions between clostridial neurotoxins (CNTs) and nerve terminal proteins that ultimately cause proteolytic cleavage of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins and functional blockade of neurotransmitter release. Although detection of cleaved SNARE proteins is routinely used as a molecular readout of CNT intoxication in cultured cells, impaired synaptic function is the pathophysiological basis of clinical disease. Work in our laboratory has suggested that the blockade of synaptic neurotransmission in networked neuron cultures offers a phenotypic readout of CNT intoxication that more closely replicates the functional endpoint of clinical disease. Here, we explore the value of measuring spontaneous neurotransmission frequencies as novel and functionally relevant readouts of CNT intoxication. The generalizability of this approach was confirmed in primary neuron cultures as well as human and mouse stem cell-derived neurons exposed to botulinum neurotoxin serotypes A–G and tetanus neurotoxin. The sensitivity and specificity of synaptic activity as a reporter of intoxication was evaluated in assays representing the principal clinical and research purposes of in vivo studies. Our findings confirm that synaptic activity offers a novel and functionally relevant readout for the in vitro characterizations of CNTs. They further suggest that the analysis of synaptic activity in neuronal cell cultures can serve as a surrogate for neuromuscular paralysis in the mouse lethal assay, and therefore is expected to significantly reduce the need for terminal animal use in toxin studies and facilitate identification of candidate therapeutics in cell-based screening assays.
Keywords: neurons, botulinum toxins, tetanus toxin, synaptic transmission, electrophysiology, spontaneous postsynaptic currents
The clostridial neurotoxins (CNTs) are a family of closely related bacterial protein toxins that include tetanus neurotoxin (TeNT) and 7 antigenically distinguishable botulinum neurotoxins (BoNTs), labeled A–G. All CNTs cause disease by interfering with the release of neurotransmitters from the presynaptic membrane, with the principal distinction between clinical manifestations of tetanus and botulism arising from their activity in central versus peripheral neurons, respectively (Simpson, 2004). Each CNT consists of a 100-kDa heavy chain and a 50-kDa light chain (LC) that remain associated through a disulfide bond (Montal, 2010). The heavy chain mediates neuron-specific binding, uptake via synaptic endocytosis and translocation into the presynaptic cytosol, while LC is a Zn2+-dependent metalloprotease that proteolytically cleaves and inactivates synaptosomal-associated protein 25 (SNAP-25; BONT/A, /C, /E), synaptobrevin-1/2 (Syb1/2; BoNT/B, /D, /F, /G, and TeNT), or syntaxin-1 (Stx1; BoNT/C). These 3 proteins comprise the neuronal SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins required for fast exocytosis of neurotransmitter-containing synaptic vesicles at the presynaptic membrane (Sudhof and Rizo, 2011). Proteolytic cleavage of neuronal SNARE proteins by LC blocks vesicle fusion, thereby preventing neurotransmitter release onto the postsynaptic membrane. This combination of efficient neuron targeting and the ability of a small number of toxin molecules to impair neurotransmission renders CNTs the most toxic substances known, with estimated human LD50 values as low as 0.1–1 ng/kg (Schiavo et al., 1994).
CNT intoxication becomes life-threatening once respiratory muscles are compromised. Although post-exposure administration of antibody-based antitoxins can neutralize CNTs in the bloodstream, there are no treatments to prevent or reverse long-term paralysis once CNTs enter the neuron (Larsen, 2009; Smith et al., 2012). Since the ability to provide supportive care to paralyzed victims for long durations is limited, treatments that can reverse paralysis are urgently needed. However, therapeutic discovery has been limited by a number of factors, including the lack of relevant and scalable assays of intoxication.
The current standard technique for testing CNTs is the mouse lethality assay (MLA), which measures the amount of intraperitoneally administered toxin sufficient to kill 50% of mice within 96 h (Brin and Aoki, 2002). Although the MLA is quantitative, evaluates all steps of toxin uptake and activation and uses clinical endpoints as output, the assay is resource-intensive and incurs significant animal distress (Pearce et al., 1994). While more humane methods have been developed to characterize toxin activity and test for symptomatic reversal of paralysis, they remain low-throughput and still require animal use (Huber et al., 2008). These limitations have motivated a search for cell-based models which exhibit physiological responses to intoxication as a replacement for animal use.
Physiological intoxication of motor nerve terminals by BoNT prevents action potential-evoked neurotransmitter release as well as action potential-independent spontaneous release (Kim et al., 1984). Since SNAP-25 and Syb1/2 are essential for both forms of neurotransmission in all known synapses (Sollner et al., 1993), we hypothesized that intoxication of synaptically active cultured neurons of CNS origin would result in synaptic blockade, which can be quantified by reduced detection of postsynaptic currents. If correct, synaptic activity would therefore represent a novel, cell-based phenotypic readout of the functional endpoint of clinical intoxication. Notably, since toxin-induced blockade of synaptic activity requires elaboration of all cellular steps of intoxication, this approach retains the key experimental advantages of the MLA while avoiding animal use.
We recently reported that synaptic neurotransmission is rapidly blocked in stem cell-derived neurons treated with BoNT/A (Beske et al., 2015). Here we expand on these findings to determine the utility of synaptic neurotransmission as a broad-spectrum, in vitro phenotypic readout of intoxication. First, to assess the generalizability of this approach, the effects of CNTs on synaptic activity were tested in 4 diverse neuron cultures, including the first evaluation of synaptic blockade in human neurons. Next, we determined the broad-spectrum potential of impaired synaptic function as a functional readout of intoxication by all known CNTs. We then determined the sensitivity of synaptic neurotransmission to 3 clinically important CNTs in concentration-response studies. Finally, the practical utility of cell-based synaptic readouts as a replacement for the MLA was evaluated in assays representative of those used in the public health and commercial sectors, including serotype determination, antitoxin specificity testing, potency determination, and toxin detection in various complex matrices (CM). Based on these studies we propose that synaptic function assays are a functionally relevant and broadly accessible method for cell-based detection and characterization of CNTs. The use of phenotypic assays to measure CNT intoxication in central neurons with sensitivities equivalent to the MLA is anticipated to significantly reduce the need for distressing and terminal in vivo studies as well as accelerate toxin characterization and therapeutic drug discovery research.
MATERIALS AND METHODS
Reagents
Supplies for embryonic stem cell maintenance, embryonic stem cell-derived neuron (ESN) differentiation and primary neuron culture and harvest were obtained from Invitrogen (Carlsbad, California), with the exception of fetal bovine serum (Applied StemCell Inc, Menlo Park, California). BoNT types A1 (with approximate specific activity of 2.5 × 108 U/mg, where 1 U is 1 mouse LD50), B1 (2.1 × 107 U/mg), C (3.5 × 107 U/mg), D (9.0 × 107 U/mg), E1 (6.0 × 107 U/mg), F1 (2.0 × 107 U/mg), and G (1.2 × 107 U/mg) as well as formalin-inactivated BoNT/A dichain were purchased from Metabiologics (Madison, Wisconsin) at 1 mg/mL in Ca2+/Mg2+-free phosphate buffered saline, pH 7.4 (PBS), and stored at −30°C. In the case of BoNT/E and /G, toxin was activated by a 60 min incubation at 37°C in 0.05 M sodium phosphate buffer (pH 6.5), 0.3 mg/ml TPCK-treated trypsin (Sigma-Aldrich, St Louis, Missouri) and 10% glycerol (Hubbard et al., 2012). Activated BoNT/E was diluted 1:4 with fetal calf serum (Sigma-Aldrich; St. Louis, Missouri) and stored at −30°C until use. TeNT from Clostridium tetani was purchased from List Biological Laboratories (1.5 × 107 U/mg; Campbell, California). BOTOX Cosmetic (onabotulinumtoxinA) was purchased from Allergan (Irvine, California), resuspended at 100 U/ml in PBS and stored at −30°C. Neutralizing rabbit anti-A IgG (187 000 neutralizing units/ml at 28.1 µM) and rabbit anti-B IgG (estimated 50 000 neutralizing units/ml at 30.2 µM) were purchased from Metabiologics and stored at 4°C. Generic brands of green beans, whole milk and non-alcoholic apple cider were purchased from a local supermarket, centrifuged for 20 min at 14 000 × g to pellet particulate matter and supernatants were aliquoted and frozen. Unless otherwise specified, reagents for electrophysiology were obtained from Sigma-Aldrich.
