

Summary
This study reveals that human papillomavirus-positive head and neck cancers are characterized by a state of chronic oxidative stress mediated by NOX2. In these cells, oxidative stress causes DNA damage and chromosomal aberration and increases the susceptibility to ionizing radiation-induced DNA damage.
Abstract
Human papillomavirus (HPV) is the causative agent of a subgroup of head and neck cancer characterized by an intrinsic radiosensitivity. HPV initiates cellular transformation through the activity of E6 and E7 proteins. E6 and E7 expression is necessary but not sufficient to transform the host cell, as genomic instability is required to acquire the malignant phenotype in HPV-initiated cells. This study reveals a key role played by oxidative stress in promoting genomic instability and radiosensitivity in HPV-positive head and neck cancer. By employing an isogenic human cell model, we observed that expression of E6 and E7 is sufficient to induce reactive oxygen species (ROS) generation in head and neck cancer cells. E6/E7-induced oxidative stress is mediated by nicotinamide adenine dinucleotide phosphate oxidases (NOXs) and causes DNA damage and chromosomal aberrations. This mechanism for genomic instability distinguishes HPV-positive from HPV-negative tumors, as we observed NOX-induced oxidative stress in HPV-positive but not HPV-negative head and neck cancer cells. We identified NOX2 as the source of HPV-induced oxidative stress as NOX2 silencing significantly reduced ROS generation, DNA damage and chromosomal aberrations in HPV-positive cells. Due to their state of chronic oxidative stress, HPV-positive cells are more susceptible to DNA damage induced by ROS and ionizing radiation (IR). Furthermore, exposure to IR results in the formation of complex lesions in HPV-positive cells as indicated by the higher amount of chromosomal breakage observed in this group of cells. These results reveal a novel mechanism for sustaining genomic instability in HPV-positive head and neck tumors and elucidate its contribution to their intrinsic radiosensitivity.
Introduction
Human papillomavirus 16 (HPV16) is an epitheliotropic virus associated with increased risk of cervical and head and neck cancer (1,2). HPV-positive head and neck cancers have become a focus of attention due to their unique biological and clinical features. This subgroup of tumors is characterized by increasing incidence and a younger population compared with HPV-negative head and neck cancers (1). Moreover, HPV-positive head and neck cancers display enhanced radiation sensitivity in vitro and in vivo (3–5). This feature probably contributes to a more favorable clinical outcome observed in patients with HPV-positive tumors compared with those with HPV-negative tumors following treatment with ionizing radiation (IR) (6–8). Due to these unique characteristics, HPV-positive head and neck cancers represent a clinically relevant model to study not only the mechanisms of viral oncogenes but also the effect of viral infection on tumor biology.
HPV16-induced cellular transformation requires the expression of two viral proteins, E6 and E7 (9). When coexpressed, E6 and E7 cooperate to transform the infected cell into a highly proliferating, immortalized cell. E7 acts primarily by inhibiting the activity of key regulators of cellular proliferation and cell-cycle progression (10,11). The best characterized of these interactions occurs with Rb protein, a master regulator of the G1-S cell-cycle checkpoint (10). Deregulated cell-cycle progression should result in cellular apoptosis by p53 activation (12). To prevent this response, E6 impairs p53 function by two different mechanisms: interfering with DNA-binding activity (13) and promoting degradation (14). Although E6/E7 are necessary to initiate and maintain the transformed phenotype, expression of these two oncoproteins is not sufficient to fully transform primary cells into cancer cells. This model is supported by the observation that HPV-immortalized cells are unable to form tumors in nude mice (15).
Development of genomic instability is considered a key enabling hallmark in HPV-induced carcinogenesis. Expression of E6/E7 in normal cells results in DNA damage and chromosomal aberrations (16). Multiple mechanisms have been proposed to explain these observations, such as replication stress and centrosome amplifications (17,18). However, these models do not consider the modulating role played by host and microenvironment-related factors. The relevance of additional factors influencing HPV-induced carcinogenesis is supported by the observation that not all HPV-infected women develop cervical cancer. Hormonal exposure, retinoid receptor deficiency, chronic inflammation, smoking history and presence of local coinfections have been identified as risk factors promoting the development of cervical cancer in HPV-infected women (19,20).
Risk factors that promote the development of cervical cancer in HPV-infected women share the capability to induce the generation of reactive oxygen species (ROS) in host cells and/or the tissue microenvironment. ROS are a family of highly reactive molecules continuously produced in the cell. The two major endogenous sources of ROS are mitochondria (21), which produce superoxide as a natural by-product of aerobic metabolism, and nicotinamide adenine dinucleotide phosphate oxidases (NOXs), which produce superoxide in response to endogenous and exogenous stimuli (22). ROS can mediate physiological processes, by functioning as cell signaling molecules as well as pathological processes, by damaging DNA, RNA, protein and lipids (23). To prevent deleterious consequences, production of ROS is balanced by complex systems of antioxidant enzymes and small molecules that scavenge such reactive species. The imbalance between ROS production and cell scavenging capability that results in intracellular oxidative damage is defined as oxidative stress.
We hypothesized that oxidative stress may act as a key cofactor in promoting HPV-induced carcinogenesis. An acute increase in ROS levels has been observed in keratinocytes upon infection with HPV16 (24). Similarly, infection by different viruses is associated with ROS generation in the host cells (25–27). There is a consensus among several groups with a model in which ROS generation occurs during the early phase of viral infection to promote viral genome amplification and integration into the host genome (25,28,29). Although the few studies that have linked ROS generation with HPV infection have focused on the acute effects of HPV-induced ROS on host cell biology, very little is known about how this response is elicited, its temporal nature (acute versus chronic) and its effect on cancer cell genomic integrity and response to therapy.
In this study, we employed a panel of HPV-positive head and neck cancer cell lines as a model system. By comparing the oxidative status and the biological characteristics of HPV-positive and HPV-negative head and neck cancer cells, we were able to (i) identify the source of ROS generation in HPV-positive tumors, (ii) elucidate the mechanism by which ROS may promote HPV-induced carcinogenesis and (iii) determine how HPV-induced ROS influences head and neck cancer cell responses to IR. Our findings not only contribute to understanding the mechanism(s) of HPV-induced carcinogenesis but may also reveal novel strategies to optimize the treatment of HPV-associated tumors.
