

Abstract
The epithelial cells of the gut form a physical barrier against the luminal contents. The collapse of this barrier causes inflammation, and its therapeutic restoration can protect the gut against inflammation. EGF enhances mucosal barrier function and increases colonocyte proliferation, thereby ameliorating inflammatory responses in the gut. Based on our previous finding that the insect peptide CopA3 promotes neuronal growth, we herein tested whether CopA3 could increase the cell proliferation of colonocytes, enhance mucosal barrier function, and ameliorate gut inflammation. Our results revealed that CopA3 significantly increased epithelial cell proliferation in mouse colonic crypts and also enhanced colonic epithelial barrier function. Moreover, CopA3 treatment ameliorated Clostridium difficile toxin As-induced inflammation responses in the mouse small intestine (acute enteritis) and completely blocked inflammatory responses and subsequent lethality in the dextran sulfate sodium-induced mouse model of chronic colitis. The marked CopA3-induced increase of colonocyte proliferation was found to require rapid protein degradation of p21Cip1/Waf1, and an in vitro ubiquitination assay revealed that CopA3 directly facilitated ubiquitin ligase activity against p21Cip1/Waf1. Taken together, our findings indicate that the insect peptide CopA3 prevents gut inflammation by increasing epithelial cell proliferation and mucosal barrier function.
Keywords: bacterial toxin, cell cycle, cell proliferation, epidermal growth factor (EGF), epithelial cell, inflammation, peptides, proteasome, protein degradation, ubiquitylation (ubiquitination)
Introduction
Numerous papers have reported that inflammatory responses in the gut are strongly associated with defects in the mucosal barrier function of the gut epithelium (1, 2). The onset of gut inflammation can also be triggered by nonsteroidal anti-inflammatory drugs or infections that transiently break the mucosal barrier, resulting in activation of the immune system (1). Clostridium difficile infection, which causes an acute gut inflammation called pseudomembranous colitis, has also been associated with mucosal cell damage and increased permeability (3, 4). Colonic epithelial cells form a physical barrier between the lumen and the submucosa, and the collapse of the colonic epithelial cell layer is considered to be a common initial cause for various inflammatory responses in the gut (1, 2). The therapeutic restoration of the mucosal barrier should therefore protect the gut against inflammation (5).
The mucosal barrier can be restored via increased epithelial cell proliferation (6). EGF is known to be a key stimulator of epithelial cell growth and remodeling (7–9), and EGF also increases the mucosal barrier function and epithelial repair/restitution processes in human and animals (6). For example, the addition of EGF enemas to mesalamine treatment in ulcerative colitis patients has been shown to increase the therapeutic response rate (6). EGF has been shown to ameliorate C. difficile toxin-induced mouse enteritis by increasing epithelial cell proliferation (10). A similar effect has been reported for FGF (11, 12). The intestinal growth hormone glucagon-like peptide 2 has been shown to reduce mucosal permeability in animal models, such as DSS-induced3 colitis; similar to the action of EGF, this effect of glucagon-like peptide 2 is associated with the enhancement of mucosal epithelial cell growth (13, 14). Furthermore, the glucagon-like peptide 2 analog teduglutide has been shown to induce mucosal healing in patients with active Crohn disease (14).
We recently found that CopA3, a 9-mer disulfide dimer peptide (LLCIALRKK-NH2, D-form) isolated from the Korean dung beetle, exhibited neurotropic effects in neuronal cell lines and mouse brain stem cells (15) and also showed antimicrobial activity (16). Although the biological activity of CopA3 has been examined in a variety of cellular systems (15, 16), no previous study has examined its actions on mucosal epithelial cells and gut inflammation. Here, we show for the first time that CopA3 strongly increases cell proliferation in colonic epithelial cells, enhances the mucosal barrier, and inhibits both C. difficile toxin A-induced enteritis and DSS-induced colitis in mice. These results indicate that CopA3 resembles EGF in its ability to enhance the epithelial barrier and induce colonic epithelial cell proliferation and thus might potently inhibit gut inflammation caused by various factors.
Experimental Procedures
Peptide Synthesis
The CopA3 peptide (from Dung beetles, Copris tripartitus) and P4 peptide (RLLLAIGRG-NH2, from the white-spotted flower chafer, Protaetia brevitarsis) were synthesized by AnyGen (Gwang-ju, South Korea) (15, 16). The peptides were purified by reverse phase HPLC using a Capcell Pak C18 column (Shiseido, Japan) and eluted with a linear gradient of water-acetonitrile (0–80%) containing 0.1% trifluoroacetic acid (45% recovery). The identity of the peptide was confirmed by electrospray ionization mass spectrometry (Platform II; Micromass, Manchester, UK). To form the interchain disulfide bond of the CopA3, the synthetic peptide was dissolved in acetonitrile-H2O (50/50) solution and then oxidized in an aqueous 0.1 m NK4HCO3 solution (pH 6.0–6.5) for 24 h.
Cell Culture and Reagents
Human colorectal adenocarcinoma HT29 cells (American Type Culture Collection, Manassas, VA) were maintained in McCoy's 5A medium containing 10% FBS, 1% penicillin, and 1% streptomycin (Invitrogen) in a 37 °C humidified incubator with a 5% CO2 atmosphere. The polyclonal antibodies against p21Cip1/Waf1, p27Kip1, Sp1, E-cadherin, GAPDH, EGF receptor, siRNAs against human p21Cip1/Waf1 (sc-29427), and control siRNA (scrambled siRNA; sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The polyclonal antibodies against caspase-3 and phospho-ERK1/2 were purchased from Cell Signaling Technology (Beverly, MA). The monoclonal antibody against Ki67 was purchased from Leica Biosystems (Buffalo Grove, IL). The β-actin antibody, propidium iodide (PI), MTT dye, EGF, cycloheximide (transcription inhibitor), and MG132 (proteasome inhibitor) were purchased from Sigma-Aldrich. The p21Cip1/Waf1-expressing adenovirus was obtained from Vector Biolabs (Philadelphia, PA). The mouse Ig blocking reagent was obtained from Vector Laboratories (Burlingame, CA).
C. difficile Toxin A Preparation
Toxin A was purified from C. difficile strain VPI 10463 (American Type Culture Collection, Manassas, VA) as described previously. The purity of native toxin A was assessed by gel electrophoresis, which confirmed a single protein at the expected molecular mass of 307 kDa (16).
BrdU Cell Proliferation Assay
The proliferation of CopA3-treated cells was measured based on the rate of DNA synthesis, using a BrdU cell proliferation assay kit (Roche Applied Science) according to the manufacturer's instructions (15). Briefly, HT29 cells (105 cells/well) were seeded in 96-well plates and incubated with or without CopA3 and then further incubated with the provided BrdU mixture for 12 h. The cells were fixed, incubated with the anti-BrdU antibody for 1 h, and then incubated with HRP-conjugated goat anti-mouse IgG for 1 h. Absorbances at 450 and 540 nm were determined using a microplate reader (model 3550; Bio-Rad). The proliferations of mouse colonic epithelial cells treated with or without CopA3 were also measured. Colonic explants were obtained from male CD1 mice (Daehan Biolink, Daejeon, South Korea); washed in PBS, treated with or without CopA3 for 36 h in McCoy's 5A medium containing 10% FBS, 1% penicillin, and 1% streptomycin; cultured in a humidified 5% CO2 atmosphere at 37 °C; and then further incubated with the BrdU mixture for 12 h. The conditioned tissues were fixed using 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 for 2 h, incubated with the anti-BrdU antibody for 1 h, and then incubated with HRP-conjugated goat anti-mouse IgG for 1 h. The tissues were washed with H2O, sonicated in PBS containing an EDTA-free protease inhibitor mixture (Roche), and centrifuged (11,000 × g for 10 min at 4 °C). Clear supernatants were collected, and the amount of BrdU in supernatants was measured.
Immunofluorescence Staining
CopA3 (2 μg/ml in the drinking water) was administered to male C57BL/6 mice for 5 days. The mice were euthanized, and their colons were isolated, fixed with 4% paraformaldehyde, and embedded in paraffin. Immunofluorescence staining was performed as previously described (17). Briefly, proliferating cells were stained using a Mouse on Mouse kit (Vector Laboratories) with a primary monoclonal antibody against Ki67 (clone MM1; diluted 1:100; Leica) in PBS with 0.3% Triton X-100, and fluorescein anti-mouse IgG (Vector Laboratories; diluted 1:100) as the detection antibody. Each slide was mounted using Prolong Gold anti-fade reagent with DAPI (Life Technologies) and analyzed with a Bio-Rad MRC 1024 confocal scanning laser microscope equipped with a krypton/argon mixed gas laser as a light source.
Cell Viability
HT29 cells treated with the various agents were incubated with 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide dye for 2 h. The solubilization (DMSO) was added, and the absorbance was determined at 570 nm in a microplate reader (model 3550; Bio-Rad) (15).
C. difficile Toxin A-induced Acute Mouse Enteritis
CopA3 at 2 μg/ml was administered to CD1 mice ad libitum in drinking water for 7 days. Control mice were given water only for this period. The mice were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg). Ileal loops (3 cm) were prepared by silk ligation and then lumenally injected with control buffer (PBS) and toxin A (3 nm) in a volume of 100 μl of PBS. After 4 h, mice were sacrificed, and ileal loop tissues were collected to evaluate acute inflammatory responses (3). This study was approved by the Animal Care and Use Committee of Daejin University (Pocheon, South Korea).
DSS-induced Chronic Mouse Colitis
Male C57BL/6 mice were pretreated with CopA3 (2 μg/ml) for 7 days and then given drinking water containing 2% DSS (MP Biomedicals, Solon, OH) plus CopA3 for 16 days. The administration of P4 was conducted at the same condition as described above. Control mice received water only for 7 days prior to DSS administration for 16 days. For this study, the weights of the mice were measured every day, and the survival rate was also recorded (18). This study was approved by the Animal Care and Use Committee of Daejin University.