Cell culture and intoxications
Four neuronal populations were used in this study: primary rat cerebellar neurons (CNs), primary rat cortical neurons (CtNs), mouse embryonic stem cell-derived neurons (ESNs), and human induced pluripotent stem cell-derived neurons (hSNs). Briefly, CNs and CtNs were harvested from P4 Wistar rats according to established protocols and plated at 150 000 cell/cm2 on 35 mm dishes or on 12 mm coverslips coated with polyethyleneimine (PEI; Sigma-Aldrich) (Beaudoin et al., 2012; Bilimoria and Bonni, 2008). Primary neurons were maintained at 5% CO2, 37°C and 95% humidity in Neurobasal-A medium supplemented with 2% B27 vitamins (NBA/B27). Cytosine beta-d-arabinofuranoside (10 µM) was added 2 d after plating (DAP 2) and half-media changes with CO2-equilibrated NBA/B27 were conducted at 3- to 4-day intervals. CN media was supplemented with 25 mM KCl from DAP 0-6. R1 embryonic stem cell lines were obtained from ATCC (Manassas, Virginia) and differentiated into ESNs as previously described (Hubbard et al., 2015; Nagy et al., 1993). ESNs were plated at 150 000 cells/cm2 in 6 cm dishes or on 18 mm coverslips coated with PEI and maintained at 5% CO2, 37°C, and 95% humidity in NBA/B27. Human iPSC-derived neural cells (MTI-GlobalStem; Gaithersburg, Maryland) were cultured on 18 mm coverslips at 150 000 cells/cm2 according to the manufacturer’s instructions with half-media changes every 3–4 days.
In most instances, toxin was prepared at a 100 × final concentration in fresh medium and diluted into neuron cultures in a tissue culture glove box (Coy Labs, Grass Lake, Michigan) maintained at 5% CO2 and 37°C. Toxin concentrations used in the initial screen of 4 neuronal cultures were: 10 pM BoNT/A, 100 pM BoNT/B and 670 pM formalin-inactivated BoNT/A toxin. Concentrations used during broad-spectrum screening were 2 pM BoNT/A, 88 pM BoNT/B, 70 pM BoNT/C, 12 pM BoNT/D, 200 pM activated BoNT/E, 670 pM activated BoNT/F, 1200 pM BoNT/G, 20 pM TeNT and 670 pM formalin-inactivated BoNT/A toxin. Neuron viabilities were determined using PrestoBlue (Invitrogen) and quantified with a Synergy MX plate reader (Biotek, Winooski, Vermont) using excitation of 535 nm and emission of 595 nm.
For antitoxin studies, 4.4 pM BoNT/A or 40 pM BoNT/B (approximately 160 U/ml each) were incubated with 280 neutralizing units/mL of anti-A or anti-B IgG for 1 h at 37°C and diluted 1:40 into ESN cultures for 20 h prior to whole cell recordings. The resulting culture media contained 7 neutralizing units/ml of IgG and 4 U/ml of toxin, corresponding to a 1.75:1 IgG:toxin ratio. For preparing nutritive samples, clarified supernatants were spiked with 5.5 pM BoNT/A and diluted 1:100 into culture media for a final concentration of 55 fM and 1% nutritive supernatant. The N-methyl-D-aspartate (NMDA) receptor antagonist MK-801 (10 µM; Tocris Bioscience, Bristol, UK) was added as described. Human serum (Sigma-Aldrich) was added directly to culture media as described. When appropriate, serum was spiked with 550 fM BoNT/A prior to addition to cultures.
Whole-cell electrophysiology
Neurons were visualized on an Olympus IX51 microscope (Shinjuku, Tokyo, Japan) equipped with a 40 × lens with differential interference contrast optics. Patch pipettes, 5–10 MΩ, were pulled from borosilicate glass (Sutter Instruments, Novato, California), filled with intracellular recording buffer and dipped in Sigmacote (Sigma-Aldrich) prior to use. Electrophysiology data were acquired at 20–22°C with an EPC10 (Heka, Lambrecht/Pfalz, Germany) and Heka Patchmaster 2.53 software. Current and voltage measurements were filtered online at 2.9 kHz and digitized at 10 kHz. Data analysis and graphing were performed in Heka Fitmaster 2.53, Igor Pro v6 (Wavemetrics, Portland, Oregon) and Prism v6.1 (Graphpad Software, La Jolla, California). Recordings were retained for analysis if the following criteria were met: series resistance was less than 50 MΩ; holding current remained between -30 to 10 pA; and resistance varied by less than 20% during the recording.
Neurons were bathed in extracellular recording buffer (ERB) containing (in mM): 140 NaCl, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 Glucose, 10 HEPES (pH = 7.3; NaOH; 315 ± 10 mOsm/kg). Recording electrodes were filled with an intracellular recording buffer (IRB) containing (in mM): 140 K-gluconate, 5 NaCl, 2 Mg-ATP, 0.5 Li-GTP, 0.1 CaCl2, 1 MgCl2, 1 EGTA and 10 HEPES (pH = 7.3; KOH; 315 ± 10 mOsm/kg). Resting membrane potential was determined using the zero-current-clamp method immediately after establishment of whole-cell configuration. Dishes were allowed to stabilize in ERB for at least 3 min prior to the start of each experiment. Whole-cell recordings were made in the presence of MK-801 (10 µM; Tocris), tetrodotoxin (TTX) (5 µM; Tocris) and bicuculline (10 µM; Tocris). These treatments enabled the postsynaptic detection of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated spontaneous synaptic events (a.k.a. excitatory minis), the frequencies of which reflect glutamate release probabilities in the absence of network activity. Recordings were made for at least 3 min in voltage-clamp mode at −84.6 mV. Miniature excitatory postsynaptic current (mEPSC) frequencies, amplitudes, and inter-event intervals were identified and quantified via MiniAnalysis v6 (Synaptosoft Inc, Fort Lee, New Jersy) using default kinetic parameters for AMPA receptor events. mEPSC frequencies were converted to Hz and normalized to age- and lot-matched vehicle-treated cultures. Mean mEPSC amplitudes were determined from 50 to 2500 events per neuron. Neurons with less than 50 events per recording were discarded from amplitude analysis. Inter-event intervals were collected from the first 50 events per recording. With the exception of CNT-intoxicated neurons, recordings with less than 50 events per recording were discarded from inter-event interval analysis. In all cases, responses to individual toxins were compared among age- and lot-matched cultures. Presented data are adjusted for a calculated liquid junction potential of −14.6 mV.
