

Abstract
BACKGROUND
The kidney plays an important role in regulating blood pressure (BP). cPLA2α in the kidney is activated by various agents including angiotensin II (Ang II) and selectively releases arachidonic acid (AA) from tissue lipids, generating pro- and antihypertensive eicosanoids. Since activation of cPLA2α is the rate-limiting step in AA release, this study was conducted to determine its contribution to renal dysfunction and end-organ damage associated with Ang II-induced hypertension.
METHODS
cPLA2α+/+ and cPLA2α−/− mice were infused with Ang II (700ng/ kg/min) or its vehicle for 13 days. Mice were placed in metabolic cages to monitor their food and water intake, and urine was collected and its volume was measured. Doppler imaging was performed to assess renal hemodynamics. On the 13th day of Ang II infusion, mice were sacrificed and their tissues and blood collected for further analysis.
RESULTS
Ang II increased renal vascular resistance, water intake, and urine output and Na+ excretion, decreased urine osmolality, and produced proteinuria in cPLA2α+/+ mice. Ang II also caused accumulation of F4/80+ macrophages and CD3+ T cells and renal fibrosis, and increased oxidative stress in the kidneys of cPLA2α+/+ mice. All these effects of Ang II were minimized in cPLA2α−/− mice.
CONCLUSION
cPLA2α contributes to renal dysfunction, inflammation, and end-organ damage, most likely via the action of pro-hypertensive eicosanoids and increased oxidative stress associated with Ang II-induced hypertension. Thus, cPLA2α could serve as a potential therapeutic target for treating renal dysfunction and end-organ damage in hypertension.
Keywords: angiotensin II, blood pressure, cPLA2α−/− mice, fibrosis; hypertension, inflammation, oxidative stress, proteinuria, renal hemodynamics.
Kidney plays an important role in regulating blood pressure (BP) by maintaining Na+ and extracellular fluid homeostasis. Alteration in kidney function promotes the development of hypertension and end-stage organ damage.1 The renin-angiotensin system contributes to the regulation of Na+ and extracellular fluid homeostasis and long-term maintenance of arterial BP via generation of angiotensin II (Ang II).1,2 Kidney also contains cPLA2 3 that releases arachidonic acid (AA) from tissue lipids4 in response to various agents including Ang II.5 AA is metabolized via cyclooxygenases (COXs) into prostaglandins (PG) E2, F2α, D2, I2, and thromboxane (Tx) A26, by lipoxygenases into leukotrienes and 5-, 12-, and 15-hydroxyeicosatrienoic acids (HETE),7 by cytochrome P450 4A through ω-hydroxylase into 20-HETE, and by epoxygenase into epoxyeicosatrienoic acids.8–10 These eicosanoids produce diverse actions and contribute to renal function as well as to the pathogenesis of renal dysfunction in various kidney diseases.11 Many of the eicosanoids generated from AA modulate and/or mediate one or more biological actions of Ang II.12 PGE2, PGI2, and epoxyeicosatrienoic acids decrease vascular tone and/or promote salt/water excretion and minimize vascular actions of Ang II,6,8–13 thus contributing to an antihypertensive mechanism. The vasodepressor effects of PGE2 are mediated via prostaglandin E (EP) 2 and EP4 receptors.14 However, PGE2 via EP1 and EP3 receptors and TxA2 via Thromboxane (TP) receptor produce vasopressor effects and contribute to Ang II-induced hypertension.14–16 12- and 20-HETE also mediate vascular effects of Ang II and contribute to its pro-hypertensive mechanism,17,18 and by increasing salt and water excretion via their tubular effects contributes to antihypertensive mechanisms.10 Cytochrome P450 1B1, expressed in cardiovascular and renal tissues, can metabolize various substrates including AA and contributes to renal dysfunction associated with Ang II-induced hypertension by generating reactive oxygen species (ROS) in male mice.19 Therefore, the release of AA by cPLA2 from tissue lipids, the rate-limiting step in the biosynthesis of eicosanoids, could be a major determinant of the cardiovascular and renal actions of Ang II. Among six cPLA2 isoforms, cPLA2α has been well studied and plays a major role in AA release. Recently, we showed that cPLA2α is critical for the development of Ang II-induced hypertension and associated cardiovascular pathogenesis.20 The present study was conducted to test the hypothesis that cPLA2α is also crucial for renal dysfunction, end-organ damage, and inflammation. The results of our study show that cPLA2α gene disruption in male mice ameliorates renal dysfunction, end-organ damage, and inflammation associated with Ang II-induced hypertension most likely due to diminished production of pro-hypertensive eicosanoids and lipid peroxides.