Measurement of Mucosal Macromolecular Permeability in the Mouse Gut
The mice were anesthetized, and both renal pedicles were ligated with 5–0 silk to prevent urinary excretion of the fluorescent probe. Silk ligation was used to prepare loops in the colons of mice exposed to CopA3 with or without DSS-induced colitis and in the ilea of the small intestine in mice subjected to toxin A-induced enteritis. Each loop was lumenally injected with normal saline (0.3 ml, PBS) containing fluorescein-labeled dextran (molecular weight, 4,000; 25 mg/ml; Sigma) using a 0.5-ml U-100 insulin syringe. To keep the animals warm and protect the dye from light exposure, each mouse was covered with an aluminum foil blanket. After 3 h, 0.5 ml of blood was collected by cardiac stab. The blood was centrifuged at 5000 rpm for 10 min, and the supernatant was diluted 1:2 in PBS (pH 7.3). The concentration of fluorescein-labeled dextran was determined with a GloMax 20/20 (Promega, Madison, WI), as previously described (19).
IL-6 and TNF-α Measurement
To evaluate colonic inflammation levels, toxin A-treated ileal loops and colonic tissues of DSS-induced colitis of the above described mice were washed in cold PBS, homogenized in cold PBS, and centrifuged (11,000 × g, 10 min at 4 °C), and the supernatants were collected. The supernatants used for measuring mouse IL-6 and TNF-α by ELISA using R&D Systems (Minneapolis, MN) (3).
Histopathology Assessment
Hematoxylin- and eosin-stainedcolonic sections were coded for blind microscopic assessment of inflammation. Histological scoring was based on two parameters. Severity of inflammation was scored as follows: 0, rare inflammatory cells in the lamina propria; 1, increased numbers of granulocytes in the lamina propria; 2, confluence of inflammatory cells extending into the submucosa; and 3, transmural extension of the inflammatory infiltrate. Epithelial damage was scored as follows: 0, intact crypts; 1, loss of the basal one-third; 2, loss of the basal two-thirds; and 3, entire crypt loss. The histologic severity of colitis was graded in a “blinded” fashion by a pathologist (J. L.) (18).
Subcellular Fractionations and Identification of Cellular Location of CopA3
HT29 cells (105 cells/well) were treated with CopA3 (20 μg/ml) for 2 h and lysed with 500 μl of subcellular fractionation buffer (250 mm sucrose, 20 mm HEPES, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, and an EDTA-free protease inhibitor mixture). The lysates were then passed through a 25-gauge needle 10 times and centrifuged at 720 × g for 5 min at 4 °C. The pellets were washed with fractionation buffer, passed through a 25-gauge needle 10 times, and centrifuged again at 720 × g for 10 min at 4 °C to obtain the nuclear pellet. The supernatants were placed in fresh 1.5-ml tubes and ultracentrifuged at 10,000 × g (32,000 rpm in an XL-90 ultracentrifuge with a SW-60 Ti rotor; Beckman). The supernatants were then centrifuged at 100,000 × g for 1 h at 4 °C. The supernatants (cytosolic fractions) were removed, and the pellets were washed with fractionation buffer, passed through a 25-gauge needle 10 times, and centrifuged for 45 min. The resulting pellets were taken as representing the membrane fractions (20). The samples were analyzed using HPLC (21). Briefly, the subcellular fractions were separated on a Kinetex C18 column (4.6 × 250 mm, 5 μm; Phenomenex, Boston, MA) using a Hewlett Packard series 1100 HPLC system equipped with a DAD detector set at 230 nm. Elution was performed with TFA in acetonitrile, as follows: 0.1% for 5 min; followed by a 5–65% gradient for 30 min; and then a column wash with 100% acetonitrile, adjusted to pH 7.0 and introduced at a flow rate of 1.0 ml/min. The column temperature was controlled at 35 °C, and the typical sample volume was 20 μl.
Reverse Transcription-PCR
Total RNA was isolated from HT29 cells using TRIzol reagent (Life Technologies), and reverse transcription was performed (15). PCR for human p21Cip1/Waf1 (370 bp) was performed using primers 5′-GGCAGACCAGCATGACAGATTT-3′ (sense) and 5′-GGCGGATTAGGGCTTCCTCT-3′ (antisense). β-Actin was amplified as the control. PCR was conducted for an optimal number of cycles at 94 °C for 1 min, 58 °C for 1 min, and 72 °C for 1 min. The amplified PCR products were fractionated by 1% agarose gel electrophoresis and visualized by ethidium bromide staining.
Cell Cycle Analysis and Flow Cytometry
For flow cytometric analysis, HT29 cells were washed with cold PBS, trypsinized, and fixed in 70% EtOH for at least 30 min on ice in 1.5-ml tubes. Fixed cells were centrifuged, pelleted, and then stained with RNase A (100 μg/ml) and propidium iodide (40 μg/ml) for 30 min at room temperature. The percentages of cells in the sub-G1, G1, S, and G2/M phases of the cell cycle were analyzed with a FACScan and CellQuest 3.0 software (BD Biosciences) (22).
Immunoprecipitation
For immunoprecipitation, HT29 cells were lysed in lysis buffer containing 20 mm Tris (pH 7.4), 1 mm EDTA, 5 mm EGTA, 10 mm MgCl2, 50 mm β-glycerophosphate, 2 mm DTT, 1 mm Na3VO4, 1 mm PMSF, and 4 μg/ml aprotinin. Cell lysates were sonicated and then centrifuged at 10,000 × g for 10 min. The supernatant was precleared with protein A/G plus-agarose (sc-2003; Santa Cruz Biotechnology) for 1 h. After centrifuge, supernatants were collected, incubated with antibodies against p21Cip1/Waf1 and p27Kip1 at 4 °C for 2 h, and additionally incubated with protein A/G Plus-agarose at 4 °C for overnight. The immune complexes precipitated by protein A/G were washed twice in lysis buffer, resuspended in 1 × SDS sample buffer, and measured by immunoblot analysis (15).
In Vitro Ubiquitination Assays
Ubiquitination assays were carried out with 400 nm S5a protein (substrate), 20 nm E1, 350 nm E2, 150 nm E3 (GST-MuRF1), and 50 μm ubiquitin in conjugation buffer (20 mm Tris-Cl, pH 7.5, 20 mm KCl, 5 mm MgCl2, and 1 mm dithiothreitol), using a MuRF1 ubiquitin ligase kit (Boston Biochem, Cambridge, MA) according to the manufacturer's instructions (23). Briefly, the reaction was initiated by adding buffer containing ATP (4 mm), and the mixtures were incubated at 37 °C for 1 h in the presence or absence of CopA3. Ubiquitinated S5a was analyzed by SDS-PAGE followed by immunoblotting with an anti-S5a antibody. Alternatively, p21Cip1/Waf1 immunoprecipitated from HT29 cells was used as a substrate (instead of S5a) for this ubiquitin ligase assay (24, 25).
Statistical Analysis
The results are presented as mean values ± S.E. The data were analyzed using the SIGMA-STAT professional statistics software package (Jandel Scientific Software, San Rafael, CA). Analyses of variance with protected t tests were used for intergroup comparisons.
Results
CopA3 Increases the Proliferation of HT29 Cells
Based on our previous finding that CopA3 promotes the growth of neurons (15), we evaluated its effect on colonic epithelial cell growth. We treated human colonocytes (HT29 cells) with different concentrations of CopA3 (1, 5, 10, 20, and 50 μg/ml) for 48 h and used BrdU uptake assays to measure cell growth. As shown in Fig. 1A, CopA3 (20 μg/ml) treatment increased cell proliferation by 20% compared with the medium-only control, and the level was similar to that observed at 50 μg/ml concentration. The increases in cell proliferation seen at 48 and 72 h in colonocytes treated with 20 μg/ml of CopA3 were similar to those seen in cells exposed to 100 ng/ml of EGF (Fig. 1B). PI staining and FACS analysis revealed that 20 μg/ml of CopA3 increased the levels of DNA synthesis at 48 h (∼11% increase in S phase cells compared with medium-only controls), similar to what was seen in 100 ng/ml of EGF-treated cultures for 48 h (∼13% increase in S phase cells) (Fig. 1C). The nine-amino acid P4 insect peptide, which was isolated from the white-spotted flower chafer (P. brevitarsis) and has an antibacterial activity similar to CopA3 (data not shown), did not affect the proliferation of HT29 cells (Fig. 1D), suggesting that the proliferative effect of CopA3 on HT29 cells is specific. Because treatment of AML-2 and U937 human leukemia cells with a high concentration of CopA3 (∼150 μg/ml) was previously reported to cause apoptosis (26), we assessed whether CopA3 was cytotoxic toward HT29 cells. However, the 20 μg/ml of CopA3 used in this study did not show any toxicity against the tested cell line (Fig. 1E).
FIGURE 1.