Materials and methods
Cell culture
Tu212 cell line was provided by G.L.Clayman in 2002 (30,31). SCC02, SCC47, JHU-012, 93-VU-147T, PCI-13 and PCI-15A cell lines were kindly given by Dr R.Ferris in 2012, SCC090 cell line by Dr S.Gollin in 2010 and SCC1483 cell line by Dr S.-Y.Sun in 2012. SCC02, SCC47, SCC090, 93-VU-147T and SCC1483 cells are positive for HPV (3,32), and HPV status was confirmed by PCR amplification of the viral genes E6 and E7 (Supplementary Figure 1, available at Carcinogenesis Online). Tu212, PCI-13 and PCI-15A cells were cultured in Dulbecco’s modified Eagle’s medium/F12 (50:50) medium; JHU-012 cells were cultured in RPMI medium; media were supplemented with 5% fetal bovine serum. SCC47, SCC02 and 93-VU-14T cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. SCC090 cells were cultured in Eagle's minimum essential medium medium with 10% fetal bovine serum, l-glutamine and minimum essential medium non-essential amino acids. Cells were grown at 37°C in 5% CO2 humidified incubators. Cells were routinely screened for mycoplasma contamination by MycoAlert Mycoplasma Detection Kit (Lonza). The authenticity of cell lines was verified through the genomic short tandem repeat profile by the Research Animal Diagnostic Laboratory, University of Missouri (Columbia, MO, USA) in September 2009 and by Emory University Integrated Genomics Core in October 2013, respectively. Authenticity of PCI-15A was not verified by the authors, but reported by Zhao et al. in 2011, using the same short tandem repeat profile (31).
ROS level measurements
Endogenous ROS levels were analysed by incubating the cells with 2′-7′ dichlorodihydrofluorescein diacetate (10 µM; Sigma–Aldrich), dihydroethidium (5 µM; Molecular Probes) or MitoSox (5 µM; Molecular Probes) for 30min at 37°C. The NOXs inhibitor diphenyleneiodonium chloride (DPI, 1 µM; Sigma–Aldrich) and the antioxidant N-acetyl-cysteine (NAC, 1mM; Sigma–Aldrich) were added to the media 1h (DPI) or 2h (NAC) before cell labeling with ROS probes. Fluorescence intensity was assessed with BD LSR II flow cytometer, and data were analysed with FlowJo Software.
Plasmid, shRNA interference and transfection
HPV-negative cells were transfected with a plasmid-driving HPV16 E6/E7 expression (Karl Munger, Addgene plasmid #13712) or the corresponding empty vector (Karl Munger, Addgene plasmid #13680) as a control (33). HPV-positive head and neck cancer cells were transfected with lentiviral plasmids carrying NOX2-targeted short hairpin RNA (shRNA) (SH88 and SH90; GE Healthcare) or non-targeted shRNA (pLKo1). A double transfection (days 1 and 3) was carried out by using Lipofectamine 3000 according to the manufacturer protocol; unless otherwise indicated, the cells were plated at day 5 and analysed at day 6.
Subcellular fractionation and western blot
Subcellular fractionation was performed by differential centrifugation. Briefly, cells were resuspended in ice-cold hypotonic buffer (10mM Tris-base pH 7.4; 10mM NaCl, 1.5 MgCl2, 1mM dithiothreitol), incubated for 15min on ice and then homogenized with a Dounce homogenizer. Cell lysate was centrifuged at 1000g for 10min to remove nuclei and cell debris. The supernatant was centrifuged at 15000g for 15min to separate mitochondrial and cytosolic fractions. The supernatant was then centrifuged at 100000g for 60min to obtain lysate enriched in membrane fraction (pellet) and soluble cytosolic fraction (supernatant).
For western blot analysis, the following primary antibodies were used: p47phox 1:1000 (Cell Signaling, cat#4312); Na, K-ATPase 1:500 (DSHB, cat#α6F); β-actin 1:1000 (Sigma–Aldrich, cat#A5441). Chemiluminescence was used to detect immunoreactive proteins, and protein abundance was quantified based on band intensities using ImageJ software.
DNA damage measurements
The alkaline comet assay was used to detect DNA single-strand breaks (SSB), double-strand breaks (DSB) and alkali-labile sites in the DNA. Following treatments with NAC, cells were mixed with 0.5% agarose and placed on a microscope slide precoated with 1% agarose. For experiments with H2O2 and IR, cells were embedded in agarose, exposed to the agents on ice and lysed after 5min (H2O2) or 30min (IR). Lysis was performed for 2h (lysis buffer: 2.5M NaCl, 10mM ethylenediaminetetraacetic acid, 10mM Tris-base, pH 10, 1% Triton X, 10% dimethyl sulfoxide; 50 µM N-tert-butyl-α-phenylnitrone added fresh) followed by incubation with electrophoresis buffer for 40min. Electrophoresis was conducted in alkaline buffer for 30min. Following neutralization, cells were stained with Sybr Green for 20min. Comets were analysed within 48h and scored with Comet Score (TriTek). Two slides per condition were analysed in each experiment and 50 comets per slide were scored.
Chromosomal aberration measurements
Cytokinesis-block micronucleus assay was used as a surrogate to detect chromosomal aberrations such as lagging whole chromosomes or acentric chromosome fragments (micronuclei) or gene amplification (nuclear buds). Cells were plated on a glass coverslip and let recover overnight. Following treatments, cytochalasin B (3 µg/ml) was added to the media for 18h and then the cells were fixed with 4% paraformaldehyde, washed with phosphate-buffered saline and stained with 4′,6-diamidino-2-phenylindoledihydrochloride. Micronuclei and nuclear buds were scored in binucleated cells according to criteria described previously (34). Each experiment was performed in duplicate, and 200 or more cells were analysed to score a minimum of 10 micronuclei/nuclear buds per sample.
PCR and RT–PCR
Total DNA was isolated by using MasterPure DNA Purification Kit (Epicentre Biotechnologies) according to the manufacturer protocol. Complementary DNA (cDNA) amplification, total RNA extraction and cDNA synthesis were performed as described previously (35). For semiquantitative PCR and RT–PCR, 1 µg of DNA or 2 µl of cDNA was amplified with Takara Ex-Taq Polymerase (Clontech) under the following cycling conditions: 30 cycles of 94°C for 30 s, 52°C for 30 s and 72°C for 60 s. No template samples were used as negative controls. Primer sequences were E6F (5′–3′ on plus strand) ATGTTTCAGGACCCACAGGA, E6R (5′–3′ on minus strand) TACAGCTGGGTTTCTCTACG (36); E7F (5′–3′ on plus strand) GCAACCAGAGACAACTGATC, E7R (5′–3′ on minus strand) ATTGTAATGGGCTCTGTCCG (37); GADPHF (5′–3′ on plus strand) GGAAGGTGAAGGTCGGAGT and GADPHR (5′–3′ on minus strand) GAAGATGGTGATGGGATTTC (35).