METHODS
The detailed experimental methods are described in the Supplementary Data.
RESULTS
cPLA2α gene disruption prevents Ang II-induced increase in cPLA2 activity and renal vascular resistance
cPLA2α gene disruption selectively prevents its expression without altering the expression of other related lipases.20 Ang II infusion increased renal cPLA2 activity as measured by its phosphorylation without altering its expression in cPLA2α+/+ mice. As expected, protein expression of cPLA2 was not observed in the cPLA2α−/− mice (Supplementary Figure S1). Recently we reported that Ang II increases BP and associated cardiac dysfunction in cPLA2α +/+ but not cPLA2α−/− mice.20 In these mice the effect of Ang II on renal hemodynamics was determined by pulse-wave Doppler method. A representative ultrasound B mode image of the kidney in transverse view is shown in Figure 1A, color Doppler to visualize blood flow in Figure 1B, and pulse-wave Doppler mode in Figure 1C. Renal artery resistive and pulsatility index were calculated as a measure of resistance and variability of blood velocity in the renal artery. Ang II infusion for 13 days increased renal vascular resistance (Figure 1D) and pulsatility index (Figure 1E) in cPLA2α+/+ but not in cPLA2α−/− mice.
Figure 1.

Angiotensin II (Ang II) infusion alters renal hemodynamics in cPLA2α+/+, But Not cPLA2α−/−, Mice. Representative ultrasound B mode image of the kidney in transverse view (A), color Doppler to visualize blood flow (B), pulse-wave Doppler mode (C), renal artery resistive index (D), and renal artery pulsitality index (E).*P < 0.05 cPLA2α+/+ vehicle vs. cPLA2α+/+ Ang II, # P < 0.05 cPLA2α+/+ Ang II vs. cPLA2α−/− Ang II (n = 3–6 for each group, data are expressed as mean ± SEM).
cPLA2α gene disruption minimizes Ang II-induced renal dysfunction
Water intake and urine output were not different between cPLA2α+/+ and cPLA2α−/− mice. Infusion of Ang II for 13 days increased water intake (Figure 2A) and urine output (Figure 2B), decreased urine osmolality (Figure 2C), did not alter plasma creatinine levels, an index of glomerular filtration rate (Figure 2D), and increased urinary Na+ excretion(Figure 2E) and caused proteinuria (Figure 2F) in cPLA2α+/+ mice; these changes were minimized in cPLA2α−/− mice except that the plasma creatinine level was not different from that observed in cPLA2α+/+ mice (Figure 2E).
Figure 2.

cPLA2α gene disruption minimizes angiotensin II (Ang II)-induced renal dysfunction. Water intake (A), urine output (B), urine osmolality (C), plasma creatinine (D), Na+ excretion (E), and protein excretion in the urine (F) were measured on day 13 of Ang II infusion, *P < 0.05 cPLA2α+/+ vehicle vs. cPLA2α+/+ Ang II, # P < 0.05 cPLA2α+/+ Ang II vs. cPLA2α−/− Ang II (n = 6–8 for each group, data are expressed as mean ± SEM).
cPLA2α gene disruption prevents renal inflammation
To determine the contribution of cPLA2α to inflammation associated with Ang II-induced end-organ damage, we examined the localization of CD3+ T lymphocytes and F4/80+ macrophages in the renal tissue of cPLA2α +/+ and cPLA2α−/− mice. Ang II caused accumulation of CD3+ T cells (Figure 3A) and infiltration of F4/80+ macrophages (Figure 3B) in the glomerulus of cPLA2α+/+ but not cPLA2α−/− mice.
Figure 3.