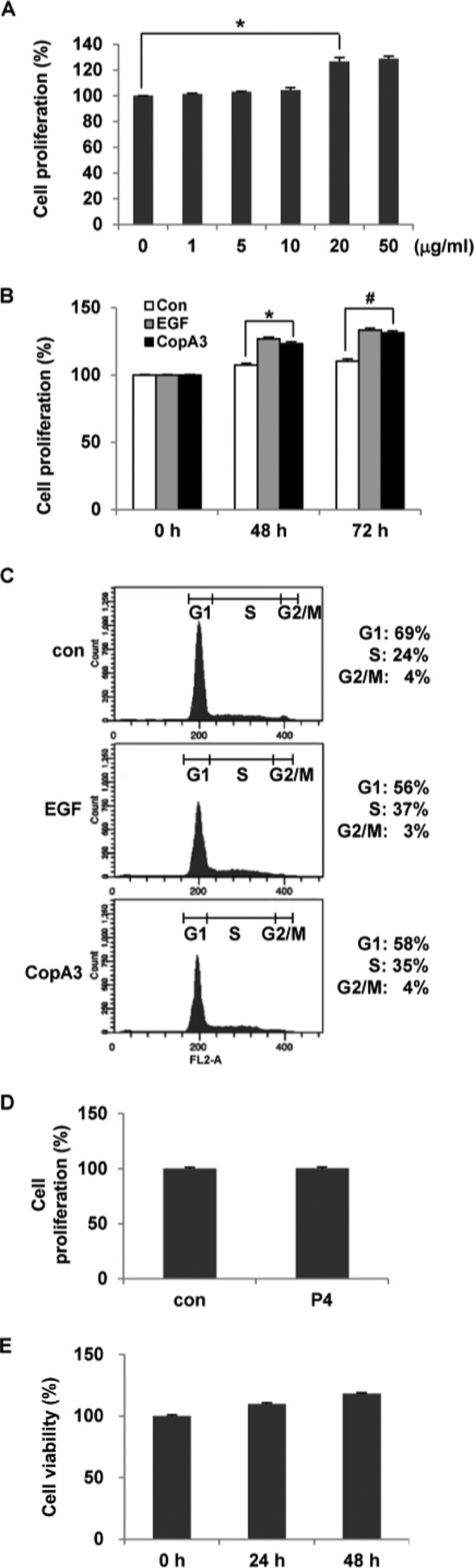
The insect peptide CopA3 enhances the proliferation of HT29 cells. A, HT29 cells (105 cells/well) were treated with different concentrations of CopA3 for 48 h, and cell proliferation was measured by BrdU cell proliferation assays. The results represent the means ± S.E. of three experiments performed in triplicate. *, p < 0.01. B, HT29 cells were treated with medium (con), EGF (100 ng/ml) or CopA3 (20 μg/ml) for 48 and 72 h, and cell proliferation was assessed by BrdU cell proliferation assays. *, p < 0.005; #, p < 0.001. C, HT29 cells were treated with medium, EGF, or CopA3 for 48 h, and DNA synthesis was measured by PI staining and FACS. D, HT29 cells were treated with P4 (20 μg/ml) for 48 h. E, HT29 cells were incubated with CopA3 (20 μg/ml) for 24 and 48 h, and cell viability was measured by MTT assay. The bars represent the means ± S.E. from three experiments performed in triplicate.
Effects of CopA3 on Epithelial Cell Proliferation and Mucosal Barrier Function in the Mouse Colon
We next assessed whether oral administration of CopA3 could increase the number of proliferating epithelial cells in the mouse colon. Male C57BL/6 mice were given drinking water containing 2 μg/ml CopA3 for 5 days and then euthanized, and proliferation of colonic epithelial cells was assayed by immunofluorescence staining with a monoclonal Ki67 antibody. As shown in Fig. 2A (top panels), the numbers of colonic crypts that showed Ki67 labeling (indicating proliferation) were significantly increased following exposure to CopA3. The numbers of apoptotic cells (TUNEL-positive cells; arrows in the middle panels) were also highly increased in colonic epithelial cells (E-cadherin positive cells; bottom panels) of CopA3-treated mice compared with control mice (Fig. 2A). The numbers of Ki67-positive proliferating cells in the crypt region were also significantly higher in CopA3-treated mice (Fig. 2B). The high rates of proliferation and apoptosis observed among CopA3-treated colonic epithelial cells indicate that they experienced increased epithelial cell turnover. This effect of CopA3 was additionally confirmed in mouse colonic explants maintained in short term organ culture and exposed to 20 μg/ml CopA3 for 48 h. The epithelial cell proliferation levels of CopA3-treated explants were 20% higher than the control levels (Fig. 2C).
FIGURE 2.

CopA3 enhances the proliferation and barrier function of mouse colonic epithelial cells. A, male C57BL/6 mice were given drinking water supplemented with or without CopA3 (2 μg/ml) for 5 days, colons were isolated, and proliferating cells were stained with a monoclonal Ki67 antibody (top panels; blue, DAPI; and green, Ki67). Apoptosis was also analyzed by TUNEL staining (arrows in the middle panels; brown, nuclei). Epithelial cells were subjected to immunohistochemical staining with an antibody specific to E-cadherin, an epithelial cell marker (bottom panels; red, epithelial cells). Representative images from each experimental group are shown. B, quantification of Ki67-positive cells per crypt (n = 20/group). *, p < 0.01. C, colonic explants were incubated with medium (con) or CopA3 (20 μg/ml) for 48 h, and the proliferation of colonic epithelial cells was assessed by a modified BrdU cell proliferation assay. The results represent the means ± S.E. from three experiments performed in triplicate. *, p < 0.005. D, mucosal macromolecular permeability assay in the guts of mice treated with CopA3 (2 μg/ml) or P4 (2 μg/ml) for 5 days (n = 8/group). *, p < 0.005.
Next, we tested whether CopA3 could also increase the mucosal barrier. Mice were treated with CopA3 (2 μg/ml) for 5 days; colonic loops were prepared and injected with fluorescein-labeled dextran; and after 3 h, the concentration of fluorescence was measured in blood. As shown in Fig. 2D, our macromolecular permeability assay indicated that the mucosal barrier function was significantly increased in CopA3-treated mice compared with control mice given drinking water alone. In contrast, P4 had no effect on the mucosal permeability. These results suggest that CopA3 stimulates both colonic epithelial cell growth and epithelial barrier function.
CopA3 Blocks C. difficile Toxin A-induced Enteritis
EGF and FGF have been shown to inhibit inflammatory responses in the colon by promoting epithelial cell growth, thereby enhancing the physical barrier function (6, 12). Because EGF was shown to markedly inhibit the acute enteritis caused by C. difficile toxin A in a mouse model (10), we herein assessed whether CopA3 could also protect against toxin A-induced gut inflammation. HT29 cells were pretreated with CopA3 (20 μg/ml) for 1 h and exposed to toxin A (3 nm) for 72 h. As shown in Fig. 3A, CopA3 significantly inhibited the apoptosis caused by toxin A: the sub-G1 population comprised 28% of toxin A-treated cells but only 3% of toxin A/CopA3-co-treated cells. In contrast, the P4 insect peptide (which had no effect on epithelial cell proliferation) did not inhibit the apoptosis caused by toxin A. CopA3 treatment also inhibited the toxin A-induced activation of the apoptosis-mediating factor, caspase-3 (Fig. 3B). To exclude the possibility that CopA3 inhibits toxin A by direct binding, we pretreated cells with CopA3 for 1 h, removed the medium, washed the cells three times with fresh medium, and then exposed the cells to toxin A for 72 h (3). As shown in Fig. 3C, CopA3 pretreatment inhibited toxin A-induced apoptosis, suggesting that CopA3 does not directly bind to toxin A. Notably, however, CopA3 pretreatment blocked the toxin A-induced reduction of cell viability (Fig. 3D).
FIGURE 3.

CopA3 inhibits C. difficile toxin A-induced cell toxicity and mouse enteritis. A, HT29 cells (105 cells/well) were pretreated with CopA3 (20 μg/ml) or P4 (20 μg/ml) for 1 h and then incubated with medium (con), toxin A alone (TxA, 3 nm), toxin A plus CopA3, or toxin A plus P4 for 72 h. Cells with fragmented DNA were quantified by PI staining and FACS. The presented results are representative of three independent experiments. B, cells were pretreated with CopA3 for 1 h and incubated with medium (con) or 3 nm toxin A for 72 h. Cell lysates were resolved by 15% SDS-PAGE, and blots were probed with antibodies against caspase-3 and β-actin. C, HT29 cells were pretreated with CopA3 for 1 h, residual CopA3 was removed by washing, and the cells were exposed to toxin A for 72 h. D, cells were pretreated with CopA3 for 1 h and then incubated with toxin A, or toxin A plus CopA3 for 48 h, and cell viability was measured by MTT assay. *, p < 0.001. E–I, male CD1 mice (n = 10/group) were given drinking water containing CopA3 (2 μg/ml) or P4 (2 μg/ml) for 7 days, and then ileal loops were prepared and exposed to 100 μl of PBS (con) or 100 μl of PBS containing toxin A (TxA, 3 nm) for 4 h. The concentrations of IL-6 (E) and TNF-α (F) were measured by ELISA. The bars represent the means ± S.E. from three independent experiments performed in triplicate (*, p < 0.005). G, light micrographs of mouse ileum (hematoxylin and eosin staining, ×200). H, histological scores. *, p < 0.005; #, p < 0.001 compared with mice that received toxin A alone. I, in the mouse enteritis models mentioned above, mucosal macromolecular permeability was measured. The results represent the means ± S.E. from three experiments performed in triplicate (n = 8/group, *, p < 0.005).
We then investigated whether CopA3 could inhibit toxin A-induced acute mouse gut inflammation in vivo. CD1 mice were given CopA3- or P4 (control)-containing drinking water for 7 days. The mice were then anesthetized, and closed ileal loops were injected with toxin A for 4 h to induce acute enteritis. The levels of IL-6 and TNF-α (two known proinflammatory cytokines) were measured in each ileal loop. As shown in Fig. 3 (E and F), toxin A strongly increased IL-6 and TNF-α production, but these increases were abolished by CopA3 co-treatment. CopA3 had a similar inhibitory effect on mucosal damage in the mouse ileal epithelium (Fig. 3G). In contrast, P4 had no effect on the inflammatory responses or mucosal damage in this model (Fig. 3, E–G). Toxin A increased the inflammation score (in terms of epithelial damage and neutrophil infiltration), whereas CopA3-co-treated mice scored significantly lower than those treated with toxin A alone (Fig. 3H).