Real-time qRT–PCR was carried out using StepOnePlus™ Real-Time PCR System (Applied Biosystems). Two microliter of cDNA was amplified by using QuantiTect SYBR Green PCR Master Mix (Qiagen) and the following cycling conditions: 10min at 95°C, 40 cycles of 15 s at 95°C, 30 s at 57°C and 30 s at 72°C. A dissociation curve analysis was performed for each sample to verify PCR specificity. Mock reverse transcription and no template samples were used as negative controls. Primer sequences for NOX2 were NOX2F (5′–3′ on plus strand) GTCACACCCTTCGCATCCATTCTCAGTCAGT and NOX2R (5′–3′ on minus strand) CTGAGACTCATCCCAGCCAGTGAGGTAG (38) and for RPLP0 gene were RPLP0F (5′–3′ on plus strand) GGGCGACCTGGAAGTCCAACT and RPLP0R (5′–3′ on minus strand) CCCATCAGCACCACAGCCTTC (35). The average NOX2 messenger RNA (mRNA) fold change was calculated by the ΔΔCt method using RPLP0 as internal control and pLKo1 cells samples as calibrator.
Data representation and statistical analysis
Unless otherwise indicated, results are presented as mean of three independent experiments, each performed in triplicate. Error bars represent ± standard deviation. Statistical analyses were performed using GraphPad Prism 6.0, and the statistical test used for each experiment is detailed in the figure legend.
Results
HPV-positive head and neck cancer cells display a state of chronic oxidative stress
To determine whether oxidative stress is associated with HPV infection in head and neck cancer, we analysed the ROS status of a panel of HPV-positive and HPV-negative head and neck cancer cell lines by utilizing a cell-permeable probe (2′-7′ dichlorodihydrofluorescein diacetate) that becomes fluorescent upon oxidation by several different ROS, thus providing a general indicator of the oxidative stress status of viable cells. HPV-positive cells display higher endogenous levels of ROS compared with HPV-negative cells (Figure 1A and Supplementary Figure 2A, available at Carcinogenesis Online), indicating that HPV-positive cells are characterized by a pro-oxidant intracellular environment. Mitochondria and NOXs are the two major intracellular sources of ROS, generating primarily superoxide; thus, we analysed the levels of superoxide in the panel of cells utilizing the superoxide-specific probe dihydroethidium. HPV-positive cells display a higher level of superoxide compared with HPV-negative cells (Figure 1B and Supplementary Figure 2B, available at Carcinogenesis Online), suggesting that elevated ROS generation is associated with HPV infection in head and neck cancer cells. Chronically elevated intracellular ROS levels may result in oxidative damage to DNA and other macromolecules if not effectively scavenged by antioxidant systems or small-molecule reducing agents. To determine whether the observed, elevated ROS cause oxidative DNA damage in HPV-positive head and neck cancer cells, we pretreated the panel of cells with the glutathione precursor NAC (1mM) that promotes ROS scavenging and then measured the level of DNA damage by the alkaline comet assay. Following NAC treatment, we observed a reduction in the amount of endogenous DNA damage present in HPV-positive cancer cells, whereas no effect was observed in HPV-negative cells (Figure 1C). Treatment with NAC did not significantly affect cell proliferation (Supplementary Table 1, available at Carcinogenesis Online); thus, the observed reduction in DNA damage levels is not due to alleviation of replication stress. These results indicate that HPV-positive head and neck cancer cells are characterized by a status of chronic oxidative stress that contributes to DNA damage.
Figure 1.
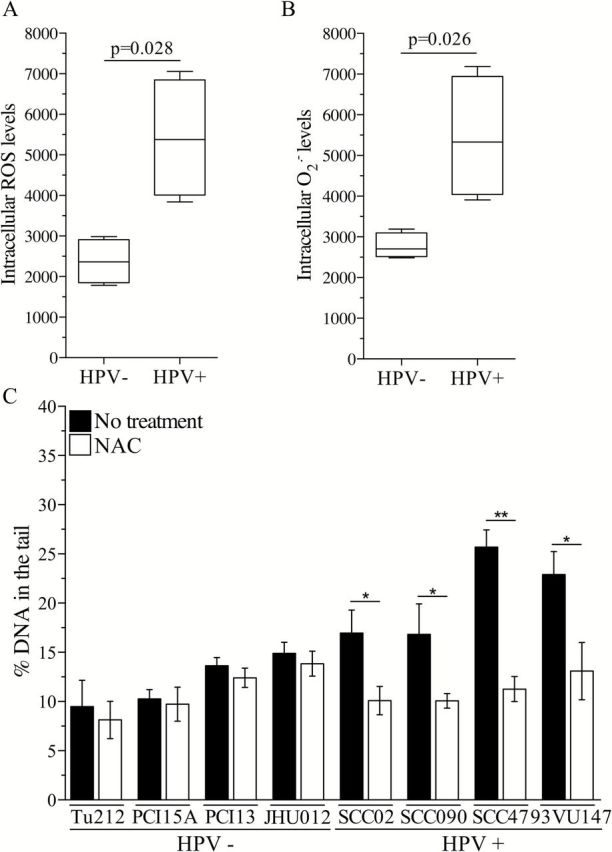
HPV-positive head and neck cancer cells are characterized by a status of chronic oxidative stress. (A) Intracellular ROS levels and (B) superoxide level in a panel of head and neck cancer cells divided in groups based on HPV status. The line in the middle of each box is plotted at the median, and whiskers represent minimum and maximum value for each group. A two-tailed Mann–Whitney test was used to compare medians between the two groups. (C) Endogenous DNA damage level in HPV-positive and HPV-negative head and neck cancer cell lines following treatment with NAC (1mM). DNA damage level in treated versus non-treated cells for each cell line was analysed by two-tailed t-test. *P < 0.05, **P < 0.005.