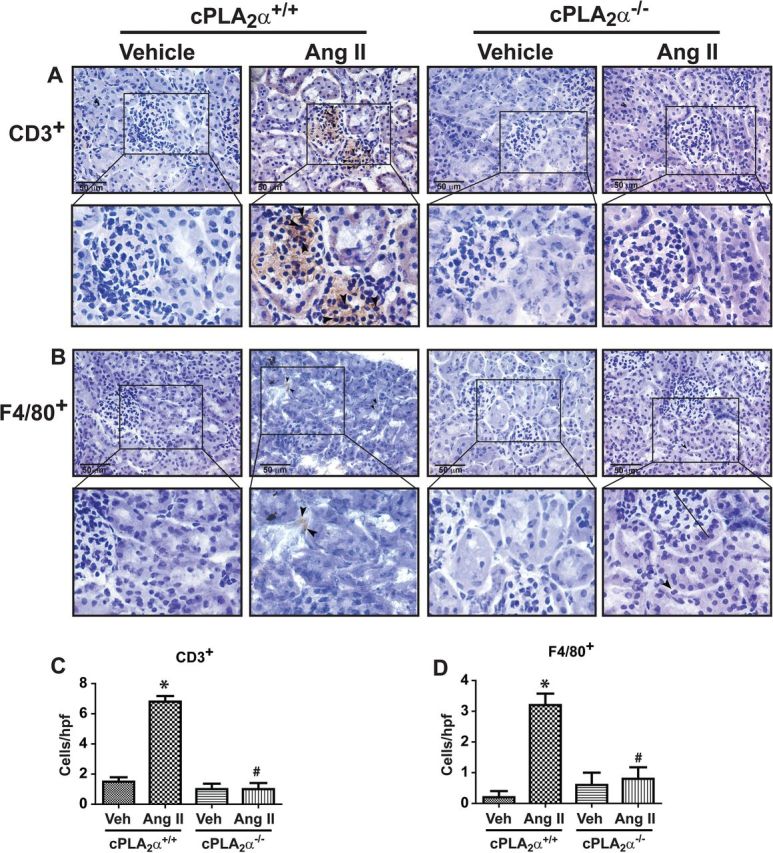
cPLA2α gene disruption prevents renal inflammation. CD3+ T cell (A) and F4/80+ macrophages (B) in glomerulus of the kidney, as determined by immunohistochemical analysis. Panels (C) and (D) represent quantified data as cells/high power field (hpf). *P < 0.05 cPLA2α+/+ vehicle vs. cPLA2α+/+ Ang II, # P < 0.05 cPLA2α+/+ Ang II vs. cPLA2α−/− Ang II (n = 4 for each group, data are expressed as mean ± SEM).
cPLA2α gene disruption prevents renal fibrosis
Increased interstitial staining of α-smooth muscle actin (Figure 4A) and transforming growth factor-β (Figure 4B), contributors of fibrosis, were observed in renal sections from Ang II-infused cPLA2α+/+, but not in cPLA2α−/− mice. Increased collagen accumulation was observed in the interstitial space in the kidney of Ang II-infused cPLA2α+/+ but not cPLA2α−/− mice (Figure 4C).
Figure 4.

cPLA2α gene disruption prevents renal fibrosis. Immunohistochemical analysis for α-smooth muscle actin (α-SMA) (A), transforming growth factor-β (B), and Masson’s trichrome staining for collagen (C) in renal sections. Panels (D), (E), and (F) represent quantified data. *P < 0.05 cPLA2α+/+ vehicle vs. cPLA2α+/+ Ang II, # P < 0.05 cPLA2α+/+ Ang II vs. cPLA2α−/− Ang II (n = 3–5 for each group, data are expressed as mean ± SEM).
cPLA2α gene disruption protects against Ang II-induced increase in renal oxidative stress
Ang II infusion for 13 days increased NADPH oxidase activity (Figure 5A) and thiobarbituric acid reactive substances (Figure 5B), a by-product of lipid peroxidation in cPLA2α+/+ but not in cPLA2α−/− mice. These data correlated with increased superoxide production in renal sections of cPLA2α+/+ mice, as indicated by increase in 2-hydroxyethidium fluorescence intensity (Figure 5C, D). This increase was not observed in cPLA2α−/− mice infused with Ang II.
Figure 5.

cPLA2α gene disruption protects against angiotensin II (Ang II)-induced increase in renal oxidative stress. NADPH oxidase activity measured in kidney homogenate (A), urinary thiobarbituric acid reactive substances (TBARS) (B), ROS production determined by DHE staining (C) quantified as fluorescence of 2-OHE (D). *P < 0.05 cPLA2α+/+ vehicle vs. cPLA2α+/+ Ang II, # P < 0.05 cPLA2α+/+ Ang II vs. cPLA2α−/− Ang II (n = 6 for each group, data are expressed as mean ± SEM).
cPLA2α gene disruption does not alter plasma levels of endothelin
Since endothelin has been implicated in Ang II-induced hypertension and some of its renal actions,21,22 we measured the plasma levels of endothelin. Ang II infusion increased plasma levels of endothelin equally in both cPLA2α+/+ and cPLA2α−/− mice, but the increase was not statistically significant (Supplementary Figure S2).