Immunoblotting
ESN cultures were lysed with denaturing cell extraction buffer (Life Technologies), and lysates were clarified by centrifugation (5 min; 16 000 × g). Total protein concentration was determined by bicinchoninic acid (BCA) analysis (Thermo Scientific, Rockford, Illinois). For immunoblot analysis of SNARE protein cleavage, 30 µg (synaptobrevin-2 detection) or 15 µg (SNAP-25 detection) of total protein was separated on a 12% Nupage gel (Life Technologies) with MOPS running buffer. Gels were transferred to PVDF and probed with a mouse anti-Syntaxin-1 (Synaptic Systems) and a mouse anti-SNAP-25 antibody (Covance, Gaithersburg, Maryland) or a mouse anti-synaptobrevin-2 antibody (Synaptic Systems, Gottingen, Germany). Primary antibodies were diluted 1:1000 in Tris-buffered saline (TBS) Superblock with 0.05% Tween-20 (TBST; Life Technologies). In some cases, blots were probed using a custom-made mouse monoclonal antibody that specifically recognizes BoNT/A-cleaved SNAP-25 (manuscript in preparation). Proteins were visualized with goat anti-mouse Alexa-488 labeled antibodies (Invitrogen) diluted 1:2500 in TBST and imaged with a Versadoc MP4000 (Bio-Rad, Hercules, California). All Western blots shown represent at least 3 biological replicates.
Immunocytochemistry
ESNs plated on 18 mm coverslips were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.1% saponin and 3% bovine serum albumin in PBS (PBSS) for 10 min. Coverslips were incubated for 1 h with various combinations of antibodies against MAP2, Syb2 (Synaptic Systems, Gottingen, Germany), SNAP-25 (Covance) and NeuN (Synaptic Systems) diluted in PBSS according to the manufacturer’s instructions. Coverslips were incubated for 1 h with Alexa-labeled secondary antibodies (Invitrogen) diluted 1:500 in PBSS and mounted with Prolong Gold DAPI mounting media (Life Technologies). Images were collected with Zeiss LSM 500 or 710 confocal microscopes (Carl Zeiss Inc, Thornwood, New York) and analyzed with Zen 2012 software (Zeiss).
Statistical analyses
mEPSC frequencies were calculated by determining the mean frequency (in Hz) of events measured during continuous voltage-clamp recordings. Rates measured in CNT-treated neurons were normalized to mean frequencies observed in age- and lot-matched, vehicle-treated neurons and presented as percentages of control values. Averaged frequency data are presented as mean ± SEM. Graphpad Prism v6.05 (Graphpad Software, La Jolla, California) was used to calculate half maximal inhibitory concentration (IC50) and half maximal effective concentration (EC50) parameters using a 4-parameter sigmoidal model. Unless otherwise indicated, statistical significances among means were determined using 1-way ANOVA testing, and P values were calculated against controls with the Dunnett’s test. Quantitative data are presented as mean ± the standard error of the mean, with the following levels of statistical significance: * indicates a P < 0.05; ** indicates a P < 0.01; *** indicates a P < 0.001.
RESULTS
Effects of BoNT/A or BoNT/B on Synaptic Transmission in Human, Mouse, and Rat Neurons
Cell-based models purporting to replicate in vivo responses to intoxication must be shown to have neurotypic synaptic behaviors that are, in turn, blocked by exposure to CNTs. One of the most commonly used techniques to measure synaptic function is whole-cell patch-clamp electrophysiology. The whole-cell patch-clamp technique involves establishment of a continuous electrical connection between a recording electrode and the cytoplasm of a neuron, allowing the detection of ion currents resulting from synaptic neurotransmission with single-synapse resolution (Hamill et al., 1981). Using whole-cell electrophysiology, we recently demonstrated that addition of BoNT/A to networked cultures of embryonic stem cell-derived neurons (ESNs) rapidly impaired evoked and spontaneous neurotransmitter release at glutamatergic and GABAergic synapses (Beske et al., 2015). To determine whether CNT-mediated blockade of spontaneous release is unique to ESNs or is a general property of synaptically active neurons, we measured the effects of BoNT/A and BoNT/B on miniature excitatory postsynaptic currents (mEPSCs or excitatory minis) in mouse ESNs, human induced pluripotent stem cell-derived neurons (hSNs), primary rat cortical neurons (CtNs) and primary rat cerebellar neurons (CNs). Spontaneous glutamatergic synaptic events were quantified from whole-cell patch-clamp recordings conducted in the presence of the voltage-gated sodium channel antagonist TTX, to block network-level activity, and the GABAA receptor antagonist bicuculline, to block the effects of inhibitory synaptic activity on mEPSC detection (Chalifoux and Carter, 2011).
We first established experimental windows during which synaptic activity could be reproducibly detected in >90% of whole-cell recordings (Fig. 1A). Excitatory minis were reliably detected at days after plating (DAP) 24–30 in ESNs, DAP 66–77 in hSNs, DAP 14–18 in CtNs, and DAP 12–16 in CNs. Average rates of spontaneous synaptic activity varied among the different neuronal models, with the following rank-order: ESNs >> CtNs ≈ CNs > hSNs. In all neuronal cultures, superfusion of the AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 µM) completely eliminated postsynaptic currents, verifying that we were measuring spontaneous activity of glutamatergic synapses (Fig. 1A). All neuronal cultures expressed and appropriately localized SNARE protein targets of CNTs, demonstrated by axolemmal distribution of SNAP-25 and punctate accumulation of Syb2 at axo-dendritic interfaces (Fig. 1B).
FIG. 1.
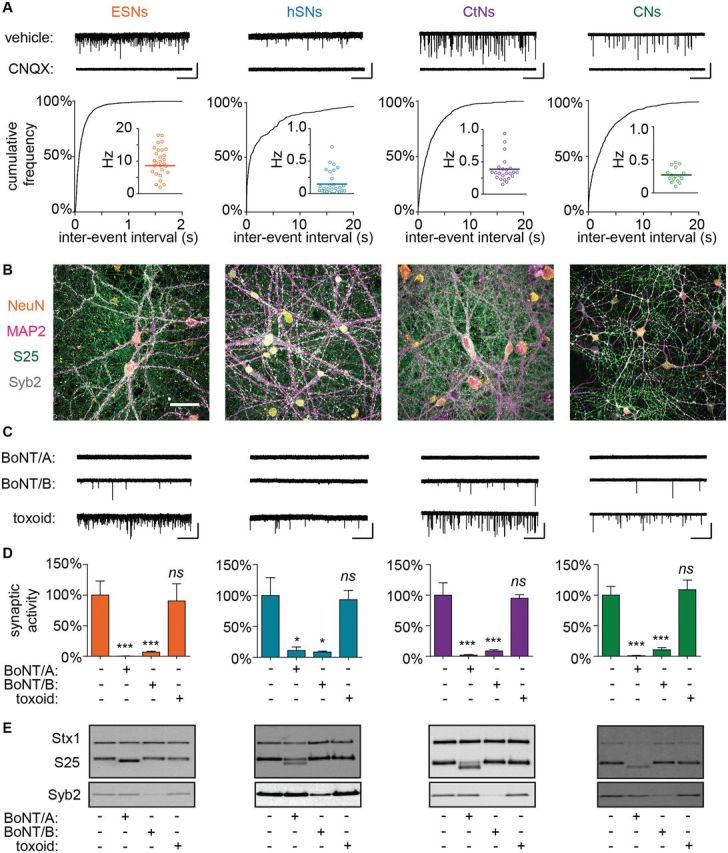
Intoxication of diverse neuron cultures with BoNT/A or BoNT/B blocks spontaneous neurotransmission and results in SNARE protein cleavage. (A) Representative whole-cell recordings, cumulative probability distributions of inter-event intervals and mean mEPSC frequencies demonstrating spontaneous synaptic activity in ESNs, hSNs, CtNs, and CNs (n = 14–30 for each). Scale bar = 2 s × 60 pA for ESNs and 20 s × 60 pA for others. (B) ICC confirming expression and localization of neurotypic markers NeuN (orange and MAP2 (magenta) as well as CNT target proteins SNAP-25 (green) and Syb2 (white). Scale = 20 µm (C) Representative mEPSC recordings and (D) normalized mEPSC frequencies demonstrating reduced spontaneous release in cultures treated with BoNT/A or BoNT/B but not formalin-inactivated toxoid (n = 8–16 each). Scale is same as panel A. Data are presented as mean ± SEM. *P < 0.05. ***P < 0.001. (E) Immunoblot demonstrating changes in SNAP-25, Syb2 and Stx1 following treatment with BoNT/A or BoNT/B but not formalin-inactivated toxoid.