Expression of E6 and E7 oncoproteins is sufficient to promote NOX-dependent ROS generation and oxidative DNA damage in head and neck cancer cells
Our results indicate a strong association between HPV infection and sustained elevated ROS in head and neck cancer. Since the oncogenic potential of HPV is mediated by E6 and E7, we evaluated whether the expression of these viral oncoproteins promotes the generation of ROS in head and neck cancer cells. An isogenic cell model was generated by transfecting PCI-15A cells (HPV-negative head and neck cancer cell line) with a plasmid-driving E6 and E7 expression (E6/E7) cells or the empty vector (CTRL cells) (Supplementary Figure 3, available at Carcinogenesis Online). We observed increased levels of ROS in E6/E7 cells compared with CTRL cells (Figure 2A and B). Such elevated ROS causes DNA damage as E6/E7 cells display higher levels of endogenous DNA damage that is significantly reduced by pretreatment with the NAC (Figure 2F). These results suggest that expression of the viral oncoproteins E6 and E7 is sufficient to initiate a cellular response resulting in sustained, elevated ROS levels that causes DNA damage in HPV-positive head and neck cancer cells. Next, we sought to identify the source of ROS generated as a result of E6 and E7 expression. To evaluate the role of mitochondria, we employed the mitochondria-targeting fluorescent probe MitoSox that detects superoxide. E6/E7 and CTRL cells display similar levels of mitochondrial superoxide (Figure 2C), indicating that mitochondria are not contributing to E6/E7-induced generation of ROS. To evaluate the role of NOXs, the cells were treated with the NOXs inhibitor DPI (1 µM). Pharmacological inhibition of NOX enzymes significantly reduced ROS levels in E6/E7 cells (Figure 2B and E) but not in CTRL cells (Figure 2B and D), indicating that NOX enzymes are the major source of E6/E7-induced generation of ROS in head and neck cancer cells.
Figure 2.
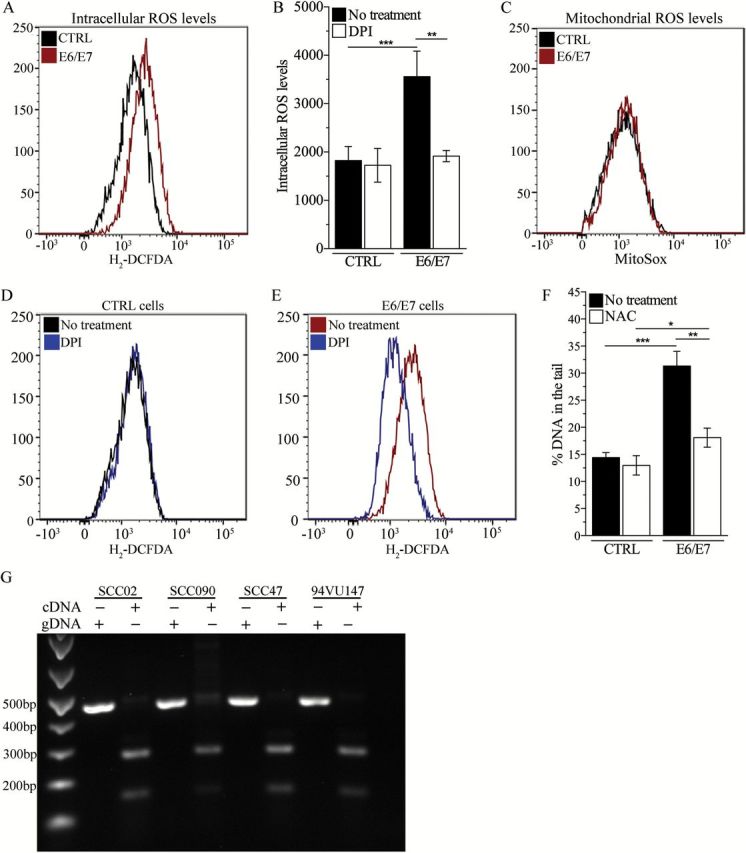
Expression of E6 ad E7 promotes NOX-dependent oxidative stress in head and neck cancer cells. (A) Representative flow cytometry curve of total intracellular ROS levels in PCI-15A cells (HPV negative) transfected with an empty plasmid (CTRL) or a plasmid expressing E6 and E7 (E6/E7). (B) Quantitative representation of previous experiment. ROS level in treated and non-treated cells for each genotype was compared by two-way analysis of variance (ANOVA; treatment × genotype interaction P = 0.0053; Bonferroni post-test for multiple comparison: **P < 0.005). (C) Representative flow cytometry curve of mitochondrial ROS levels in CTRL and E6/E7 cells. (D–E) Representative flow cytometry curves of ROS levels in (D) CTRL and (E) E6/E7 cells following treatment with DPI (1 µM). (F) DNA damage levels in E6/E7 transfected and isogenic CTRL cells following treatment with NAC (1mM). DNA damage level in treated and non-treated cells for each genotype was compared by two-way ANOVA (treatment × genotype interaction P < 0.005; Bonferroni post-test for multiple comparison: *P < 0.05, **P < 0.005; ***P < 0.0005). (G) Expression of full-length (E6) and truncated (E6* and E6**) mRNA transcripts in HPV-positive head and neck cancer cell lines. Genomic DNA for each cell line was used as control.
We observed that E6/E7 cells expressed both the full-length (E6) and the truncated (E6* and E6**) forms of E6 mRNA transcripts (Supplementary Figure 3, available at Carcinogenesis Online). A previous report showed that expression of E6* induces ROS generation and acute oxidative stress in keratinocytes (24), suggesting a conserved mechanism for HPV-induced ROS generation across cell types. To address this possibility, we determined whether the truncated form of E6 is expressed in naturally infected HPV-positive head and neck cells. By performing RT–PCR, we observed that HPV-positive head and neck cancer cells express both the full-length and the truncated forms of E6 transcripts (Figure 2G), supporting the role of E6* in HPV-induced ROS generation regardless of the cell type.
NOX2 is the source of HPV-induced ROS in naturally infected HPV-positive head and neck cancer cells
The NOX-dependent ROS response observed in our isogenic cell model is probably sustaining and promoting the chronic oxidative stress state observed in naturally infected HPV-positive head and neck cancer cells. To investigate this possibility, we analysed the effect of DPI, a NOXs inhibitor, on the endogenous ROS levels of the panel of patient-derived head and neck cancer cells. Independent exposures to NAC and H2O2 were employed as negative and positive controls, respectively. Inhibition of NOXs with DPI significantly reduced the levels of ROS in HPV-positive cells (Figure 3A), indicating that NOX enzymes are a source of the elevated ROS levels observed in this subgroup of head and neck cancer cells. To identify the specific NOX involved in HPV-induced ROS generation, we analysed the expression of the catalytic subunits of NOX enzymes (NOX1 to NOX5) in the panel of cells. NOX2 mRNA transcript is expressed in all the HPV-positive cells and in three of the HPV-negative cells (Supplementary Figure 4, available at Carcinogenesis Online), whereas no other NOX-encoding gene was transcribed in our cells (data not shown). These results suggest that HPV-induced ROS generation probably occurs via NOX2 activity. To further address this possibility, we silenced NOX2 in a HPV+ head and neck cancer cell line (SCC47) with NOX2-targeted shRNAs (SH88 and SH90). Control cells were transfected with non-targeted shRNA (pLKo1). Transfection of SCC47 with either SH88 or SH90 shRNA resulted in ~50% reduction in NOX2 mRNA transcript (Figure 3B), which resulted in a significant reduction in the levels of ROS (Figure 3C) and DNA damage (Figure 3D and E). Collectively, these results demonstrate that the HPV-induced ROS response and subsequent DNA damage occur via NOX2 oxidase activation.