cPLA2α gene disruption reduces mRNA expression of AT1A, AT2, and Mas (Mas 1 protooncogene, G-protein-coupled) receptors, angiotensin-converting enzyme (ACE) and ACE2 in the kidney
To determine if Ang II-induced renal dysfunction, damage, and inflammation in cPLA2α+/+ but not in cPLA2α−/− mice are due to alterations in one or more components of the renin-angiotensin system, we examined renal expression of AT1A, AT2, Mas receptors, ACE, and ACE2. In each case the basal mRNA expression was not different in the kidneys of cPLA2α+/+ and cPLA2α−/− mice. Ang II infusion increased mRNA expression of AT1AR (Supplementary Figure S3A) and AT2R (Supplementary Figure S3B) receptors in cPLA2α+/+ but not cPLA2α-−/− mice. Ang II also increased Mas receptor mRNA expression, although it did not reach statistical significance in cPLA2α+/+ mice. Mas receptor mRNA was diminished in cPLA2α−/− compared to cPLA2α+/+ mice (Supplementary Figure S3C). Expression of ACE mRNA was decreased in cPLA2α+/+ mice and was further reduced in cPLA2α−/− mice (Supplementary Figure S3D). ACE2 mRNA expression was increased in cPLA2α+/+ but not in cPLA2α−/− mice (Supplementary Figure S3E).
DISCUSSION
This study demonstrates that cPLA2α, that selectively releases AA from tissue lipids, is crucial for renal dysfunction, inflammation, and end-organ damage in Ang II-induced hypertension, most likely due to increased production and activity of predominantly pro-hypertensive eicosanoids and generation of ROS. In a previous study we showed that Ang II increased BP in cPLA2α+/+ but not cPLA2α−/− mice.20 The effect of Ang II examined in the kidneys of these mice showed increased renal cPLA2 activity without altering its expression in cPLA2α+/+ mice. This observation together with our previous demonstration that Ang II increases urinary output of AA metabolites, PGE2, TxB2, and 20-HETE in cPLA2α+/+ but not cPLA2α−/− mice,20 and that cPLA2α gene disruption also inhibits the basal, and furosemide-induced increase in urinary PGE2 excretion,3 suggest that eicosanoids generated by Ang II in the kidney are most likely due to AA release consequent to activation of cPLA2α.
It has been shown that cPLA2 gene disruption does not alter basal renal function; but it produces a concentration defect in older mice (>45 weeks).3 In the present study, cPLA2 gene disruption also did not alter basal renal function in younger mice. However, Ang II increased renal arterial resistance and pulsatility in cPLA2α+/+ but not in cPLA2α−/− mice. This observation suggests that one or more AA metabolites, with pro-hypertensive effects, mediate Ang II-induced increase in renal vascular resistance in cPLA2α+/+ mice. Supporting this view, is the report that an inhibitor of AA metabolism, 5,8,11,14-eicosatetraynoic acid, attenuates Ang II-induced renal vasoconstriction.23 PGE2 through EP1 and EP3 receptors14 and PGH2 and TxA2 via Thromboxane receptor contribute to the pressor action of Ang II.15,16 The demonstration that chronic blockade of thromboxane synthase attenuates Ang II-induced mesenteric artery vasosconstriction,24 and distruption of Thromboxane receptor attenuates Ang II- induced hypertension and cardiac hypertrophy,25 suggests that TxA2 is a significant component of pro-hypertensive eicosanoids that contribute to Ang II-induced hypertension. A recent study showed that PGE2 via activation of EP4 receptor increased the expression of (pro) renin receptor in rat renal medulla, and renin activity in medulla and urine, that partly contributes to Ang II-induced hypertension.26 Products of AA generated via lipoxygenase (12-HETE) and cytochrome P450 4A (20-HETE) also contribute to Ang II-induced renal vasoconstriction.17,18,27 The increase in pulsatility and the resistive index in renal arteries correlates with long-term progression in chronic renal failure,28 suggesting that pro-hypertensive products of AA contribute to renal dysfunction in Ang II-induced hypertension.