After establishing that neuronal cultures exhibited synaptic activity and expressed target SNARE proteins, each population was treated with bath applications of BoNT/A (10 pM), BoNT/B (100 pM) or formalin-inactivated BoNT/A toxoid (670 pM) for 20 h, and mEPSC frequencies were quantified in whole-cell recordings and normalized to vehicle-treated controls. In all cultures, addition of BoNT/A or BoNT/B reduced spontaneous release by over 95%, whereas addition of toxoid had no effect on mEPSC frequencies (Fig. 1C and D). Blockade of spontaneous release was specifically associated with production of cleaved SNAP-25 (BoNT/A) or loss of Syb2 immunoreactivity (BoNT/B), correlating molecular products of LC catalytic activity with phenotypic readouts of intoxication (Fig. 1E). Note that while the LC/A-cleaved product of SNAP-25 can be distinguished by a 1 kDa gel mobility shift, the cytosolic domain of Syb2 is rapidly cleared upon proteolysis, and therefore loss of Syb2 immunoreactivity is used to indicate LC/B activity (Hubbard et al., 2012). These data support the hypothesis that quantal analysis of spontaneous release frequencies can be used as a phenotypic readout of CNT intoxication in diverse populations of synaptically active neurons.
Synaptic Neurotransmission Is Impaired in ESNs Treated with Each CNT
Further exploration of the effects of CNT intoxication on spontaneous neurotransmission was conducted in ESNs. This selection was based on the comparatively long experimental window during which ESNs can be used; the robust levels of excitatory minis, which increases sensitivity and dynamic range; and the highly scalable nature of ESN production, which enables detailed concentration-response studies (Hubbard et al., 2012).
To determine whether spontaneous neurotransmission can serve as a broad-spectrum readout of CNT intoxication, we evaluated the effects of BoNT/A-G or TeNT on mEPSC frequencies. BoNT serotypes A–G were applied to ESN cultures at approximately 3 times their median effective concentration (EC50), as previously determined in SNARE protein cleavage assays (Hubbard et al., 2012). TeNT, which has not been previously evaluated in ESNs, was added at 20 pM based on a specific activity that is one-tenth that of BoNT/A. Whole-cell recordings collected at 20 h after toxin addition demonstrated that each CNT reduced spontaneous neurotransmission rates by over 99%, with the exception of BoNT/B and TeNT, which continued to produce mEPSCs at 6.7 ± 1.5% and 4.1 ± 1.1% of control frequencies, respectively (Fig. 2A and B; n = 8–12 for each CNT). Immunoblot analysis of whole-cell lysates harvested from parallel cultures revealed proteolytic conversion of the majority of SNAP-25 to the cleaved form (cSNAP-25) in cultures treated with BoNT serotypes A, C, or E; loss of Stx1 immunoreactivity in cultures treated with BoNT/C; and loss of Syb2 immunoreactivity in cultures treated with TeNT or BoNT serotypes B, D, F, or G (Fig. 2C and D). Intoxicated neurons did not undergo acute cytotoxicity and had unchanged resting membrane potentials compared to controls (Supplementary Fig. 1), consistent with previous studies demonstrating that CNT-induced reductions in spontaneous neurotransmission were not attributable to cell death (Beske et al., 2015).
FIG. 2.

BoNT/A-G or TeNT intoxication blocks synaptic neurotransmission in ESNs. Treatment with BoNT/A-G or TeNT reduces or eliminates spontaneous release after 20 h, demonstrated in (A) representative whole-cell recordings and (B) mean normalized mEPSC frequencies. Scale bar = 2.5 s × 60 pA (n = 8–16 each). ***P < 0.001. (C) Representative immunoblot demonstrating cleavage of SNAP-25 (S25) and loss of immunoreactivity of Stx1 in cultures treated with BoNT/A, /C or /E (n ≥ 4). (D) Representative immunoblot demonstrating loss of Syb2 immunoreactivity following treatment with BoNT/B, /D, /F, /G, and TeNT (n ≥ 4).
Concentration-Response Analysis of Spontaneous Release Frequencies and SNARE Protein Cleavage in ESNs Exposed to 3 Clinically Relevant CNTs
The above data suggested that quantal analysis offers a broad-spectrum method to measure the effects of intoxication on synaptic function. While this approach has the potential to serve as a highly relevant platform for phenotypic screening of countermeasures, it could also be used to reduce animal use in testing of pharmaceutical preparations and forensic characterization of toxin. Thus we focused on the potential use of synaptic neurotransmission to detect and characterize 3 clinically important CNTs: BoNT/A, BoNT/B, and TeNT. BoNT/A and BoNT/B were responsible for 95% of clinical cases of botulism in the United States from 2004-2014 (Centers for Disease Control, 2015), while TeNT had the most significant effect of all CNTs on global health, with an estimated 58,900 fatalities in 2013 alone (Mortality and Causes of Death, 2015).
ESNs exposed to BoNT/A, BoNT/B, or TeNT exhibited concentration-dependent reductions in mean mEPSC frequencies (Fig. 3A–C) and cleavage of cognate SNARE proteins (Fig. 3D–F). Concentration-response curves were used to estimate toxin concentrations that caused proteolysis of 50% of cognate SNARE proteins (EC50 values; Fig. 3G–I) or 50% reduction in spontaneous release (IC50 values; Fig. 3J). Comparison of the EC50 and IC50 values indicate that spontaneous neurotransmission is disproportionately sensitive to SNARE protein cleavage, with reductions in mEPSC frequencies apparent at concentrations that did not produce significant SNARE protein cleavage (Fig. 3J).
FIG. 3.

Concentration–response comparisons of synaptic neurotransmission and SNARE protein cleavage in ESNs exposed to BoNT/A, BoNT/B or TeNT. Representative whole-cell recordings (A–C) and immunoblots (D–F) demonstrating impaired synaptic neurotransmission and SNARE protein cleavage following exposure to BoNT/A, BoNT/B or TeNT. (G–I) Concentration-response curves of normalized mEPSC frequencies (n = 12–20 each) and SNARE protein cleavage (n = 4–5 each) to each CNT. Scale bar = 5 s × 60 pA. Data are presented as mean ± SEM. (J) Tabular summary of median concentrations for each toxin determined from whole-cell recordings and immunoblot data.
According to the quantal theory of neurotransmitter release, a change in quantal amplitude is interpreted as a change in postsynaptic function, whereas a change in quantal frequency represents altered presynaptic neurotransmitter release (Katz, 1971). To confirm that reductions in mEPSC frequencies were due to reduced presynaptic release probabilities as opposed to attenuated postsynaptic detection, mean mEPSC amplitudes and inter-event intervals were compared between partially intoxicated and control neurons (Supplementary Fig. 2). Partially intoxicated cultures exhibited no reductions in mean mEPSC amplitudes, whereas inter-event intervals increased proportionally to toxin concentration, confirming that toxin-induced reduction of spontaneous neurotransmission is a consequence of presynaptic blockade.
Synaptic Function Assays Can Substitute for the MLA in Determining Toxin Serotype and Assessing Antitoxin Specificities
The preceding experiments suggested that spontaneous synaptic release frequencies represent a novel, broad-spectrum and sensitive method to functionally test for presynaptic blockade by CNTs. To evaluate whether synaptic activity in cell-based assays could serve as an in vitro surrogate for the MLA, we assessed the sensitivity and specificity of spontaneous release in a variety of assays representing clinical and forensic functions of the MLA.