We also tested whether CopA3 blocked the toxin A-induced increase of mucosal permeability. Our results revealed that toxin A caused a massive increase in the macromolecular permeability of the ileal epithelium; this was significantly recovered by CopA3 co-treatment but not by P4 co-treatment (Fig. 3I). These results suggest that, similar to EGF, CopA3 can increase epithelial cell growth and mucosal barrier function, thereby inhibiting toxin A-induced acute inflammation in the gut.
CopA3 Ameliorates Disease Symptoms in DSS-induced Mice Colitis
We next investigated the inhibitory effect of CopA3 on the DSS-induced colitis model. The mice were given drinking water containing CopA3 (2 μg/ml) or P4 (2 μg/ml) for 7 days, and then their drinking water was further supplemented with 2% DSS for the indicated periods. DSS-treated mice showed gradual decreases in body weight that were not recovered by the end of the experiment. However, mice co-treated with CopA3 showed a marked preservation of body weight from 9 to 16 days (Fig. 4A). As expected, P4 did not affect the body weight loss caused by DSS (Fig. 4A). Moreover, mice co-treated with DSS plus water or P4 exhibited sharply decreased survival beginning on day 9, and all of these mice died with severe colitis by day 16. In contrast, DSS/CopA3-co-treated mice exhibited 100% survival, similar to the nontreated control group (Fig. 4B). The colonic lengths in the water/DSS- and DSS/P4-co-treated mice (i.e. those with DSS-induced colitis) were significantly shorter (<5 cm) than that in nontreated control mice (7.8 cm), whereas the colonic length in CopA3/DSS-treated mice (6.7 cm) was significantly greater than that in mice with DSS-induced colitis (Fig. 4C). Moreover, substantial increases in TNF-α, IL-6, and mucosal damage were seen in DSS/water- and DSS/P4-treated mice compared with nontreated control mice, but these parameters were significantly rescued in CopA3/DSS-treated mice (Fig. 4, D–F). Histological scoring clearly revealed that co-treatment of mice with DSS plus CopA3 (but not P4) inhibited all DSS-induced inflammatory parameters, including epithelial damage and neutrophil infiltration (Fig. 4G). Our mucosal macromolecular permeability assay revealed that CopA3, but not P4, significantly inhibited the DSS-induced increase in mucosal permeability (Fig. 4H). These results suggest that the ability of CopA3 to increase epithelial cell growth and epithelial barrier function may be critical for its anti-inflammatory effects in the colon.
FIGURE 4.

Therapeutic effects of CopA3 in the DSS-induced mouse model of colitis. C57BL/6 mice (n = 30/group) were given drinking water containing 2% DSS with or without CopA3 (2 μg/ml) or P4 (2 μg/ml) for 16 days. Control mice received plain drinking water. A, body weight was measured daily throughout the experiment and is expressed as the mean percent change from the starting body weight. The presented results represent the means ± S.E. Closed squares, control mice; open circles, mice treated with DSS and CopA3; open triangles, mice treated with DSS and P4; closed diamonds, mice treated with DSS alone. B, survival was measured throughout the experiment. C, colon lengths were measured upon death or at the end of the experimental period. The bars represent the means ± S.E. from three independent experiments performed in triplicate. *, p < 0.001. D and E, the concentrations of TNF-α (D) and IL-6 (E) were measured from colon tissues obtained from each group. *, p < 0.005. F, light micrographs of mouse colon (hematoxylin and eosin staining, ×200). G, histological scores. *, p < 0.005; #, p < 0.001 compared with mice that received DSS alone. H, mucosal macromolecular permeability (n = 8/group). *, p < 0.005.
The CopA3-dependent Induction of Colonic Epithelial Cell Proliferation Is Associated with Down-regulation of p21Cip1/Waf1
Given our observation that CopA3 enhances colonic epithelial cell growth, we sought to identify some of the intracellular mechanisms responsible for mediating this effect. We previously reported that CopA3 promotes the growth of neuronal cells by down-regulating the cyclin-dependent kinase inhibitor, p27Kip1 (15). In the present study, we observed that CopA3 blocked toxin A-induced cytotoxicity, which is known to increase the expression of the other cyclin-dependent kinase inhibitor, p21Cip1/Waf1, which in colonic epithelial cells triggers cell cycle arrest and apoptosis (4). Therefore, we assessed whether the ability of CopA3 to block toxin A cytotoxicity and enhance colonic epithelial cell growth might be associated with diminished expression of p21Cip1/Waf1 or p27Kip1. We first tested whether CopA3 could inhibit the toxin A-induced up-regulation of p21Cip1/Waf1 in vitro. As shown in Fig. 5A, toxin A strongly induced p21Cip1/Waf1 protein expression, but this effect was significantly decreased by co-treatment with CopA3 (20 μg/ml). In contrast, there was no significant change in the levels of p27Kip1 and phospho-ERK1/2 (which is involved in cell survival and proliferation) (15). P4 had no effect on the tested parameters. Next, we investigated whether p21Cip1/Waf1 was up-regulated in toxin A-induced acute mouse enteritis (Fig. 5B) and DSS-induced chronic mouse colitis in vivo (Fig. 5C). The expression of p21Cip1/Waf1 was markedly increased in both mouse models of gut inflammation but was significantly decreased by CopA3 co-treatment (Fig. 5, B and C). The expression levels of p27Kip1 did not differ in either mouse model (Fig. 5, B and C). These results suggest that p21Cip1/Waf1 may be a critical mediator for various inflammatory responses in the gut and that its suppression by CopA3 inhibits inflammation. HT29 cells infected with adeno-p21Cip1/Waf1 virus (1 × 107 PFU/ml) successfully overexpressed the p21Cip1/Waf1 protein (Fig. 5D, lower panels), and their cell proliferation was markedly decreased compared with control cells infected with a control GFP-expressing adenovirus (Fig. 5D, upper panel). Cells overexpressing p21Cip1/Waf1 also exhibited fewer cells in the S phase (proliferative cells, ∼18%) compared with control GFP-expressing cells (∼28%) (Fig. 5E). As expected, the cell proliferation induced by CopA3 treatment was blocked in p21Cip1/Waf1-overexpressing cells but not control GFP-expressing cells (Fig. 5F). This suggests that p21Cip1/Waf1 is essential for CopA3-dependent colonic epithelial cell proliferation. As shown in Fig. 5G, there were more apoptosis-positive cells in toxin A-treated cultures overexpressing p21Cip1/Waf1 compared with toxin A-treated cultures expressing GFP (Fig. 5G). Interestingly, overexpression of p21Cip1/Waf1 (but not GFP) blocked the ability of CopA3 to inhibit toxin A-induced apoptosis (Fig. 5G, lower panels), and the toxin A-induced activation of caspase-3 was inhibited by CopA3 in GFP-expressing cells but not in p21Cip1/Waf1-overexpressing cells (Fig. 5H). Notably, the expression pattern of p21Cip1/Waf1 after challenging toxin A was consistent with the levels of active caspase-3 (Fig. 5H). This is consistent with a previous report that p21Cip1/Waf1 overexpression markedly activates caspase-3 in colonic epithelial cells (27). Next, HT29 cells were transfected with a p21Cip1/Waf1-targeting siRNA for 24 h to reduce the level of endogenous p21Cip1/Waf1 protein (Fig. 5I) and then treated with toxin A. Toxin A-induced apoptosis and cytotoxicity were significantly decreased in p21Cip1/Waf1 knockdown cells compared with control cells (Fig. 5, I and J). Together, these results suggest that colonocyte-expressed p21Cip1/Waf1 may be a key regulator of the epithelial cell proliferation and cytotoxicity seen in inflamed gut tissues.
FIGURE 5.

CopA3 reduces p21Cip1/Waf1 expression in colonic epithelial cells. A, HT29 cells were pretreated with CopA3 (10 or 20 μg/ml) or P4 (20 μg/ml) for 1 h and incubated with medium (con), toxin A (TxA, 3 nm) alone, toxin A plus P4, or toxin A plus CopA3 for 72 h. Cell lysates were resolved by 15% SDS-PAGE, and blots were probed with antibodies against p21Cip1/Waf1, p27Kip1, phospho-ERK1/2, and β-actin. B, immunoblot analysis was also performed using proteins extracted from ileal loops of the C. difficile toxin A-induced enteritis model described in Fig. 3. C, immunoblot analysis was performed using proteins extracted from colon samples of the DSS-induced colitis model described in Fig. 4. The presented results are representative of three independent experiments. D, HT29 cells (105 cells/well) were infected with a p21Cip1/Waf1-expressing adenovirus (1 × 107 PFU/ml) or a control GFP adenovirus (1 × 107 PFU/ml) for 24 h, and cell proliferation was assessed using BrdU cell proliferation assays. *, p < 0.001. E, cell cycle distribution was analyzed by PI staining and FACS. F, cells were infected with the above-described viruses for 24 h and then incubated with or without CopA3 (20 μg/ml) for 48 h, and cell proliferation was measured by PI staining and FACS analysis. *, p < 0.01. G, cells were infected with the viruses and then incubated with medium, toxin A alone, or toxin A plus CopA3 (20 μg/ml) for 48 h. Apoptosis was measured by PI staining and FACS analysis. H, cells were infected with the viruses for 24 h and then incubated with medium, toxin A, or toxin A plus CopA3 for 48 h. I, cells were transfected with control siRNA or p21Cip1/Waf1 siRNA for 24 h and then incubated with toxin A for 48 h. J, the viability of the cells described in I was measured by MTT assay. *, p < 0.001.