Ang II infusion for 13 days did not alter kidney: body weight ratio, food intake, or glomerular filtration rate as indicated by lack of change in plasma creatinine clearance but increased water intake, urine output, and Na+ excretion, and decreased urinary osmolality and caused proteinuria in cPLA2α+/+ mice. Each of these effects, however, were minimized in cPLA2α−/− mice, suggesting that AA metabolites most likely contribute to these actions of Ang II. The cPLA2α-dependent dipsogenic effect of Ang II could be mediated by the central actions of TxA2, because TxA2 receptor blockade inhibits while its activation enhances this effect of intracerebroventricularly administered Ang II.29 Ang II stimulates the production of eicosanoids with both pro- and antihypertensive actions, and the balance between their vascular and tubular actions most likely maintains renal homeostasis which can become maladaptive with persistently higher levels of Ang II.6,8–13,27,30 Therefore, it appears that renal dysfunction associated with Ang II-induced hypertension in cPLA2α+/+ mice, minimized in cPLA2α−/− mice, is primarily mediated by pro-hypertensive eicosanoids. Urinary Na+ excretion was increased by Ang II in cPLA2α+/+ mice, most likely due to pressure diuresis. cPLA2α gene disruption, however, prevented the effect of Ang II to increase urinary Na+ excretion, most likely due to decrease in BP and renal levels of natriuretic eicosanoids including PGE2, 12-HETE, 20-HETE, and 11,12-epoxyeicosatrienoic acid.9–13 Further studies on the effect of various eicosanoids on renal function in cPLA2α−/− mice would be required to address this issue. Since nonsteroidal anti-inflammatory COX-2 inhibitors or COX gene disruption produce renal dysfunction and hypertension in mice on a high salt diet due to loss of antihypertensive and renoprotective effect of PGI2,30 studies in cPLA2α−/− mice on low and high salt diets and other models of hypertension should allow further assessment of the role of eicosanoids with pro- and antihypertensive effects on renal function.
cPLA2α is also present in immune cells. AA, released by its activation, is metabolized into eicosanoids with pro- and anti-inflammatory properties and is able to generate cytokines/chemokines, which regulate both innate and adaptive immune responses.31,32 In our study, Ang II caused renal inflammation as demonstrated by increased F4/80+ macrophages and CD3+ T lymphocytes infiltration and damage as indicated by accumulation of α-smooth muscle actin+ myofibroblasts, transforming growth factor-β, and collagen in the kidneys of cPLA2α+/+ mice. The demonstration that Ang II infusion failed to produce these effects in cPLA2α−/− mice suggests that AA released by cPLA2α activation results in production of eicosanoids that stimulate infiltration of macrophages and CD3+ T lymphocytes in the kidney resulting in inflammation and renal fibrosis.
The effect of cPLA2α gene disruption to minimize Ang II-induced renal dysfunction, damage, and inflammation without altering basal renal function suggests that cPLA2α activity, and the subsequent release of AA under basal physiological conditions, is low and does not appear to generate sufficient amount of eicosanoids to affect renal function. In the present study, Ang II increased renal cPLA2 activity. Taken together with our recent finding that Ang II increases urinary excretion of AA metabolites in cPLA2α+/+ but not cPLA2α−/− mice20 suggests that the increased cPLA2 activity observed in diabetic nephropathy,33 glomerular nephritis,34 and poly cystic kidney disease35 is most likely due to associated increase in the activity of various vasoactive systems including the renin-angiotensin system.36 Increased levels of Ang II would result in increased calcium influx in various renal cell types, AA release, and generation of eicosanoids that favor renal pro-hypertensive mechanisms and contribute to renal dysfunction, inflammation, and end-organ damage.
The mechanism by which cPLA2α gene disruption minimizes Ang II-induced renal dysfunction, inflammation, and damage could be related in part to decreased BP.37 The mechanical stretch and inflammation that are associated with hypertension promote aortic stiffening via activation of p38 mitogen-activated protein kinase.38 The mechanical stretch increases Ca2+ influx via stress-operated Ca2+ channels, which is known to increase cPLA2 activity and generation of eicosanoids,39 and metabolites of AA increase p38 mitogen-activated protein kinase activity.40 Therefore, it is possible that the effect of mechanical stretch caused by high BP on cardiovascular remodeling, activation of immune cells, and end-organ damage might be mediated in part by the effect of pro-hypertensive eicosanoids. However, Ang II also produces cardiovascular and renal pathophysiological changes independent of increased BP.41 In transgenic rats carrying both human renin and angiotensinogen genes, treatment with triple therapy (hydralazine, reserpine, and hydrochlorothiazide) prevented increase in BP but not end-organ damage, inflammation, or cellular growth in the kidney.41 Therefore, the protection against Ang II-induced renal dysfunction, inflammation, and damage could also result from a pressure-independent mechanism. Endothelin has been implicated in Ang II-induced hypertension and some of its renal actions.21,22 However, in our study, endothelin does not appear to mediate cPLA2α-dependent actions of Ang II as its serum levels increased insignificantly to the same degree in both cPLA2α+/+ and cPLA2α−/− mice.