The first 2 major uses of the MLA tested were identification of toxin serotypes and determination of antitoxin specificities. Both techniques involve pre-incubation of toxin with antitoxin antibodies prior to intraperitoneal injection. For serotype determination, serotype-specific neutralizing antibodies are given to mice in conjunction with the toxin sample (AOAC International, 2001). Mice receiving the cognate antitoxin remain asymptomatic, while mice receiving antitoxins for other serotypes develop symptoms of botulism. For antitoxin specificity studies, the MLA is used to determine the ability of antitoxin to protect mice against the lethal effects of cognate BoNT serotypes as well as to measure antitoxin potency against standardized BoNT preparations (Bowmer, 1963).
To determine whether spontaneous neurotransmission was protected from intoxication by neutralizing antitoxin, 110 fM of BoNT/A or 1 pM of BoNT/B (approximately 4 U/ml each) was pre-incubated with 7 neutralizing units/mL of rabbit anti-A IgG or anti-B IgG for 1 h and added to ESNs. Spontaneous activity was quantified in whole-cell recordings 20 h later. Each antitoxin prevented impairment of synaptic function (Fig. 4A–D) and cleavage of SNARE protein (Fig. 4E and F) by cognate BoNT serotypes. In contrast, anti-A IgG did not protect against BoNT/B intoxication and vice versa. Since one neutralizing unit is the quantity of antitoxin that protects against 1 mouse lethal unit in the MLA, frequencies of spontaneous neurotransmission can be used to determine antitoxin specificity and test protection against intoxication at antitoxin:toxin ratios that are at least 1.75:1.
FIG. 4.

Use of synaptic neurotransmission to determine toxin serotypes and evaluate antitoxin specificity. Pre-incubation of toxin with serotype-specific antitoxin protects spontaneous release frequencies and SNARE protein integrity at 20 h after intoxication. A, B, Representative whole-cell recordings. C, D, Normalized mEPSC frequencies. E, F, Representative immunoblots. Scale bar = 2.5 s × 60 pA. Normalized data are from 12 to 20 recordings and presented as mean ± SEM. ***P < 0.001.
Synaptic Function Assays Provide Ultra-Sensitive Determination of the Potency of Pharmaceutical Toxin Preparations
Another important use of the MLA is determination of toxin potency in pharmaceutical formulations. BOTOX is the most prescribed botulinum neurotoxin pharmaceutical on the market. Each BOTOX vial contains stabilizing excipients with 100 or 200 U of “complex” toxin, consisting of BoNT/A associated with naturally associated proteins that are normally produced by the bacteria and believed to facilitate transfer of the toxin from the gut to the vascular system (Gu and Jin, 2013). To determine whether the presence of pharmaceutical excipients impairs functional, cell-based detection of toxin, we evaluated synaptic activity in ESNs after addition of 0.1, 1, or 2 U/ml of BOTOX. Significant reductions in mEPSC frequencies (Fig. 5A and B) and production of the LC/A-cleaved form of SNAP-25 (cSNAP-25; Fig. 5C) were apparent 20 h after addition of either 1 or 2 U/ml, but not 0.1 U/ml.
FIG. 5.

Exposure to clinical concentrations of BOTOX block spontaneous release and produce cleaved SNAP-25 in ESNs. Representative whole-cell recordings (A), quantitation of normalized mEPSC frequencies (B) and representative immunoblot (C) demonstrating that exposure to 1 or 2 U/mL of BOTOX results in reduced neurotransmission and production of cleaved SNAP-25 after 20 h. Scale bar = 2 s × 60 pA. Normalized data are from 12 to 20 recordings and presented as mean ± SEM. ** P < 0.01, *** P < 0.001.
Potencies of pharmaceutical preparations are rigorously benchmarked using the MLA. Since a 25 g mouse has a serum volume of approximately 1 ml, a median lethal dose of BOTOX therefore corresponds to serum concentrations of 1 U/ml BoNT/A, allowing a direct correspondence between the MLA and cell-based assays. Treatment of ESNs with 1 U/ml of BOTOX resulted in a significant reduction in spontaneous release, indicating that quantal analysis is approximately as sensitive as the MLA in detecting the presence of BoNT/A. Notably, the calculated IC50 value for BoNT/A in concentration-response studies was 55 fM (Fig. 3J), which corresponds to approximately 2 U/ml of toxin based on its reported specific activity. Thus purified dichain BoNT/A and BOTOX mutually corroborate that quantal release is as sensitive as the MLA in detecting the presence of active toxin, with a limit of detection that is at least 1 U/ml. Furthermore, these experiments were completed within 24 h of intoxication, whereas 48-96 h are required to detect equivalent doses in the MLA. These data suggest that quantal analysis offers an equally sensitive and more rapid alternative to the MLA for quantifying physiological concentrations of active toxin in clinically reconstituted drug products, without interference from excipients.
Detection of BoNT/A Activity in Clinically Important Complex Matrices Using Synaptic Function Assays
The final important function of the MLA is direct detection of toxin in CMs, such as food products or human clinical samples. Unlike the defined formulations of pharmaceutical preparations, the compositions of nutritive and forensic samples are highly unpredictable and may include bioactive components that confound cell-based or biochemical assays. To mitigate this problem, toxin is often purified from complex samples prior to analysis or produced from isolated bacteria and then tested (Kalb et al., 2015). Here we were interested in determining whether complex toxin samples could be directly analyzed in cell-based assays, which would be more rapid and cost-effective than testing of purified analytes or re-expressed toxin, or alternatively whether a small degree of contamination during the purification process might interfere with toxin detection in cultured neurons.
In preliminary feasibility studies, neat samples of centrifugation-clarified milk, green bean juice, or non-alcoholic apple cider were added to ESN cultures at a final concentration of 1%. In all cases, cells were visibly degenerated after 20 h and could not be patch-clamped (data not shown), suggesting that each sample contained acutely cytotoxic components. One well-described mechanism of acute neurotoxicity is excitogenic Ca2+ signaling from overstimulated NMDA receptors (NMDARs) (Aarts and Tymianski, 2004). We have previously reported that ESNs are highly sensitive to glutamate excitotoxicity, with a median cytotoxic concentration of 0.44 µM glutamate (Gut et al., 2013). Based on published values, cell culture media supplemented with 1% milk or non-alcoholic cider contain 0.68 µM or 2.73 µM free glutamate, respectively (Giacometti, 1979; Riekstina-Dolge et al., 2014; Ventura et al., 2012). To test whether free glutamate was responsible for the acute loss of cell viability, the irreversible NMDAR antagonist MK-801 (10 µM) was administered to ESNs immediately prior to addition of each CM. MK-801 protected ESNs from acute toxicity without altering mEPSC frequencies (Supplemental Fig. 3A) and co-administration of MK-801 and CMs had no effect on mEPSC frequencies compared to control cultures, confirming that MK-801 preserved cell viability and protected synaptic function from CM-induced cytotoxicity without interfering with detection of spontaneous events (Supplemental Fig. 3B). Consequently MK-801 was co-administered in all subsequent studies.
ESNs were then treated with a final concentration of 1% CMs and 55 fM BoNT/A in the presence of MK-801 and excitatory minis were quantified after 20 h in comparison to controls. The presence of nutritive samples did not alter the effects of BoNT/A on synaptic neurotransmission (Fig. 6A–D) or cSNAP-25 production (Fig. 6E). Thus by blocking NMDAR function, spontaneous release in neuronal cultures can be used to directly evaluate 55 fM BoNT/A in the presence of 1% nutritive samples without requiring additional purification.
FIG. 6.