Figure 3.
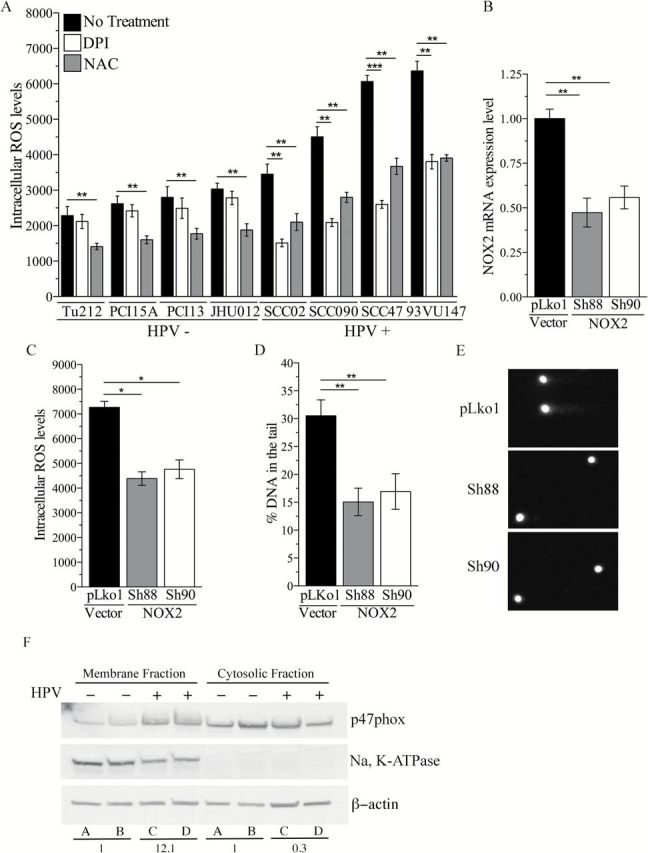
NOX2 is the source of HV16-induced chronic oxidative stress in head and neck cancer cells. (A) Intracellular ROS level in HPV-positive and HPV-negative head and neck cancer cell lines following treatment with DPI (1 µM) or NAC (1mM). ROS level in treated and non-treated cells for each cell line was analysed by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test. *P < 0.05, **P < 0.005, ***P < 0.0005. (B) NOX2 mRNA expression levels and (C) intracellular ROS levels in SCC47 cells (HPV positive) transfected with plasmids carrying non-targeted shRNA (pLKo1 cells) or NOX2-targeted shRNA (SH88 and SH90). (D) Amount of DNA damage in pLKo1, SH88 or SH90 cells. (E) Representative comets in each cell line. NOX2 mRNA expression, intracellular ROS levels and DNA damage amount in pLKo1, SH88 and SH90 cells were analysed by one-way ANOVA followed by Dunnett’s multiple comparisons test. *P < 0.05, **P < 0.005. (F) Representative western blot analysis of p47phox expression in the membrane enriched and the soluble cytosolic fraction of HPV-negative (A: PCI-15A; B: PCI-13) and HPV-positive (C: SCC47; D: 93VU147) cells lysate. Na, K-ATPase and β-actin were used as loading control for membrane-enriched fraction and soluble cytosolic fraction, respectively. Average of p47phox expression for each group (A + B versus C + D) was compared and numbers represent normalized fold changes.
The active form of NOX2 is comprised of a multiprotein complex in which the catalytic subunit (NOX2) is activated following the recruitment of multiple cytoplasmic subunits (p47phox, p67phox, p40phox, p22phox and Rac) (22). Previous reports demonstrate that activation of NOX oxidases can occur via two distinct mechanisms: (i) increased transcription of the catalytic subunit of NOX2 oxidases (NOX2) (27) or (ii) increased recruitment of regulatory subunits at the plasma membrane (39). Since we did not observe changes in NOX2 mRNA levels following expression of E6 and E7 in PCI-15A cells (data not shown), we excluded transcriptional activation of NOX2 in our model. Therefore, we determined whether activation of NOX2 oxidases in HPV-positive cells is due to increase recruitment of regulatory subunits. By analysing the expression levels of the p47phox subunit in the membrane fraction of HPV-positive and HPV-negative head and neck cancer cells, we observed an increase in membrane localization of p47phox in HPV-positive cells compared with HPV-negative cells (Figure 3F), indicating that activation of NOX2 oxidase occurs via increased assembly of active complex at the membrane.
Chronic oxidative stress sustains genomic instability in HPV-positive but not HPV-negative head and neck cancer cells
Development of genomic instability constitutes a crucial event during HPV-induced carcinogenesis (9). Previous reports demonstrated that expression of E6/E7 in human keratinocytes results not only in increased DNA damage but also in increased frequency of structural chromosomal aberrations (16). Since we found that ROS is a major source of DNA damage in HPV-positive head and neck cancer cells (Figure 1C), we next examined whether HPV-induced oxidative stress contributes to genomic instability. To detect and quantify chromosomal aberrations in our cells, we performed a micronucleus assay and scored micronuclei and nuclear buds. Micronuclei are the result of either aneugenic events (i.e. lagging chromosomes and subsequent whole chromosomal loss) or clastogenic events (chromosome breakage and subsequent chromosome fragments loss) (34), and nuclear buds are markers of DNA amplification (40,41). Expression of E6/E7 in HPV-positive head and neck cancer cells resulted in a significant increase in micronuclei frequency compared with control cells (Figure 4B) and was partially reduced by treatment with NAC. These data suggest that oxidative stress may contribute to HPV-induced chromosomal aberrations. Supporting this mechanism, silencing NOX2 expression reduced the number of micronuclei in HPV-positive SCC47 cells (Figure 4A). Similarly, pretreatment with NAC reduced the amount of micronuclei in naturally infected HPV-positive (Figure 4C) but not HPV-negative head and neck cancer cells. Therefore, our results indicate that oxidative stress is a major factor in the promotion and establishment of genomic instability in HPV-positive head and neck tumors, whereas its contribution to genomic instability in HPV-negative head and neck tumors appears to be negligible.