The protective effect of cPLA2α gene disruption against renal dysfunction, inflammation, and end-organ damage could be due to alterations by eicosanoids in expression of one or more Ang II receptors, Mas receptor, or ACE and ACE2. Ang II increased renal mRNA expression of AT1AR and AT2R and decreased ACE and increased ACE2 expression in cPLA2α+/+ mice. Since these effects of Ang II were minimized in cPLA2α−/− mice, they are most likely mediated by eicosanoids. The increase in expression of AT1AR receptors could contribute to the renal effects of Ang II in cPLA2α+/+ mice. The increase in mRNA expression of AT2R and ACE2 and decrease in ACE in cPLA2α+/+ mice could be a compensatory mechanism activated by Ang II. Since expression of AT2R, MAS receptor and ACE2 were reduced in the kidneys of cPLA2α−/− compared to cPLA2α+/+ mice, it appears that they are unlikely to contribute to reno-protection against the effects of Ang II in cPLA2α−/− mice.
Ang II is known to increase oxidative stress, activate immune cells that release cytokines, and promote inflammation that have been implicated in the development of hypertension and end-organ damage.42,43 Our finding that cPLA2α gene disruption prevented Ang II-induced increase in NADPH oxidase activity, generation of ROS and thiobarbituric acid reactive substances, suggests that ROS/lipid peroxides generated by renal and immune cells by AA and/or its metabolites most likely contribute to renal dysfunction, inflammation, and end-organ damage. Supporting this view are the reports that (i) monocytes/macrophages and T cells express cPLA2α44 and NADPH oxidase,45–47 (ii) AA is required for activation of NADPH oxidase and generation of ROS in monocyte/macrophages and vascular smooth muscle cells,45,46,48 and (iii) ROS and/or one or more AA metabolites stimulate release of cytokines that promote inflammation.49
In conclusion, this study demonstrates that cPLA2α is essential for the development of renal dysfunction, inflammation, and end-organ damage associated with Ang II-induced hypertension, most likely via generation of eicosanoids with pro-hypertensive effects and ROS/lipid peroxides. Therefore, cPLA2α could serve as a potential novel target for developing therapeutic agents for treating renal dysfunction and end-organ damage associated with hypertension. Moreover, the development of water-soluble selective cPLA2α inhibitors would allow further assessment of its physiological and pathophysiological significance in kidney diseases in other models of hypertension.
SUPPLEMENTARY MATERIAL
Supplementary materials are available at American Journal of Hypertension (http://ajh.oxfordjournals.org).
DISCLOSURE
The authors declared no conflict of interest.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr David Armbruster for editorial assistance. We also thank the Laboratory of Dr Gary Cline at Yale University for their assistance with measurements and analysis of sodium and creatinine (grant U24 DK059635).
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institutes, grant R01-HL-19134–40 (K.U.M.) and National Institute of Diabetes and Digestive and Kidney Diseases-R37 (DK39773) and DK072381 (J.V.B). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute.
REFERENCES
- 1. Navar LG. The kidney in blood pressure regulation and development of hypertension. Med Clin North Am 1997; 81:1165–1198. [DOI] [PubMed] [Google Scholar]
- 2. Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol 1986; 250:R960–R972. [DOI] [PubMed] [Google Scholar]
- 3. Downey P, Sapirstein A, O’Leary E, Sun TX, Brown D, Bonventre JV. Renal concentrating defect in mice lacking group IV cytosolic phospholipase A(2). Am J Physiol Renal Physiol 2001; 280:F607–F618. [DOI] [PubMed] [Google Scholar]
- 4. Lin LL, Lin AY, Knopf JL. Cytosolic phospholipase A2 is coupled to hormonally regulated release of arachidonic acid. Proc Natl Acad Sci USA 1992; 89:6147–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muthalif MM, Benter IF, Uddin MR, Harper JL, Malik KU. Signal transduction mechanisms involved in angiotensin-(1-7)-stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth muscle cells. J Pharmacol Exp Ther 1998; 284:388–398. [PubMed] [Google Scholar]
- 6. McGiff JC. Prostaglandins, prostacyclin, and thromboxanes. Annu Rev Pharmacol Toxicol 1981; 21:479–509. [DOI] [PubMed] [Google Scholar]