Detection of BoNT/A prepared in nutritive samples using synaptic activity. Representative whole-cell recordings and normalized mEPSC frequencies demonstrating reduced synaptic transmission and cSNAP-25 production at 20 h after treatment of ESNs with final concentrations of 55 fM BoNT/A and 1% of CMs. (A) Vehicle, (B) whole milk, (C) green bean liquid or (D) apple cider. Scale bar = 5 s × 50 pA. Normalized data are from 12 to 20 recordings and presented as mean ± SEM. *P < 0.05, ** indicates P < 0.01. (E) Representative immunoblot.
Human serum collected after the onset of clinical symptoms is often analyzed to estimate the severity of exposure and to determine toxin serotype (Sheth et al., 2008). In preliminary experiments, human serum was added to ESNs at final concentrations of 10 and 50% in the presence of MK-801, and neuronal viability and synaptic activity were assessed after 20 h. While neurons in 10% human serum retained intact processes and round soma, cultures treated with 50% serum exhibited significant evidence of neuronal degeneration, including fragmented processes and amorphous soma (not shown). Spontaneous release frequencies were not altered in neurons treated with 10% serum compared to control dishes, indicating that addition of 10% human serum to tissue culture media is well tolerated by ESNs (Supplementary Fig. 3C). ESNs were then treated with a final concentration of 10% human serum and 55 fM BoNT/A, and excitatory minis were quantified after 20 h in comparison to controls. Synaptic activity was impaired by ∼50% in cultures treated with serum and BoNT/A (Fig. 7A and B) consistent with detection of cSNAP-25 by immunoblot (Fig. 7C). Collectively these data demonstrate that detection of LC/A impairment of spontaneous release is not altered by the presence of 10% human serum. Since the median mammalian toxicity is estimated to be 27 fM in serum (corresponding to 1 U/ml in serum), these data suggest that direct detection of BoNT/A in clinical serum samples is limited to doses that exceed 10 U/ml. Plasma concentrations of BoNT/A were reported to range from 20 to 1800 U/ml when measured days after ingestion in a well-characterized mass foodborne exposure (Sheth et al., 2008), suggesting that synaptic neurotransmission can be used to directly evaluate the presence and serotype of CNTs in clinical samples collected from victims of moderate-to-severe exposures. However, at toxin doses less than 10 U/ml, toxin will have to be concentrated or purified prior to application to neuronal cultures.
FIG. 7.

Detection of BoNT/A prepared in human serum using synaptic activity. Representative whole-cell recordings (A) and normalized mEPSC frequencies (B) demonstrating reduced synaptic neurotransmission at 20 h after addition of final concentrations of 55 fM BoNT/A and 10% human serum to ESNs. Scale bar = 1 s × 60 pA. Normalized data are from 12 to 20 recordings and presented as mean ± SEM. *** indicates P < 0.001. (C) Representative immunoblot demonstrating production of cSNAP-25 for above treatments.
DISCUSSION
In this study, we demonstrate that spontaneous synaptic transmission in networked neuron cultures can serve as a physiologically relevant, cell-based model of neuromuscular junction (NMJ) intoxication. Toxin-induced blockade of synaptic release represents a physiologically relevant phenotypic readout that corresponds to the functional endpoint of NMJ paralysis in vivo. While blockade of miniature excitatory end-plate potentials in rodent muscle preparations (Kim et al., 1984) and human extensor digitorum brevis muscles (Van Putten et al., 2002) treated with BoNT/A provides physiological support for the use of spontaneous synaptic activity as a functional readout of intoxication, the work described here represents the first characterization of the effects of CNTs on synaptic neurotransmission in cultured neurons.
We previously demonstrated that BoNT/A blocks spontaneous neurotransmission as well as network-level behaviors in CNS neurons (Beske et al., 2015). In this study, we focused on spontaneous neurotransmission as a readout of intoxication. Spontaneous neurotransmission is caused by exocytosis of neurotransmitter quanta independent of an action potential, presumably due to stochastic fluctuations in active zone Ca2+ levels (Fatt and Katz, 1952). Unlike the cooperative and thresholded nature of evoked synaptic release, spontaneous neurotransmission is discrete and stochastic, such that changes in postsynaptic detection of spontaneous events are considered indicative of altered presynaptic release probabilities (Wyllie et al., 1994). Spontaneous release is a salient feature of all synapses, and the large number of afferent synapses per neuron (102–104) results in spontaneous postsynaptic events with aggregate frequencies that typically range from 0.1 to 10 Hz (Kaeser and Regehr, 2014). Thus, whole-cell recordings from a single neuron allow simultaneous evaluation of release probabilities from hundreds to thousands of nerve terminals with single-synapse resolution.
All neuron cultures underwent synaptic blockade following intoxication by CNTs, suggesting that the effects of CNTs on neurotransmission are generalizable to neurons from diverse origins and species. This finding dramatically expands the availability of neuron sources for CNT studies and reduces the motivations for use of non-neurotypic models (Eubanks et al., 2007; Larsen, 2009). In particular, researchers will have the flexibility to select neuron platforms that are optimized for their particular objectives. For example, ESNs of defined lineages can be produced in vast quantities with high lot-to-lot consistency, and therefore may be well-suited for therapeutic screening or diagnostic testing (Hubbard et al., 2012). However, widespread use of ESNs is limited by the expertise needed for embryonic stem cell culture and neuronal differentiation. In contrast, while primary neuron harvest is less scalable, primary culture is a widely used technique, and cultures of defined composition are routinely obtained by microdissection of neonatal brain tissues. While hSNs have the potential to offer direct translational relevance, onset of synaptic activity occurs over months (Bradford and McNutt, 2015). Consequently, there will be limited utility for hSNs in function-based toxin studies until reproducible methods are available to rapidly produce cultures with robust levels of synaptic activity.
Glutamatergic neurons comprise about 80% of CNS neurons and are, in many preparations, the dominant neuron population in primary cultures. For this reason, we focused on AMPA receptor-mediated synaptic neurotransmission. However, the development of protocols to derive synaptically active neurons of defined lineages from stem cells raises the possibility of studying intoxication in derived neuron cultures that are predominantly non-glutamatergic (Bradford and McNutt, 2015; Pellett, 2013). In principle the method described here can be extended to other neuron subtypes, as long as they exhibit synaptic activity and produce postsynaptic currents that are mediated by ionotropic receptors. For example, in vitro-derived NMJs would be an ideal model for CNT detection and therapeutic testing. However, while the in vitro production of NMJs comprising derived cholinergic neurons and muscle fibers has been reported, there is currently no scalable method to generate large numbers for diagnostic or therapeutic testing (Toma et al., 2015).
Although functional detection of reduced synaptic activity in CNT-treated neurons is a definitive readout of intoxication, patch-clamp electrophysiology is a low-throughput and specialized technique that may not be routinely available to all laboratories. However, once culture methods have been demonstrated to produce neurons with active synapses, SNARE protein cleavage assays can subsequently be used at corresponding time points as a more accessible method of approximating synaptic intoxication, albeit with reduced sensitivity and specificity. Culture methods that facilitate the development of synaptic activity have been well-described for various primary neuron populations and, more recently, for ESNs (Beaudoin et al., 2012; Bilimoria and Bonni, 2008; Hubbard et al., 2015), and therefore a transition to use of synaptically active neuron populations should be relatively simple for most researchers.