CopA3 Promotes the Growth of Serum-starved Cells by Down-regulating p21Cip1/Waf1
Previous reports found that serum starvation causes growth arrest at the G0/G1 phase of the cell cycle (28) and that serum starvation-dependent growth arrest is associated with p21Cip1/Waf1 induction (29). In addition, p21Cip1/Waf1 has been shown to inhibit colonocyte proliferation (30, 31). Given our observation that CopA3 enhanced colonic epithelial cell proliferation by down-regulating p21Cip1/Waf1, we next assessed whether CopA3 could reverse serum starvation-induced growth arrest and p21Cip1/Waf1 induction. We cultured HT29 cells in serum-free medium for 24, 48, and 72 and then measured the levels of p21Cip1/Waf1. As shown in Fig. 6A, colonocytes exposed to serum starvation for at least 24 h exhibited significant up-regulation of p21Cip1/Waf1, but this effect was markedly reduced by CopA3 co-treatment for all time periods (Fig. 6B). In contrast, p27Kip1 (which was also increased by serum starvation) was not affected by CopA3 treatment (Fig. 6B). These findings suggest that p21Cip1/Waf1 is a unique target for CopA3 in colonic epithelial cells. CopA3 treatment also significantly restored the serum starvation-induced decrease in the number of S phase cells (∼11% serum starvation versus 19% serum starvation + CopA3) (Fig. 6C). We did not observe any CopA3-mediated enhancement of proliferation in p21Cip1/Waf1-overexpressing cells (Fig. 6D, lower panels), whereas GFP-expressing control cells showed significant recovery (to levels comparable with those in serum-exposed cells) of the serum starvation-induced decrease in S phase cells following CopA3 treatment (∼13% no serum versus 21% no serum + CopA3) (Fig. 6D, upper panels). Next, we investigated the effect of CopA3 in mouse colonic explants in an ex vivo model. Colonic biopsies were excised from CD1 mice and incubated with CopA3 (20 μg/ml) in the absence of serum for 48 h. As shown in Fig. 6E, the p21Cip1/Waf1 and p27Kip1 protein levels of colonic explants were increased by serum starvation, and CopA3 treatment markedly reduced this effect for p21Cip1/Waf1 but not p27Kip1 (which showed a further slight increase). These results suggest that the CopA3-dependent induction of colonic epithelial cell proliferation is strongly associated with the down-regulation of p21Cip1/Waf1, which is known to arrest cell growth.
FIGURE 6.
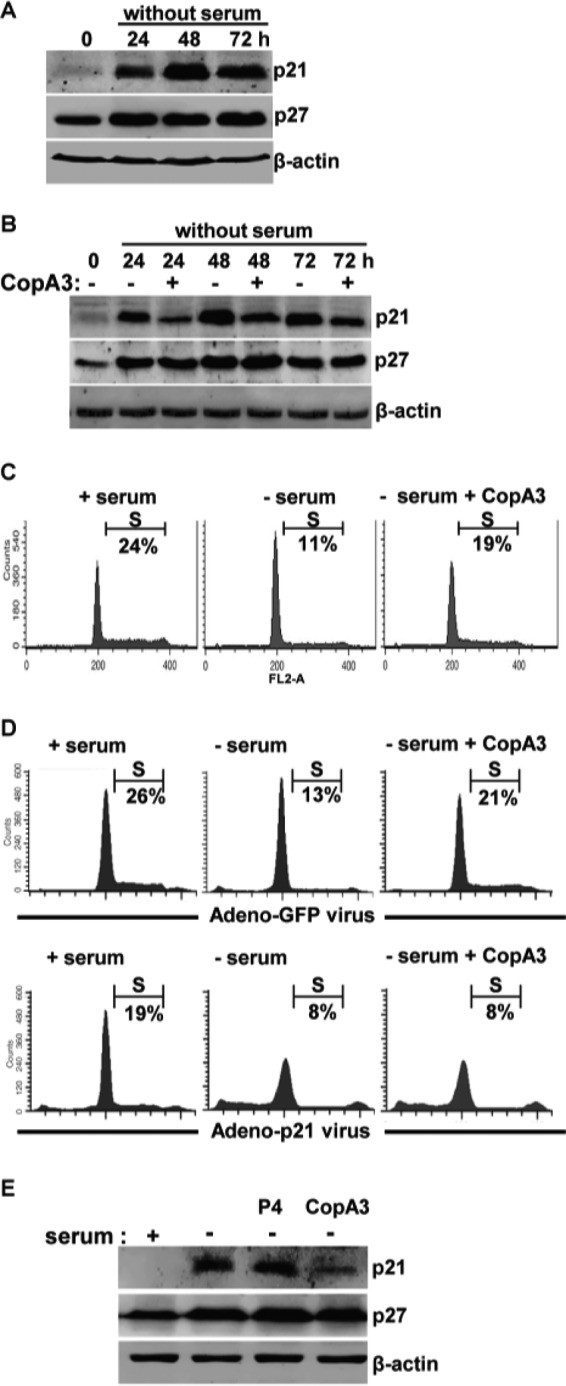
CopA3 restores the growth inhibition caused by serum starvation. A, HT29 cells were incubated in serum-free medium for 24, 48, and 72 h. Cell lysates were resolved by 15% SDS-PAGE, and blots were probed with antibodies against p21Cip1/Waf1, p27Kip1, and β-actin. B, HT29 cells were incubated with medium (0 h), serum-free medium, or serum-free medium plus CopA3 (20 μg/ml) for 24, 48, and 72 h. C, HT29 cells were incubated with medium (+ serum), serum-free medium (− serum), or serum-free medium plus CopA3 (20 μg/ml; − serum + CopA3) for 48 h, and the cell cycle distribution was analyzed by PI staining and FACS. D, HT29 cells were infected with a p21Cip1/Waf1-expressing adenovirus (lower panels) or a control GFP-expressing adenovirus (upper panels) for 24 h and then incubated with medium, serum-free medium, or serum-free medium plus CopA3 for 48 h. The presented results represent the means ± S.E. from three experiments performed in triplicate. E, colonic explants of mice were incubated with medium, serum-free medium, serum-free medium plus P4 (20 μg/ml), or serum-free medium plus CopA3 (20 μg/ml) for 48 h. Cell lysates were resolved by 15% SDS-PAGE, and blots were probed with antibodies against p21Cip1/Waf1, p27Kip1, and β-actin.
CopA3 Down-regulates p21Cip1/Waf1 through Ubiquitination and Proteasomal Degradation
Next, we investigated the mechanism(s) through which CopA3 reduces p21Cip1/Waf1 protein levels in HT29 cells and the mouse colon. We first assessed whether CopA3 inhibited the transcription of p21Cip1/Waf1 but found that CopA3 did not affect the level of the p21Cip1/Waf1-encoding mRNA in HT29 cells (Fig. 7A). We then tested whether CopA3 caused the protein degradation of p21Cip1/Waf1.HT29 cells (which have a low basal expression level of p21Cip1/Waf1) were treated with toxin A to induce p21Cip1/Waf1 protein expression in the presence or absence of the proteasome inhibitor MG132 (10 μm) (15), and p21Cip1/Waf1 degradation was monitored by immunoblot analysis. As shown in Fig. 7B, MG132 co-treatment highly increased toxin A-induced p21Cip1/Waf1 protein levels compared with that in cells treated with toxin A alone. Furthermore, MG132 treatment completely blocked the ability of CopA3 to down-regulate the toxin A-induced increase of p21Cip1/Waf1 protein levels. This indicates that the CopA3-mediated down-regulation of p21Cip1/Waf1 is strongly associated with the proteasomal protein degradation pathway. Next, we measured alterations in the protein half-life of p21Cip1/Waf1 after CopA3 exposure. HT29 cells were treated with 3 nm toxin A for 6 h and then incubated with the translation inhibitor cycloheximide (100 μm) (15) in the presence or absence of CopA3 for the indicated times, and the protein levels of p21Cip1/Waf1 were measured. As shown in Fig. 7C, the half-life of the p21Cip1/Waf1 protein in CopA3-treated cells was shorter than that in cells that did not receive CopA3. In contrast, CopA3 was not found to alter the protein degradation of p27Kip1 (Fig. 7D).
FIGURE 7.

The CopA3-dependent enhancement of ubiquitination shortens the protein half-life of p21Cip1/Waf1. A, HT29 cells (105 cells/well) were pretreated with CopA3 (20 μg/ml) for 1 h and then incubated with medium (con), CopA3 alone, toxin A (TxA, 3 nm) alone, or toxin A plus CopA3 for 6 h. Total RNA was isolated, cDNA was synthesized, and p21Cip1/Waf1 and β-actin were amplified by PCR. The results shown are representative of three separate experiments. B, cells were pretreated with CopA3 for 1 h in the presence or absence of MG132 (10 μm) and then incubated with medium (con), toxin A (TxA) alone, or toxin A plus CopA3 for 24 h. Cell lysates were resolved by 15% SDS-PAGE, and the blots were probed with antibodies against p21Cip1/Waf1 and β-actin. C, cells were treated with toxin A for 6 h (to increase the levels of p21Cip1/Waf1) and then incubated with cycloheximide alone (CHX, 100 μm) or cycloheximide plus CopA3 for the indicate times. The results shown are representative of three separate experiments. D, the protein half-life of p27Kip1 was assessed in cells exposed to CopA3. E and F, cells were pretreated with CopA3 for 1 h in the presence or absence of MG132 and further incubated with medium, toxin A alone, or toxin A plus CopA3. After 24 h, cell extracts were immunoprecipitated (IP) with an anti-p21Cip1/Waf1 antibody (p21) and resolved by 10% SDS-PAGE, and blots were probed (IB) with antibodies against ubiquitin or p21Cip1/Waf1. G, the ubiquitination levels of p27Kip1 (p27) were assessed in cells exposed to CopA3. H, HT29 cells were transiently transfected with a vector expressing HA-tagged ubiquitin and incubated with MG132 and toxin A alone or toxin A plus CopA3. After 24 h, cell extracts were IP with an anti-p21Cip1/Waf1 antibody, the immunoprecipitates were resolved by 10% SDS-PAGE, and the blots were probed with antibodies against HA (for ubiquitinated p21Cip1/Waf1) and p21Cip1/Waf1.