- 7. Kuhn H, Chaitidis P, Roffeis J, Walther M. Arachidonic acid metabolites in the cardiovascular system: the role of lipoxygenase isoforms in atherogenesis with particular emphasis on vascular remodeling. J Cardiovasc Pharmacol 2007; 50:609–620. [DOI] [PubMed] [Google Scholar]
- 8. Capdevila JH, Falck JR, Imig JD. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int 2007; 72:683–689. [DOI] [PubMed] [Google Scholar]
- 9. Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 2012; 92:101–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 2002; 82:131–185. [DOI] [PubMed] [Google Scholar]
- 11. Hao CM, Breyer MD. Roles of lipid mediators in kidney injury. Semin Nephrol 2007; 27:338–351. [DOI] [PubMed] [Google Scholar]
- 12. Nasjletti A. Arthur C. Corcoran Memorial Lecture. The role of eicosanoids in angiotensin-dependent hypertension. Hypertension 1998; 31:194–200. [DOI] [PubMed] [Google Scholar]
- 13. Raymond KH, Lifschitz MD. Effect of prostaglandins on renal salt and water excretion. Am J Med 1986; 80:22–33. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y, Guan Y, Schneider A, Brandon S, Breyer RM, Breyer MD. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension 2000; 35:1129–1134. [DOI] [PubMed] [Google Scholar]
- 15. Guan Y, Zhang Y, Wu J, Qi Z, Yang G, Dou D, Gao Y, Chen L, Zhang X, Davis LS, Wei M, Fan X, Carmosino M, Hao C, Imig JD, Breyer RM, Breyer MD. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J Clin Invest 2007; 117:2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen L, Miao Y, Zhang Y, Dou D, Liu L, Tian X, Yang G, Pu D, Zhang X, Kang J, Gao Y, Wang S, Breyer MD, Wang N, Zhu Y, Huang Y, Breyer RM, Guan Y. Inactivation of the E-prostanoid 3 receptor attenuates the angiotensin II pressor response via decreasing arterial contractility. Arterioscler Thromb Vasc Biol 2012; 32:3024–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yiu SS, Zhao X, Inscho EW, Imig JD. 12-Hydroxyeicosatetraenoic acid participates in angiotensin II afferent arteriolar vasoconstriction by activating L-type calcium channels. J Lipid Res 2003; 44:2391–2399. [DOI] [PubMed] [Google Scholar]
- 18. Alonso-Galicia M, Maier KG, Greene AS, Cowley AW, Jr, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 2002; 283:R60–R68. [DOI] [PubMed] [Google Scholar]
- 19. Jennings BL, Anderson LJ, Estes AM, Yaghini FA, Fang XR, Porter J, Gonzalez FJ, Campbell WB, Malik KU. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension 2012; 59:348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khan NS, Song CY, Jennings BL, Estes AM, Fang XR, Bonventre JV, Malik KU. Cytosolic phospholipase A2alpha is critical for angiotensin II-induced hypertension and associated cardiovascular pathophysiology. Hypertension 2015; 9:04803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajagopalan S, Laursen JB, Borthayre A, Kurz S, Keiser J, Haleen S, Giaid A, Harrison DG. Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension 1997; 30:29–34. [DOI] [PubMed] [Google Scholar]
- 22. Riggleman A, Harvey J, Baylis C. Endothelin mediates some of the renal actions of acutely administered angiotensin II. Hypertension 2001; 38:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oyekan A, Balazy M, McGiff JC. Renal oxygenases: differential contribution to vasoconstriction induced by ET-1 and ANG II. Am J Physiol 1997; 273:R293–R300. [DOI] [PubMed] [Google Scholar]
- 24. Jackson EK. Effects of thromboxane synthase inhibition on vascular responsiveness in the in vivo rat mesentery. J Clin Invest 1985; 76:2286–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Francois H, Athirakul K, Mao L, Rockman H, Coffman TM. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension 2004; 43:364–369. [DOI] [PubMed] [Google Scholar]
- 26. Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A, Yang T. Prostaglandin E-prostanoid4 receptor mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Hypertension 2014; 64:369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev 2014; 22:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petersen LJ, Petersen JR, Talleruphuus U, Ladefoged SD, Mehlsen J, Jensen HA. The pulsatility index and the resistive index in renal arteries. Associations with long-term progression in chronic renal failure. Nephrol Dial Transplant 1997; 12:1376–1380. [DOI] [PubMed] [Google Scholar]