Phenotypic detection of synaptic responses to CNTs has distinct advantages over SNARE protein cleavage assays that extend beyond improved specificity and sensitivity. For example, the use of phenotypic assays promotes the selection of cell models with functioning synaptic junctions, increasing the probability of physiologically relevant host:toxin interactions. Second, all biological steps of intoxication are required for impaired neurotransmission, thereby comprising a testing platform that is amenable to a broad range of therapeutic interventions. Third, this approach allows for the development of novel methods to exploit synapse- or network-level behaviors as readouts of intoxication, including live imaging of Ca2+ oscillations (Li et al., 2014), extracellular electrophysiological recordings using planar multielectrode arrays (Pancrazio et al., 2014) and evoked neurotransmission via localized field stimulation (Maximov et al., 2007) or at autaptic synapses (Bekkers and Stevens, 1991). Many of these methods are non-destructive and therefore will facilitate longitudinal studies testing for the functional reversal of paralysis.
Mechanisms of neurotransmission are highly conserved among all synapse types (Sollner et al., 1993), supporting the hypothesis that central synapses are a relevant model for studying CNTs. Here we found that CNTs evoked SNARE protein cleavage and reduced postsynaptic detection of neurotransmission in CNS neurons at physiological concentrations. Other distinctive host:toxin behaviors observed in vivo have also been reported in CNS neuron cultures, including depolarization-enhanced toxin uptake, serotype-dependent persistences and serotype-dependent potencies (Foran et al., 2003; Hubbard et al., 2012; Keller et al., 1999). Collectively these data suggest that toxin:host interactions are conserved between central and peripheral synapses, supporting the argument that intoxication of central synapses is a relevant surrogate for NMJ paralysis. Nonetheless, there are structural differences between peripheral and central synapses that could potentially interfere with translational studies, such as overall synapse size, number of release sites per synapse and differences in SNARE protein isoforms (Tarr et al., 2013).
Our data corroborate in vivo findings that synaptic release is disproportionately sensitive to SNARE protein cleavage (Kalandakanond and Coffield, 2001; Keller, et al., 1999; Raciborska et al., 1998). In the case of SNAP-25, this can be explained by the fact that SNAP-25 is distributed along the axolemma and does not accumulate in the presynaptic compartment (Tao-Cheng et al., 2000). Since the collective active zone area comprises a trivial fraction of the axonal membrane (Schikorski and Stevens, 1997), the sensitivity of synaptic release to SNAP-25 cleavage would therefore be a consequence of the specific cleavage of the small but functionally important pool of active zone SNAP-25 that is actively engaged in vesicle fusion and neurotransmitter release. Alternatively, Syb1/2 is predominantly localized to several populations of synaptic vesicles, each of which is replenished by recycling endosomes at different rates and therefore may have varying likelihoods of encountering LC after intoxication (Guo et al., 2015; Watanabe et al., 2013). Elucidating the mechanistic basis for the increased sensitivity of synaptic release over Syb cleavage will require improved understanding of the trafficking of LC and synaptic vesicles within the nerve terminal.
Millions of mice are used annually to detect or quantify CNTs in a variety of contexts, including pharmaceutical preparations, nutritive samples and clinical samples. Since blockade of synaptic release requires the successful negotiation of toxin uptake and processing, quantal analysis implicitly tests for physiologically appropriate host:toxin interactions and reproduces the physiological endpoint of intoxication, thereby replicating the principal advantages of the MLA. Based on these findings, we propose that a tiered approach using preliminary studies conducted in vitro with subsequent validation in the MLA would significantly reduce the number of mice required per experimental or clinical sample. Such an approach would be especially important for determining potencies of toxin formulations and antitoxin preparations. Thus, quantitation of neurotransmission in networked neuron cultures has the potential to dramatically reduce the need for distressful and terminal animal studies while providing relevance and utility comparable to the MLA.
In conclusion, we have found that electrophysiological measures of synaptic activity provide a robust and physiologically relevant readout of CNT intoxication in diverse cultures of synaptically active CNS neurons. Analysis of synaptic neurotransmission directly evaluates functional endpoints of intoxication, thereby implicitly testing for successful negotiation of all stages of toxin uptake and processing. Quantitation of spontaneous release frequencies was found to match or exceed all current functions of the MLA, with improved speed and similar sensitivities. Our data suggest that diverse populations of synaptically active neurons are suitable for cell-based studies, significantly increasing the ability of researchers to optimize assays for particular research objectives. Collectively, these data suggest that quantitation of synaptic activity in networked neuron cultures has the potential to dramatically reduce terminal animal use in diagnostic and forensic testing and accelerate therapeutic countermeasure screening.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
This work was conducted under funding from the Defense Threat Reduction Agency – Joint Science and Technology Office, Medical S&T Division (grant number CBM.THRTOX.01.10.RC.021); National Institute of Allergy and Infectious Diseases (IAA AOD12058-0001-0000 and R01 5R01AI093504). This research was performed while P.B. held a Defense Threat Reduction Agency-National Research Council Research Associateship award; K.H. held a National Research Council Research Associateship award; and J.O., A.B., E.G., K.M., and K.T. held Oak Ridge Institute of Science and Engineering Fellowship awards.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Megan Lyman, Angela Adkins, Chelsea Andres, and Marian Nelson for technical assistance; and Cindy Kronman for editorial and administrative assistance. The views expressed in this article are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government.
REFERENCES
- Aarts M. M., Tymianski M. (2004). Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr. Mol. Med. 4, 137–147. [DOI] [PubMed] [Google Scholar]
- AOAC International (2001). AOAC official method 977.26 (sec. 17.7.01). Clostridium botulinum and its toxins in foods. Off. Methods Anal. [Google Scholar]
- Beaudoin G. M., 3rd, Lee S. H., Singh D., Yuan Y., Ng Y. G., Reichardt L. F., Arikkath J. (2012). Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 7, 1741–1754. [DOI] [PubMed] [Google Scholar]
- Bekkers J. M., Stevens C. F. (1991). Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc. Natl. Acad. Sci. 88, 7834–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beske P. H., Scheeler S., Adler M., McNutt P. M. (2015). Accelerated intoxication of GABAergic synapses by botulinum neurotoxin A disinhibits stem cell-derived neuron networks prior to network silencing. Front. Cell. Neurosci. 9, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilimoria P. M., Bonni A. (2008). Cultures of cerebellar granule neurons. Cold Spring Harbor Protoc. 2008, prot5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowmer E. J. (1963). Preparation and assay of the international standards for Clostridium Botulinum types a, B, C, D and E antitoxins. Bull. World Health Organ. 29, 701–709. [PMC free article] [PubMed] [Google Scholar]
- Bradford A. B., McNutt P. M. (2015). Importance of being Nernst: Synaptic activity and functional relevance in stem cell-derived neurons. World J. Stem Cells 7, 899–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brin M., Aoki K. (2002). Botulinum toxin type A: Pharmacology. In Spasticity: Etiology, evaluation, management and the role of botulinum toxin. (Simpson D., Mayer N., Eds.), pp. 100–109. We Move Publications, New York, NY. [Google Scholar]
- Centers for Disease Control (2015). National Botulism Surveillance, Annual Summaries. Available at: http://www.cdc.gov/nationalsurveillance/botulism-surveillance.html doi.