Figure 4.
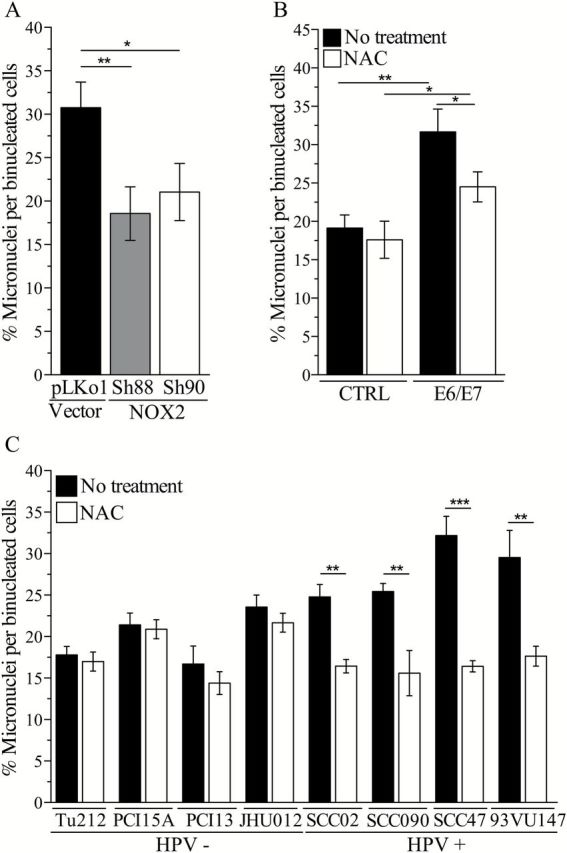
Chronic oxidative stress contributes to chromosomal instability in HPV-positive head and neck cancer cells. (A) Micronuclei frequency in SCC47 cells (HPV positive) transfected with non-targeted shRNA (pLKo1 cells) or NOX2-targeted shRNA (SH88 and SH90 cells). Micronuclei frequency was compared by one-way analysis of variance (ANOVA; P = 0.006; Dunnett’s post-test for multiple comparison: *P < 0.05, **P < 0.005. (B) Micronuclei frequency in PCI-15A cells (HPV negative) transfected with empty plasmid (CTRL) or plasmid expressing HPV16 E6 and E7 (E6/E7). Micronuclei frequency was compared by two-way ANOVA (Bonferroni post-test for multiple comparison: *P < 0.05, **P < 0.005). (C) Micronuclei frequency in HPV-positive and HPV-negative head and neck cancer cells following treatment with NAC (1mM). Micronuclei amount in treated and non-treated cells for each cell line was analysed by two-tailed t-test. *P < 0.05, **P < 0.005.
HPV-positive head and neck cancer cells are more susceptible to ROS-induced DNA damage
We have demonstrated that HPV-positive head and neck cancer cells are characterized by a state of chronic oxidative stress due to continuous endogenous ROS production. As a result, HPV-positive head and neck cancer cells may have limited residual antioxidant capacities, compared with HPV-negative head and neck cancer cells. As a consequence of impaired antioxidant capacity, HPV-positive head and neck cancer cells may be highly susceptible to DNA damage induced by ROS. To address this possibility, we exposed our cells to increasing doses of H2O2 on ice and immediately evaluated the amount of DNA damage induced by each dose. This experiment was designed to minimize any influence of cellular repair on the amount of DNA damage detected. Increasing doses of H2O2 caused higher levels of DNA damage in HPV-positive cells compared with HPV-negative cells (Figure 5A), indicating that HPV-positive cells are more susceptible to ROS-induced DNA damage.
Figure 5.

HPV-positive head and neck cancer cells are more susceptible to ROS-induced DNA damage. (A) DNA damage level following exposure to a dose range of H2O2 in HPV-positive and HPV-negative head and neck cancer cell lines. Column represents mean for each group, whereas black dots represent individual values for each cell line. DNA damage amount in treated versus non-treated cells for each group was analysed by two-way analysis of variance (ANOVA; treatment × HPV status interaction P < 0.005; Bonferroni post-test for multiple comparison: **P < 0.005). (B) DNA damage levels and (C) micronuclei frequency following exposure to IR (2 or 6 Gy) in HPV-positive and HPV-negative head and neck cancer cell lines. DNA damage and micronuclei amount in treated versus non-treated cells for each group were analysed by two-way ANOVA followed by Bonferroni’s multiple comparison test. **P < 0.005.
Regardless of their HPV status, the majority of head and neck cancer patients receive IR as an integral component of their therapy. IR kills cancer cells by inducing DNA damage directly via generation of strand breaks or indirectly via generation of intracellular ROS (42). Thus, based on our findings, HPV-positive head and neck cancer cells may be more susceptible to IR-induced DNA damage. Accordingly, we observed a higher amount of DNA damage per dose of IR in HPV-positive head and neck cancer cells compared with HPV-negative head and neck cancer cells (Figure 5B).
IR induces a variety of DNA damage ranging from isolated to clustered DNA lesions (42,43). Exposure to IR may result in the generation of clustered lesions, especially in HPV-positive head and neck cancer cells as these cells are chronically exposed to elevated ROS and are more prone to ROS-induced DNA damage. Since unrepaired, clustered DNA lesions result in chromosomal breakage in human cells (43), we determined the amount of chromosomal aberrations induced by IR in the panel of cells as a marker of non-repairable DNA lesions. Supporting our hypothesis, we observed that each dose of IR induces a higher frequency of chromosomal aberrations in HPV-positive cells compared with HPV-negative cells when measured at the first mitosis following exposure (Figure 5C).
Discussion
The aim of our study was to determine the role of oxidative stress in promoting HPV-induced carcinogenesis and modulating cell phenotype and response to therapy. We employed HPV-positive head and neck cancer cell lines as a model of HPV-associated tumors that display unusual phenotypic characteristics and elevated sensitivity to IR.
Our study reveals that chronic oxidative stress is an inherent property of HPV-positive head and neck cancer cells and contributes to the establishment of genomic instability in this subgroup of cancers. By employing an isogenic cell model, we elucidated the mechanism by which oxidative stress is generated and sustained in head and neck cancer cells. We demonstrated that expression of E6/E7 evokes a ROS response that occurs via NOX2 oxidase activation. This ROS response, in turn, causes DNA damage and chromosomal aberrations. We also demonstrated that this mechanism for ROS generation and subsequent genomic instability recapitulates what occurs in naturally infected head and neck cancer cells as we validated our findings in a panel of HPV-positive head and neck cancer cells isolated from patients.