- 29. Kitiyakara C, Welch WJ, Verbalis JG, Wilcox CS. Role of thromboxane receptors in the dipsogenic response to central angiotensin II. Am J Physiol Regul Integr Comp Physiol 2002; 282:R865–R869. [DOI] [PubMed] [Google Scholar]
- 30. Imig JD. Eicosanoid regulation of the renal vasculature. Am J Physiol Renal Physiol 2000; 279:F965–F981. [DOI] [PubMed] [Google Scholar]
- 31. Alvarez Y, Valera I, Municio C, Hugo E, Padron F, Blanco L, Rodriguez M, Fernandez N, Crespo MS. Eicosanoids in the innate immune response: TLR and non-TLR routes. Mediators Inflamm 2010; 201929:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sadik CD, Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol 2012; 91:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furuya Y, Tagami S, Hasegawa A, Ishii J, Hirokawa J, Yoshimura H, Honda T, Sakaue S, Aoki K, Murakami M, Kudo I, Kawakami Y. Increased glomerular cytosolic phospholipase A2 activity of OLETF rats with early diabetes. Exp Clin Endocrinol Diabetes 1999; 107:299–305. [DOI] [PubMed] [Google Scholar]
- 34. Cybulsky AV, Takano T, Papillon J, McTavish AJ. Complement-induced phospholipase A2 activation in experimental membranous nephropathy. Kidney Int 2000; 57:1052–1062. [DOI] [PubMed] [Google Scholar]
- 35. Aukema HM, Adolphe J, Mishra S, Jiang J, Cuozzo FP, Ogborn MR. Alterations in renal cytosolic phospholipase A2 and cyclooxygenases in polycystic kidney disease. FASEB J 2003; 17:298–300. [DOI] [PubMed] [Google Scholar]
- 36. Urushihara M, Kinoshita Y, Kondo S, Kagami S. Involvement of the intrarenal renin-angiotensin system in experimental models of glomerulonephritis. J Biomed Biotechnol 2012; 601786: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Polichnowski AJ, Cowley AW., Jr Pressure-induced renal injury in angiotensin II versus norepinephrine-induced hypertensive rats. Hypertension 2009; 54:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 2014; 114:616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alexander LD, Alagarsamy S, Douglas JG. Cyclic stretch-induced cPLA2 mediates ERK ½ signaling in rabbit proximal tubule cells. Kidney Int 2004; 65:551–563. [DOI] [PubMed] [Google Scholar]
- 40. Kalyankrishna S, Malik KU. Norepinephrine-induced stimulation of p38 mitogen-activated protein kinase is mediated by arachidonic acid metabolites generated by activation of cytosolic phospholipase A(2) in vascular smooth muscle cells. J Pharmacol Exp Ther 2003; 304:761–772. [DOI] [PubMed] [Google Scholar]
- 41. Mervaala E, Müller DN, Schmidt F, Park JK, Gross V, Bader M, Breu V, Ganten D, Haller H, Luft FC. Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension 2000; 35:587–594. [DOI] [PubMed] [Google Scholar]
- 42. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension 2011; 57:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schiffrin EL. T lymphocytes: a role in hypertension? Curr Opin Nephrol Hypertens 2010; 19:181–186. [DOI] [PubMed] [Google Scholar]
- 44. Burgermeister E, Endl J, Scheuer WV. Activation of cytosolic phospholipase A2 in human T-lymphocytes involves inhibitor-kappaB and mitogen-activated protein kinases. Eur J Pharmacol 2003; 466:169–180. [DOI] [PubMed] [Google Scholar]
- 45. Shmelzer Z, Karter M, Eisenstein M, Leto TL, Hadad N, Ben-Menahem D, Gitler D, Banani S, Wolach B, Rotem M, Levy R. Cytosolic phospholipase A2alpha is targeted to the p47phox-PX domain of the assembled NADPH oxidase via a novel binding site in its C2 domain. J Biol Chem 2008; 283:31898–31908. [DOI] [PubMed] [Google Scholar]
- 46. Zhao X, Bey EA, Wientjes FB, Cathcart MK. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phosphorylation of p67(phox) and p47(phox). J Biol Chem 2002; 277:25385–25392. [DOI] [PubMed] [Google Scholar]
- 47. Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 2009; 296:R208–R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yaghini FA, Song CY, Lavrentyev EN, Ghafoor HU, Fang XR, Estes AM, Campbell WB, Malik KU. Angiotensin II-induced vascular smooth muscle cell migration and growth are mediated by cytochrome P450 1B1-dependent superoxide generation. Hypertension 2010; 55:1461–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim N, Luster AD. Regulation of immune cells by eicosanoid receptors. ScientificWorldJournal 2007; 7:1307–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.