Protein degradation is dependent on the ubiquitin-mediated proteasomal system (32–34). To test whether CopA3 could promote p21Cip1/Waf1 ubiquitination, we pretreated cells with CopA3 for 1 h in the presence or absence of MG132, treated the cells with toxin A for 24 h to induce p21Cip1/Waf1 expression, and then immunoprecipitated cell lysates with an anti-p21Cip1/Waf1 antibody. As shown in Fig. 7E (three leftmost lanes), cells treated with CopA3 in the absence of MG132 exhibited a marked increase in the ubiquitination of p21Cip1/Waf1 (indicated by a ≧30-kDa band reflecting the 21-kDa p21Cip1/Waf1 protein plus the 9-kDa ubiquitin complex) compared with medium-control and toxin A-treated cells. Cells that were treated with CopA3 in the presence of MG132 showed highly increased ubiquitination (Fig. 7E, three rightmost lanes), as confirmed by immunoblotting with an antibody against p21Cip1/Waf1 (Fig. 7F). The expression patterns of p21Cip1/Waf1 and ubiquitinated p21Cip1/Waf1 were inversely correlated (Fig. 7F, three leftmost lanes). In MG132-untreated cells, toxin A exposure was associated with increased p21Cip1/Waf1 protein levels and reduced ubiquitination, and CopA3/toxin A treatment ameliorated these effects; in MG132-pretreated cells, furthermore, these patterns were strongly increased (Fig. 7F, three rightmost lanes). In contrast, CopA3 was not found to alter the ubiquitination of p27Kip1 (Fig. 7G). To further confirm our findings, we transfected HT29 cells with an expression vector for HA-tagged ubiquitin, immunoprecipitated the p21Cip1/Waf1 proteins, and probed them with an antibody against HA. As shown in Fig. 7H, the co-precipitation of p21Cip1/Waf1 with HA-ubiquitin was much higher in CopA3/toxin A co-treated cells compared with those exposed to toxin A alone (Fig. 7H). These results suggest that CopA3 increases the ubiquitination of p21Cip1/Waf1, resulting in its rapid protein degradation and the subsequent enhancement of colonic epithelial cell proliferation.
CopA3 Directly Enhances Ubiquitin Ligase Activity
Given that CopA3 was found to increase the ubiquitination of p21Cip1/Waf1, we assessed whether it could directly enhance ubiquitin ligase activity. We performed an in vitro ligase activity assay by combining CopA3 with the E1, E2, and RING finger E3 ligases, their S5a substrate, ubiquitin, and ATP, as per the utilized ubiquitin ligase kit (35). After 1 h of incubation, the levels of ubiquitinated S5a were measured and compared with those obtained in the presence of P4 or the absence of both. As shown in Fig. 8A, the ubiquitination of S5a by the RING finger E3 ligase was strongly increased by CopA3 but not P4. This suggests that CopA3 directly enhances ubiquitin ligase activity. Next, the p21Cip1/Waf1 protein was isolated from HT29 cell extracts by immunoprecipitation and added into the reaction mixture instead of the S5a substrate. Complexes of the expected molecular mass (∼40 and ∼30 kDa, representing p21Cip1/Waf1 + ubiquitin) were detected (Fig. 8B), suggesting that the human p21Cip1/Waf1 protein could be a substrate for the RING finger E3 ligase included in the assay kit. Moreover, CopA3 markedly increased the ubiquitination of p21Cip1/Waf1, whereas P4 did not (Fig. 8B). As expected, the levels of p21Cip1/Waf1 and ubiquitinated p21Cip1/Waf1 were completely antiparallel: increases in the ubiquitination of p21Cip1/Waf1 were associated with decreases in naïve p21Cip1/Waf1 and vice versa. Immunoblot analysis with an antibody against ubiquitin also revealed that the addition of CopA3 (but not P4) markedly increased the ubiquitination of p21Cip1/Waf1 (Fig. 8C). These results indicate that CopA3 directly enhances ubiquitin ligase activity, increasing the ubiquitination and degradation of p21Cip1/Waf1.
FIGURE 8.
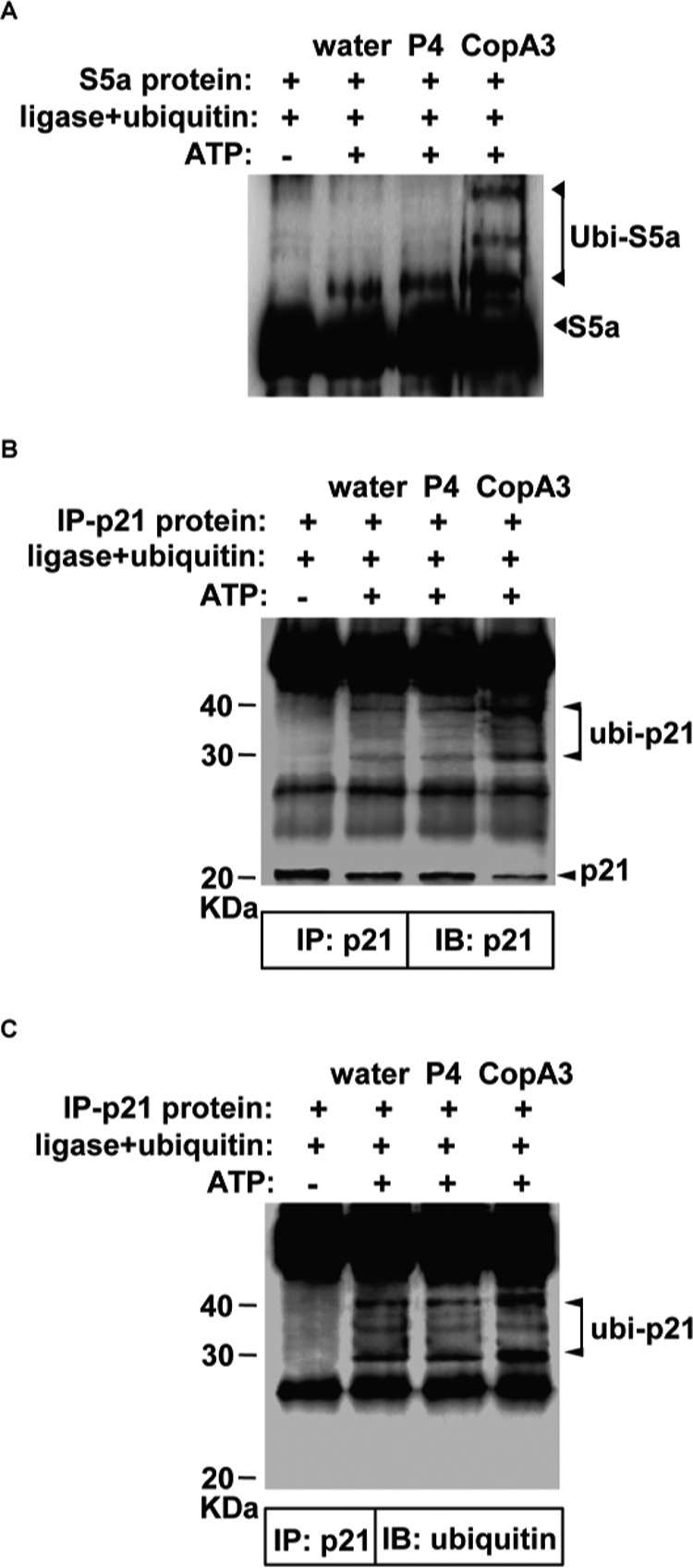
CopA3 directly enhances ubiquitin ligase activity. A, in vitro ubiquitination assays were carried out with the S5a substrate, the E1, E2, and RING E3 ligases, and ubiquitin in the presence or absence of CopA3 (20 μg/ml) or P4 (20 μg/ml). Reactions were initiated by the addition of ATP and samples were incubated at 37 °C for 1 h. The presented results are representative of three independent experiments. B, HT29 cell lysates were prepared and immunoprecipitated with an anti-p21Cip1/Waf1 antibody. C, immunoprecipitated p21Cip1/Waf1 was added to the ubiquitination assay reaction buffer in place of S5a, and ubiquitinated p21Cip1/Waf1 was visualized with antibodies against p21Cip1/Waf1 or ubiquitin. IB, immunoblot; IP, immunoprecipitation.
Cytosolic Location of CopA3 in Human Colonic Epithelial Cells
CopA3 is known to have various biological effects, but its location within cells is not known. To clarify the intracellular localization of CopA3, we first attempted to attach a fluorophore or conjugated biotin to CopA3 but found that CopA3 was easily crystallized (data not shown). Thus, we attempted to determine the intracellular localization of CopA3 by subcellular fractionation of CopA3 (20 μg/ml, 2 h)-treated cells, followed by HPLC analysis. Normalized portions of each extract were subjected to immunoblot analysis using antibodies against protein markers for the cytosol (GAPDH), nucleus (Sp1), and membrane (EGF receptor) (20). The results demonstrated that we obtained clear fractionation with low cross-contamination (Fig. 9A). HPLC analysis (Fig. 9B) revealed that CopA3 had a retention time of 13.9 min; it was found as a dimerized form in the cytosolic fraction (∼14.7 μg/ml) but was not present in the membrane or nuclear fractions. We also detected residual CopA3 in the cell culture medium (∼4.8 μg/ml). These results suggest that CopA3 directly enters the cells rather than regulation of cell growth by binding to membrane receptors on the cell surface. Because proteasomes and ubiquitin ligase are located in the cytosol (32–34), this finding is consistent with the notion that CopA3 directly enhances ubiquitin ligase activity, leading to p21Cip1/Waf1 degradation and consequent enhancement of colonic epithelial cell growth.