Based on these results, we propose a model in which HPV16 E6 and E7 establish a sustained ROS response in head and neck cancer cells by promoting the translocation of NOX2 oxidase regulatory subunits to the membrane and thus activating the enzyme by increasing the formation of active complexes (Figure 6). Once established, chronic oxidative stress may act as a promoting factor in HPV16-associated carcinogenesis. This model is also consistent with earlier reports of elevated markers for oxidative stress in HPV-positive head and neck and cervical cancer tumors (44,45).
Figure 6.

Proposed model for establishment of NOX-induced chronic oxidative stress and genomic instability in HPV-positive head and neck cancer cells. Expression of HPV16 E6/E7 generates a ROS response in host cell via NOX2 oxidase activation. Activation of NOX2 oxidase occurs via increased formation of NOX2 active complex at membrane. NOX2-generated ROS causes DNA damage, such as bases oxidation, single DNA strand breaks and double DNA strand breaks, leading to chromosomal aberration and genomic instability. This state of chronic oxidative stress contributes to the increased sensitivity to IR of HPV-positive head and neck cancer cells. Additional mechanisms for HPV16-induced genomic instability comprise replication stress and centrosome amplification.
Additional cofactors could contribute to the establishment of the sustained chronic activation of NOX2 oxidases in HPV-positive head and neck cancer cells. E6 and E7 cause genomic instability in host cells by multiple mechanisms not directly related to oxidative stress, such as stalled replication forks, replication stress (17) and centrosome amplification (16,18). Our results are consistent with a multifactorial model as antioxidant treatment reduces but does not totally abrogate DNA damage (Figures 1C and 2F) and chromosomal aberrations (Figure 4B) induced by E6/E7 expression in HPV-negative head and neck cancer cells. Several types of DNA damage induce a ROS response that is conserved from yeast (46) to mammals and in mammals is mediated by NOX (47). Thus, E6/E7-induced ROS generation may also occur as a part of a general intracellular response to DNA damage.
Another finding of our study is that oxidative stress causes chromosomal instability in HPV-positive cells. Although other groups have reported that HPV16 infection is associated with increases in oxidative base damage (e.g. 8-oxodG) in DNA (24), our results support and expand these observations suggesting a more pervasive effect of ROS on the genomic integrity of the infected cells. How might oxidative DNA damage cause chromosomal instability in HPV-positive cells? ROS damages DNA by inducing oxidative base lesions and DNA SSB (23). The cell must efficiently repair SSB before entering the S-phase of the cell cycle to avoid their conversion into DNA DSB during replication. A conversion of SSB to DSB by this mechanism is probably to occur in HPV-positive cells as they harbor an elevated level of endogenous DNA damage and have impaired cell-cycle checkpoints due to Rb and p53 inactivation (10,13). Moreover, a previous report demonstrated that HPV-positive cells have impaired homologous recombination, the intracellular pathway that repairs DNA DSB in an error-free manner (4). Therefore, HPV-positive cells will probably repair DNA DSB through non-homologous end-joining, the intracellular pathway that repairs these lesions in an error-prone manner, potentially resulting in chromosomal aberrations. This ROS-mediated mechanism for genomic instability distinguishes HPV-positive from HPV-negative head and neck tumors as our results indicate that the contribution of oxidative stress to genomic instability in HPV-negative cells is negligible. Interestingly, no significant difference was observed in the endogenous levels of DNA damage and chromosomal aberration between the two groups (Figure 5), suggesting that other mechanisms are sustaining genomic instability in HPV-negative cells.
Several clinical studies report that patients with HPV-positive head and neck tumors have a more favorable clinical outcome following treatment with IR, compared with patients with HPV-negative tumors. Previous in vitro and in vivo studies have supported these clinical observations by demonstrating that HPV-positive head and neck cancer cells display enhanced sensitivity to the cell killing effects of IR (5,7,48). Several models have been proposed to explain the elevated radiosensitivity of HPV-positive tumors such as defects in DSB repair (4,48), impaired recovery of stalled replication fork (49) and/or activation of the remaining, functional p53 (5). Our results suggest an additional, major mechanism that may contribute to the radiosensitivity of HPV-positive tumors. The endogenous ROS generated in HPV-positive head and neck cancer cells may synergize with IR by creating clustered lesions that are highly lethal, especially in cells with impaired DNA damage repair systems such as homologous recombination. Supporting this model, a previous report demonstrates that expression of E6*, the truncated form of E6 associated with ROS generation and highly expressed in our panel of HPV-positive cells, is sufficient to sensitize oropharyngeal squamous cancer cells to IR (50). This mechanism is probably to be amplified in in vivo, where cells of the tumor microenvironment may enhance the generation of ROS both directly, as a part of the immune system response to viral infection, and indirectly, by releasing inflammatory cytokines. Further in vivo studies are necessary to elucidate the effect of oxidative stress and tumor microenvironment on HPV-positive head and neck cancer cells sensitivity to IR.
In conclusion, this is the first report that delineates the contribution of ROS to HPV-induced genomic instability and radiosensitivity in head and neck cancer cells. Our findings also have translational implications for HPV-negative head and neck patients as we speculate that altering the cellular redox balance toward a pro-oxidant status may sensitize HPV-negative head and neck cancer to the cell killing effects of IR.
Supplementary material
Supplementary Table 1 and Figures 1–4 can be found at http://carcin.oxfordjournals.org/
Funding
Pilot funding for this project was provided by a grant from the V Foundation. This work was also supported by Specialized Program of Excellence (SPORE) in Head and Neck Cancer (P50CA128613) — Developmental Research Program. Additional support for this project was from the Department of Hematology & Medical Oncology, Emory University School of Medicine.