- Chalifoux J. R., Carter A. G. (2011). GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 21, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubanks L. M., Hixon M. S., Jin W., Hong S., Clancy C. M., Tepp W. H., Baldwin M. R., Malizio C. J., Goodnough M. C., Barbieri J. T., et al. (2007). An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc. Natl. Acad. Sci. 104, 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P., Katz B. (1952). Spontaneous subthreshold activity at motor nerve endings. J. Physiol. 117, 109–128. [PMC free article] [PubMed] [Google Scholar]
- Foran P. G., Mohammed N., Lisk G. O., Nagwaney S., Lawrence G. W., Johnson E., Smith L., Aoki K. R., Dolly J. O. (2003). Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 278, 1363–1671. [DOI] [PubMed] [Google Scholar]
- Giacometti T. (1979). Free and bound glutamate in natural products. In Glutamic Acid: Advances in Biochemistry and Physiology (Filer L., Garattini S., Kare M., Reynolds W., Wurtman R., Eds.), pp. 25–34. Raven Press, New York, NY. [Google Scholar]
- Gu S., Jin R. (2013). Assembly and function of the botulinum neurotoxin progenitor complex. Curr. Topics Microbiol. Immunol. 364, 21–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Ge J. L., Hao M., Sun Z. C., Wu X. S., Zhu J. B., Wang W., Yao P. T., Lin W., Xue L. (2015). A three-pool model dissecting readily releasable pool replenishment at the calyx of held. Scientific Reports 5, 9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut I. M., Beske P. H., Hubbard K. S., Lyman M. E., Hamilton T. A., McNutt P. M. (2013). Novel application of stem cell-derived neurons to evaluate the time- and dose-dependent progression of excitotoxic injury. PloS One 8, e64423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981). Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Hubbard K., Beske P., Lyman M., McNutt P. (2015). Functional evaluation of biological neurotoxins in networked cultures of stem cell-derived central nervous system neurons. J. Visual Exp. 96, e52361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard K. S., Gut I. M., Lyman M. E., Tuznik K. M., Mesngon M. T., McNutt P. M. (2012). High yield derivation of enriched glutamatergic neurons from suspension-cultured mouse ESCs for neurotoxicology research. BMC Neurosci. 13, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A., France R. M., Riccalton-Banks L., McLaren J., Cox H., Quirk R. A., Shakesheff K. M., Thompson D., Panjwani N., Shipley S., et al. (2008). The Intercostal NMJ Assay: a new alternative to the conventional LD50 assay for the determination of the therapeutic potency of botulinum toxin preparations. Alternat. Lab. Anim. 36, 141–152. [DOI] [PubMed] [Google Scholar]
- Kaeser P. S., Regehr W. G. (2014). Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu. Rev. Physiol. 76, 333–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalandakanond S., Coffield J. A. (2001). Cleavage of SNAP-25 by botulinum toxin type A requires receptor-mediated endocytosis, pH-dependent translocation, and zinc. J. Pharmacol. Exp. Therap. 296, 980–986. [PubMed] [Google Scholar]
- Kalb S. R., Baudys J., Wang D., Barr J. R. (2015). Recommended mass spectrometry-based strategies to identify botulinum neurotoxin-containing samples. Toxins (Basel) 7, 1765–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. (1971). Quantal mechanism of neural transmitter release. Science 173, 123–126. [DOI] [PubMed] [Google Scholar]
- Keller J. E., Neale E. A., Oyler G., Adler M. (1999). Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 456, 137–142. [DOI] [PubMed] [Google Scholar]
- Kim Y. I., Lomo T., Lupa M. T., Thesleff S. (1984). Miniature end-plate potentials in rat skeletal muscle poisoned with botulinum toxin. J. Physiol. 356, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J. C. (2009). US Army botulinum neurotoxin (BoNT) medical therapeutics research program: past accomplishments and future directions. Drug Dev. Res. 70, 266–278. [Google Scholar]
- Li W., Xu Z., Huang J., Lin X., Luo R., Chen C. H., Shi P. (2014). NeuroArray: a universal interface for patterning and interrogating neural circuitry with single cell resolution. Scientific Reports 4, 4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A., Pang Z. P., Tervo D. G., Sudhof T. C. (2007). Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J. Neurosci. Methods 161, 75–87. [DOI] [PubMed] [Google Scholar]
- Montal M. (2010). Botulinum neurotoxin: a marvel of protein design. Annu. Rev. Biochem. 79, 591–617. [DOI] [PubMed] [Google Scholar]
- Mortality and Causes of Death. (2015). Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385, 117–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Rossant J., Nagy R., Abramow-Newerly W., Roder J. C. (1993). Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc .Natl. Acad. Sci. 90, 8424–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancrazio J. J., Gopal K., Keefer E. W., Gross G. W. (2014). Botulinum toxin suppression of CNS network activity in vitro. J. Toxicol. 2014, 732913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce L. B., Borodic G. E., First E. R., MacCallum R. D. (1994). Measurement of botulinum toxin activity: evaluation of the lethality assay. Toxicol. Appl. Pharmacol. 128, 69–77. [DOI] [PubMed] [Google Scholar]
- Pellett S. (2013). Progress in cell based assays for botulinum neurotoxin detection. Curr. Topics Microbiol. Immunol. 364, 257–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raciborska D. A., Trimble W. S., Charlton M. P. (1998). Presynaptic protein interactions in vivo: evidence from botulinum A, C, D and E action at frog neuromuscular junction. Eur. J. Neurosci. 10, 2617–2628. [DOI] [PubMed] [Google Scholar]
- Riekstina-Dolge R., Kruma Z., Dimins F., Straumite E., Karklina D. (2014). Phenolic composition and sensory properties of ciders produced from Latvian apples. Proc. Latvia Univ. Agric. 31, 39–45. [Google Scholar]
- Schiavo G., Rossetto O., Montecucco C. (1994). Clostridial neurotoxins as tools to investigate the molecular events of neurotransmitter release. Semin. Cell Biol. 5, 221–229. [DOI] [PubMed] [Google Scholar]
- Schikorski T., Stevens C. F. (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 17, 5858–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth A. N., Wiersma P., Atrubin D., Dubey V., Zink D., Skinner G., Doerr F., Juliao P., Gonzalez G., Burnett C., et al. (2008). International outbreak of severe botulism with prolonged toxemia caused by commercial carrot juice. Clin. Infect. Dis. 47, 1245–1251. [DOI] [PubMed] [Google Scholar]
- Simpson L. L. (2004). Identification of the major steps in botulinum toxin action. Annu. Rev. Pharmacol. Toxicol. 44, 167–193. [DOI] [PubMed] [Google Scholar]
- Smith T. J., Roxas-Duncan V. I., Smith L. A. (2012). Botulinum neurotoxins as biothreat agents. J. Bioterrorism Biodefense S2, 003. [Google Scholar]
- Sollner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J. E. (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. [DOI] [PubMed] [Google Scholar]
- Sudhof T. C., Rizo J. (2011). Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng J. H., Du J., McBain C. J. (2000). SNAP-25 is polarized to axons and abundant along the axolemma: an immunogold study of intact neurons. J. Neurocytol. 29, 67–77. [DOI] [PubMed] [Google Scholar]
- Tarr T. B., Dittrich M., Meriney S. D. (2013). Are unreliable release mechanisms conserved from NMJ to CNS? Trends Neurosci. 36, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma J. S., Shettar B. C., Chipman P. H., Pinto D. M., Borowska J. P., Ichida J. K., Fawcett J. P., Zhang Y., Eggan K., Rafuse V. F. (2015). Motoneurons derived from induced pluripotent stem cells develop mature phenotypes typical of endogenous spinal motoneurons. J. Neurosci. 35, 1291–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Putten M. J., Padberg M., Tavy D. L. (2002). In vivo analysis of end-plate noise of human extensor digitorum brevis muscle after intramuscularly injected botulinum toxin type A. Muscle Nerv. 26, 784–790. [DOI] [PubMed] [Google Scholar]
- Ventura A. K., Beauchamp G. K., Mennella J. A. (2012). Infant regulation of intake: the effect of free glutamate content in infant formulas. Am. J. Clin. Nutr. 95, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Rost B. R., Camacho-Perez M., Davis M. W., Sohl-Kielczynski B., Rosenmund C., Jorgensen E. M. (2013). Ultrafast endocytosis at mouse hippocampal synapses. Nature 504, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie D. J., Manabe T., Nicoll R. A. (1994). A rise in postsynaptic Ca2 + potentiates miniature excitatory postsynaptic currents and AMPA responses in hippocampal neurons. Neuron 12, 127–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.