FIGURE 9.
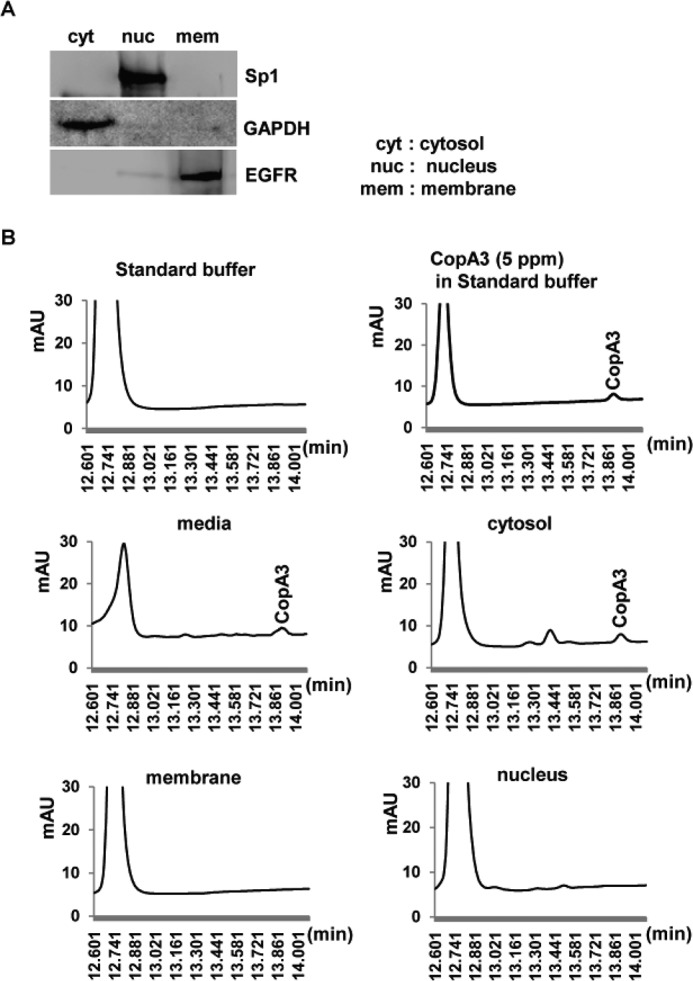
Dimerized CopA3 is detected in the cytosol of human colonocytes. A, HT29 cells were treated with CopA3 (20 μg/ml) for 2 h and separated into cytosolic (cyt), nuclear (nuc), and membrane (mem) fractions, which were probed for purity using primary antibodies against Sp1, GAPDH, and EGF receptor (EGFR), respectively. The presented results are representative of three separate experiments. B, HPLC analysis revealing the cytosolic location of CopA3.
Discussion
Gut epithelial cells form a physical barrier between the lumen and the submucosa (1, 2). Damage to the colonic epithelial cell layer and loss of barrier function is observed in humans with severe gut inflammation, whereas preservation of the cell barrier is believed to have potential for inhibiting gut inflammation (5). Consistent with this, EGF, which increases colonic epithelial cell growth and enhances barrier function, shows high therapeutic efficacy against various gut inflammations (7–9). Here, we report for the first time that the insect peptide, CopA3, acts against gut inflammation by increasing the proliferation of epithelial cells and enhancing mucosal barrier function in the gastrointestinal (GI) tract. We found that CopA3 increased cell division in a human colonocyte cell line, and its oral administration enhanced the number of proliferating crypt cells in the mouse colon. This increased proliferation of epithelial cells was accompanied by accelerated cell apoptosis, suggesting that CopA3 triggers a marked increase in epithelial cell turnover in the mouse colon. Moreover, CopA3 co-treatment significantly reduced the mucosal macromolecular permeability caused by toxin A or DSS. These results suggest that, similar to the case of EGF, the anti-inflammatory effect of CopA3 is associated with its ability to enhance epithelial barrier function.
We previously reported that expression of the cell cycle inhibitor, p21Cip1/Waf1, is significantly correlated with disease activity in C. difficile toxin A-induced mouse enteritis (4). Here, we examined the involvement of p21Cip1/Waf1 in these effects of CopA3. In patients with active ulcerative colitis, 57% of the crypt cells in inflamed tissues reportedly exhibited high p21Cip1/Waf1 expression compared with those in normal colorectal tissues (36). Given that up-regulation of p21Cip1/Waf1 causes epithelial cell apoptosis (27), these results indicate that pathological up-regulation of p21Cip1/Waf1 appears to cause cell death, loss of barrier function, and eventual aggravation of the inflammatory response. Because accelerated epithelial cell turnover is accompanied by an increase in epithelial cell apoptosis, high p21CIP1/WAF1 expression has been associated with the active phase of ulcerative colitis (37, 38). These observations indicate that p21Cip1/Waf1 plays a key role in the epithelial cell turnover of the GI tract, further suggesting that the strict regulation of p21Cip1/Waf1 is essential for controlling inflammatory responses in the gut. Interestingly, our present results show for the first time that CopA3 selectively targets p21Cip1/Waf1 and inhibits its up-regulation during gut epithelial cell injury.
The ubiquitin/proteasome pathway mediates the degradation of proteins, reducing their half-life to impact many cellular events (32–34). Ubiquitin ligase has two critical domains: a RING (really interesting new gene) domain and the tyrosine kinase-binding domain (TKBD) (39). The RING domain binds the ubiquitin-donor protein, E2, whereas the TKBD binds substrates for ubiquitination (39). Phosphorylation of ubiquitin ligase triggers conformational changes that enable it to bind with the substrate and E2, thereby initiating ligase activity (23). We observed that CopA3 directly enhances the activity of ubiquitin ligase. Furthermore, we found that CopA3 is internalized to the cytosol of human colonocytes where ubiquitin ligase is found. We speculated that CopA3 may directly bind to the TKBD domain of E3 in the cytosol, triggering the conformational change needed to reveal the substrate-binding region and increase ubiquitin ligase activity. Indeed, it has been shown that the TKBD domain of ubiquitin ligase binds to a peptide of receptor tyrosine kinase to act as an adapter protein (40, 41).
We detected CopA3 in a dimerized form in the cytosolic fraction but not in the membrane or nuclear fractions (Fig. 9B). This strongly suggests that even though the disulfide bridge of CopA3 (which is formed through a medial cysteine) is vulnerable to changes in the pH and oxidation/reduction conditions (data not shown), the structure of CopA3 is preserved after its internalization into cells. We further observed that monomeric CopA3 peptides readily form dimers in cell culture medium but not water (data not shown). Our previous report showed that the D-form of the CopA3 peptide exhibits antimicrobial activity in the mouse colon following oral administration (16). In this study, we found that form of CopA3, which was of the D-type structure and synthesized so as to not biodegrade in the gastrointestinal tract, reaches the colon and promote proliferation. These results suggest that the peptide structure of CopA3 is very stable, rendering it able to confer therapeutic effects in the GI tract after oral delivery.
In conclusion, we herein report that the CopA3 insect peptide increases colonic epithelial cell proliferation by down-regulating p21Cip1/Waf1 via changes in ubiquitin ligase activity and proteasomal protein degradation. CopA3 also enhances mucosal barrier function in the gut. These abilities of CopA3 significantly ameliorate both toxin A-induced enteritis and DSS-induced mouse colitis in vivo. Collectively, these findings suggest that CopA3 could potently inhibit GI tract inflammation in a clinically relevant manner similar to that seen for EGF.
Author Contributions
D. H. K., J. S. H., I. H. L., S. T. N., J. H., P. Z., and L. F. L. conducted most of the study concept/design and acquisition of data and analysis of data. H. S., C. P., J. T. L., and H. K. conducted the drafting of the manuscript and critical correction of the manuscript. J. L. conducted the pathological scoring of mouse gut inflammation model.
This work was supported by a grant from the Next-Generation BioGreen 21 Program (no, PJ011043022015), Rural Development Administration, Republic of Korea. The authors declare that they have no conflicts of interest with the contents of this article.
- DSS
- dextran sulfate sodium
- PI
- propidium iodide
- MTT
- 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide
- GI
- gastrointestinal
- TKBD
- tyrosine kinase-binding domain.