Supplementary Material
Acknowledgements
We would like to thank all the members of the Doetsch lab for their helpful input and support.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- cDNA
complementary DNA
- DPI
diphenyleneiodonium chloride
- DSB
double-strand breaks
- HPV
human papillomavirus
- IR
ionizing radiation
- mRNA
messenger RNA
- NAC
N-acetyl-cysteine
- NOXs
nicotinamide adenine dinucleotide phosphate oxidases
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- SSB
single-strand breaks
References
- 1. Marur S., et al. (2010). HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol., 11, 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giuliano A.R., et al. (2015) EUROGIN 2014 roadmap: differences in human papillomavirus infection natural history, transmission and human papillomavirus-related cancer incidence by gender and anatomic site of infection. Int. J. Cancer, 136, 2752–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kimple R.J., et al. (2013) Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res., 73, 4791–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dok R., et al. (2014) p16INK4a impairs homologous recombination-mediated DNA repair in human papillomavirus-positive head and neck tumors. Cancer Res., 74, 1739–1751. [DOI] [PubMed] [Google Scholar]
- 5. Sørensen B.S., et al. (2014) Effect of radiation on cell proliferation and tumor hypoxia in HPV-positive head and neck cancer in vivo models. Anticancer Res., 34, 6297–6304. [PubMed] [Google Scholar]
- 6. Ang K.K., et al. (2010) Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med., 363, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lindel K., et al. (2001) Human papillomavirus positive squamous cell carcinoma of the oropharynx: a radiosensitive subgroup of head and neck carcinoma. Cancer, 92, 805–813. [DOI] [PubMed] [Google Scholar]
- 8. Lassen P., et al. (2009) Effect of HPV-associated p16INK4A expression on response to radiotherapy and survival in squamous cell carcinoma of the head and neck. J. Clin. Oncol., 27, 1992–1998. [DOI] [PubMed] [Google Scholar]
- 9. Moody C.A., et al. (2010) Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer, 10, 550–560. [DOI] [PubMed] [Google Scholar]
- 10. McLaughlin-Drubin M.E., et al. (2009) The human papillomavirus E7 oncoprotein. Virology, 384, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pang C.L., et al. (2014) A functional interaction of E7 with B-Myb-MuvB complex promotes acute cooperative transcriptional activation of both S- and M-phase genes (129 c). Oncogene, 33, 4039–4049. [DOI] [PubMed] [Google Scholar]
- 12. Jones D.L., et al. (1997) Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology, 239, 97–107. [DOI] [PubMed] [Google Scholar]
- 13. Mantovani F., et al. (2001) The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene, 20, 7874–7887. [DOI] [PubMed] [Google Scholar]
- 14. Hengstermann A., et al. (2001) Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc. Natl Acad. Sci. USA, 98, 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaur P., et al. (1988) Characterization of primary human keratinocytes transformed by human papillomavirus type 18. J. Virol., 62, 1917–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duensing S., et al. (2002) The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res., 62, 7075–7082. [PubMed] [Google Scholar]
- 17. Bester A.C., et al. (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell, 145, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duensing S., et al. (2001) Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res., 61, 2356–2360. [PubMed] [Google Scholar]
- 19. Castellasague X., et al. (2002). Environmental co-factors in HPV carcinogenesis. Virus Res., 89, 191–199. [DOI] [PubMed] [Google Scholar]
- 20. Gariglio P., et al. (2009). The role of retinoid deficiency and estrogen as cofactors in cervical cancer. Arch. Med. Res., 40, 449–465. [DOI] [PubMed] [Google Scholar]
- 21. Poyton R.O., et al. (2009). Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrin. Metab., 20, 332–340. [DOI] [PubMed] [Google Scholar]
- 22. Bedard K., et al. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev., 87, 245–313. [DOI] [PubMed] [Google Scholar]
- 23. Hussain S.P., et al. (2003) Radical causes of cancer. Nat. Rev. Cancer, 3, 276–285. [DOI] [PubMed] [Google Scholar]
- 24. Williams V.M., et al. (2014) Human papillomavirus type 16 E6* induces oxidative stress and DNA damage. J. Virol., 88, 6751–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bottero V., et al. (2013) Reactive oxygen species are induced by Kaposi’s sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. J. Virol., 87, 1733–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. To E.E., et al. (2014) Influenza A virus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production in macrophages. Free Radic. Res., 48, 940–947. [DOI] [PubMed] [Google Scholar]
- 27. Gruhne B., et al. (2009) The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl Acad. Sci. USA, 106, 2313–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim S.J., et al. (2013) ROS upregulation during the early phase of retroviral infection plays an important role in viral establishment in the host cell. J. Gen. Virol., 94(Pt 10), 2309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams V.M., et al. (2011) HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Future Virol., 6, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang X., et al. (2008) Synergistic inhibition of head and neck tumor growth by green tea (−)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer, 123, 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao M., et al. (2011) Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin. Cancer Res., 17, 7248–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wald A.I., et al. (2011) Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck, 33, 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Münger K., et al. (1989) The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol., 63, 4417–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fenech M. (2007) Cytokinesis-block micronucleus cytome assay. Nat. Protoc., 2, 1084–1104. [DOI] [PubMed] [Google Scholar]
- 35. Marullo R., et al. (2013) Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One, 8, e81162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamato K., et al. (2008) New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther., 15, 140–153. [DOI] [PubMed] [Google Scholar]
- 37. Kamio M., et al. SOC1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene, 23, 3107–3115. [DOI] [PubMed] [Google Scholar]
- 38. Kim Y.M., et al. (2005) A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res., 97, 629–636. [DOI] [PubMed] [Google Scholar]
- 39. Bousetta T., et al. (2010). The prolyl isomerase Pin1 acts as a novel molecular switch for TNF-α–induced priming of the NADPH oxidase in human neutrophils. Blood, 116, 5795–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shimizu N., et al. (1998) Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J. Cell Biol., 140, 1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gisselsson D., et al. (2001) Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol., 158, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blaisdell J.O., et al. (2001) Base excision repair processing of radiation-induced clustered DNA lesions. Radiat. Prot. Dosim., 97, 25–31. [DOI] [PubMed] [Google Scholar]
- 43. Asaithamby A., et al. (2011) Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl Acad. Sci. USA, 108, 8293–8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Marco F., et al. (2012) Oxidative stress in HPV-driven viral carcinogenesis: redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One, 7, e34366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Looi M.L., et al. (2008) Oxidative damage and antioxidant status in patients with cervical intraepithelial neoplasia and carcinoma of the cervix. Eur. J. Cancer Prev., 17, 555–560. [DOI] [PubMed] [Google Scholar]
- 46. Rowe L.A., et al. (2008) DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae . Free Radic. Biol. Med., 45, 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang M.A., et al. (2012) DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis., 3, e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rieckmann T., et al. (2013) HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol., 107, 242–246. [DOI] [PubMed] [Google Scholar]
- 49. Day T., et al. (2009) HPV E6 oncoprotein prevents recovery of stalled replication forks independently of p53 degradation. Cell Cycle, 8, 2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pang E., et al. (2011). Radiosensitization of oropharyngeal squamous cell carcinoma cells by human papillomavirus 16 oncoprotein E6*I. Int. J. Radiat. Oncol., 79, 860–865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.