References
- 1. Newman B., and Siminovitch K. A. (2005) Recent advances in the genetics of inflammatory bowel disease. Curr. Opin. Gastroenterol. 21, 401–407 [PubMed] [Google Scholar]
- 2. Khor B., Gardet A., and Xavier R. J. (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim H., Rhee S. H., Pothoulakis C., and Lamont J. T. (2007) Inflammation and apoptosis in Clostridium difficile enteritis is mediated by PGE2 up-regulation of Fas ligand. Gastroenterology 133, 875–886 [DOI] [PubMed] [Google Scholar]
- 4. Kim H., Kokkotou E., Na X., Rhee S. H., Moyer M. P., Pothoulakis C., and Lamont J. T. (2005) Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology 129, 1875–1888 [DOI] [PubMed] [Google Scholar]
- 5. Sartor R. B. (2006) Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 390–407 [DOI] [PubMed] [Google Scholar]
- 6. Sinha A., Nightingale J., West K. P., Berlanga-Acosta J., and Playford R. J. (2003) Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N. Engl. J. Med. 349, 350–357 [DOI] [PubMed] [Google Scholar]
- 7. Frey M. R., Golovin A., and Polk D. B. (2004) Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J. Biol. Chem. 279, 44513–44521 [DOI] [PubMed] [Google Scholar]
- 8. Brown G. L., Nanney L. B., Griffen J., Cramer A. B., Yancey J. M., Curtsinger L. J. 3rd, Holtzin L., Schultz G. S., Jurkiewicz M. J., and Lynch J. B. (1989) Enhancement of wound healing by topical treatment with epidermal growth factor. N. Engl. J. Med. 321, 76–79 [DOI] [PubMed] [Google Scholar]
- 9. Playford R. J. (1995) Peptides and gastrointestinal mucosal integrity. Gut 37, 595–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riegler M., Sedivy R., Sogukoglu T., Castagliuolo I., Pothoulakis C., Cosentini E., Bischof G., Hamilton G., Teleky B., Feil W., Lamont J. T., and Wenzl E. (1997) Epidermal growth factor attenuates Clostridium difficile toxin A- and B-induced damage of human colonic mucosa. Am. J. Physiol. 273, G1014–G1022 [DOI] [PubMed] [Google Scholar]
- 11. Kanazawa S., Tsunoda T., Onuma E., Majima T., Kagiyama M., and Kikuchi K. (2001) VEGF, basic-FGF, and TGF-β in Crohn's disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am. J. Gastroenterol. 96, 822–828 [DOI] [PubMed] [Google Scholar]
- 12. Jeffers M., McDonald W. F., Chillakuru R. A., Yang M., Nakase H., Deegler L. L., Sylander E. D., Rittman B., Bendele A., Sartor R. B., and Lichenstein H. S. (2002) A novel human fibroblast growth factor treats experimental intestinal inflammation. Gastroenterology 123, 1151–1162 [DOI] [PubMed] [Google Scholar]
- 13. Drucker D. J., Yusta B., Boushey R. P., DeForest L., and Brubaker P. L. (1999) Human [Gly2]GLP-2 reduces the severity of colonic injury in a murine model of experimental colitis. Am. J. Physiol. 276, G79–G91 [DOI] [PubMed] [Google Scholar]
- 14. Tsai C. H., Hill M., Asa S. L., Brubaker P. L., and Drucker D. J. (1997) Intestinal growth-promoting properties of glucagon-like peptide-2 in mice. Am. J. Physiol. 273, E77–E84 [DOI] [PubMed] [Google Scholar]
- 15. Nam S. T., Kim D. H., Lee M. B., Nam H. J., Kang J. K., Park M. J., Lee I. H., Seok H., Lee D. G., Hwang J. S., and Kim H. (2013) Insect peptide CopA3-induced protein degradation of p27Kip1 stimulates proliferation and protects neuronal cells from apoptosis. Biochem. Biophys. Res. Commun. 437, 35–40 [DOI] [PubMed] [Google Scholar]
- 16. Kang J. K., Hwang J. S., Nam H. J., Ahn K. J., Seok H., Kim S. K., Yun E. Y., Pothoulakis C., Lamont J. T., and Kim H. (2011) The insect peptide coprisin prevents Clostridium difficile-mediated acute inflammation and mucosal damage through selective antimicrobial activity. Antimicrob. Agents. Chemother. 55, 4850–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yui S., Nakamura T., Sato T., Nemoto Y., Mizutani T., Zheng X., Ichinose S., Nagaishi T., Okamoto R., Tsuchiya K., Clevers H., and Watanabe M. (2012) Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat. Med. 18, 618–623 [DOI] [PubMed] [Google Scholar]
- 18. Rhee S. H., Im E., Riegler M., Kokkotou E., O'Brien M., and Pothoulakis C. (2005) Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc. Natl. Acad. Sci. U.S.A. 102, 13610–13615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alscher K. T., Phang P. T., McDonald T. E., and Walley K. R. (2001) Enteral feeding decreases gut apoptosis, permeability, and lung inflammation during murine endotoxemia. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G569–G576 [DOI] [PubMed] [Google Scholar]
- 20. Petiot A., Ogier-Denis E., Bauvy C., Cluzeaud F., Vandewalle A., and Codogno P. (1999) Subcellular localization of the Gαi3 protein and Gα interacting protein, two proteins involved in the control of macroautophagy in human colon cancer HT-29 cells. Biochem. J. 337, 289–295 [PMC free article] [PubMed] [Google Scholar]
- 21. Literakova P., Lochman J., Zdrahal Z., Prokop Z., Mikes V., and Kasparovsky T. (2010) Determination of capsidiol in tobacco cells culture by HPLC. J. Chromatogr. Sci. 48, 436–440 [DOI] [PubMed] [Google Scholar]
- 22. Kim D. H., Lee I. H., Nam S. T., Hong J., Zhang P., Hwang J. S., Seok H., Choi H., Lee D. G., Kim J. I., and Kim H. (2014) Neurotropic and neuroprotective activities of the earthworm peptide Lumbricusin. Biochem. Biophys. Res. Commun. 448, 292–297 [DOI] [PubMed] [Google Scholar]
- 23. Levkowitz G., Waterman H., Ettenberg S. A., Katz M., Tsygankov A. Y., Alroy I., Lavi S., Iwai K., Reiss Y., Ciechanover A., Lipkowitz S., and Yarden Y. (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 4, 1029–1040 [DOI] [PubMed] [Google Scholar]
- 24. Fielitz J., Kim M. S., Shelton J. M., Latif S., Spencer J. A., Glass D. J., Richardson J. A., Bassel-Duby R., and Olson E. N. (2007) Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Invest. 117, 2486–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shibata E., Abbas T., Huang X., Wohlschlegel J. A., and Dutta A. (2011) Selective ubiquitylation of p21 and Cdt1 by UBCH8 and UBE2G ubiquitin-conjugating enzymes via the CRL4Cdt2 ubiquitin ligase complex. Mol. Cell. Biol. 31, 3136–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang B. R., Kim H., Nam S. H., Yun E. Y., Kim S. R., Ahn M. Y., Chang J. S., and Hwang J. S. (2012) CopA3 peptide from Copris tripartitus induces apoptosis in human leukemia cells via a caspase-independent pathway. BMB Rep. 45, 85–90 [DOI] [PubMed] [Google Scholar]
- 27. Izawa H., Yamamoto H., Damdinsuren B., Ikeda K., Tsujie M., Suzuki R., Kitani K., Seki Y., Hayashi T., Takemasa I., Ikeda M., Ohue M., Sekimoto M., Monden T., and Monden M. (2005) Effects of p21cip1/waf1 overexpression on growth, apoptosis and differentiation in human colon carcinoma cells. Int. J. Oncol. 27, 69–76 [PubMed] [Google Scholar]
- 28. Shin J. S., Hong S. W., Lee S. L., Kim T. H., Park I. C., An S. K., Lee W. K., Lim J. S., Kim K. I., Yang Y., Lee S. S., Jin D. H., and Lee M. S. (2008) Serum starvation induces G1 arrest through suppression of Skp2-CDK2 and CDK4 in SK-OV-3 cells. Int. J. Oncol. 32, 435–439 [PubMed] [Google Scholar]
- 29. Braun F., Bertin-Ciftci J., Gallouet A. S., Millour J., and Juin P. (2011) Serum-nutrient starvation induces cell death mediated by Bax and Puma that is counteracted by p21 and unmasked by Bcl-xL inhibition. PLoS One 6, e23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harper J. W., Adami G. R., Wei N., Keyomarsi K., and Elledge S. J. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 [DOI] [PubMed] [Google Scholar]
- 31. el-Deiry W. S., Harper J. W., O'Connor P. M., Velculescu V. E., Canman C. E., Jackman J., Pietenpol J. A., Burrell M., Hill D. E., and Wang Y. (1994) WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 54, 1169–1174 [PubMed] [Google Scholar]
- 32. Sarmento L. M., Huang H., Limon A., Gordon W., Fernandes J., Tavares M. J., Miele L., Cardoso A. A., Classon M., and Carlesso N. (2005) Notch1 modulates timing of G1-S progression by inducing SKP2 transcription and p27 Kip1 degradation. J. Exp. Med. 202, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bornstein G., Bloom J., Sitry-Shevah D., Nakayama K., Pagano M., and Hershko A. (2003) Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 278, 25752–25757 [DOI] [PubMed] [Google Scholar]
- 34. Yu Z. K., Gervais J. L., and Zhang H. (1998) Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 11324–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uchiki T., Kim H. T., Zhai B., Gygi S. P., Johnston J. A., O'Bryan J. P., and Goldberg A. L. (2009) The ubiquitin-interacting motif protein, S5a, is ubiquitinated by all types of ubiquitin ligases by a mechanism different from typical substrate recognition. J. Biol. Chem. 284, 12622–12632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wong N. A., Mayer N. J., Anderson C. E., McKenzie H. C., Morris R. G., Diebold J., Mayr D., Brock I. W., Royds J. A., Gilmour H. M., and Harrison D. J. (2003) Cyclin D1 and p21 in ulcerative colitis-related inflammation and epithelial neoplasia: a study of aberrant expression and underlying mechanisms. Hum. Pathol. 34, 580–588 [DOI] [PubMed] [Google Scholar]
- 37. Iwamoto M., Koji T., Makiyama K., Kobayashi N., and Nakane P. K. (1996) Apoptosis of crypt epithelial cells in ulcerative colitis. J. Pathol. 180, 152–159 [DOI] [PubMed] [Google Scholar]
- 38. Serafini E. P., Kirk A. P., and Chambers T. J. (1981) Rate and pattern of epithelial cell proliferation in ulcerative colitis. Gut 22, 648–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ng C., Jackson R. A., Buschdorf J. P., Sun Q., Guy G. R., and Sivaraman J. (2008) Structural basis for a novel intrapeptidyl H-bond and reverse binding of c-Cbl-TKB domain substrates. EMBO J. 27, 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmidt M. H., and Dikic I. (2005) The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 6, 907–918 [DOI] [PubMed] [Google Scholar]
- 41. Swaminathan G., and Tsygankov A. Y. (2006) The Cbl family proteins: ring leaders in regulation of cell signaling. J. Cell Physiol. 209, 21–43 [DOI] [PubMed] [Google Scholar]