

Abstract
We identify two heteroallelic mutations in the acetylcholine receptor δ-subunit from a patient with severe myasthenic symptoms since birth: a novel δD140N mutation in the signature Cys-loop and a mutation in intron 7 of the δ-subunit gene that disrupts splicing of exon 8. The mutated Asp residue, which determines the disease phenotype, is conserved in all eukaryotic members of the Cys-loop receptor superfamily. Studies of the mutant acetylcholine receptor expressed in HEK 293 cells reveal that δD140N attenuates cell surface expression and apparent channel gating, predicting a reduced magnitude and an accelerated decay of the synaptic response, thus reducing the safety margin for neuromuscular transmission. Substituting Asn for Asp at equivalent positions in the α-, β-, and ϵ-subunits also suppresses apparent channel gating, but the suppression is much greater in the α-subunit. Mutant cycle analysis applied to single and pairwise mutations reveals that αAsp-138 is energetically coupled to αArg-209 in the neighboring pre-M1 domain. Our findings suggest that the conserved αAsp-138 and αArg-209 contribute to a principal pathway that functionally links the ligand binding and pore domains.
Keywords: ion channel, nicotinic acetylcholine receptors (nAChR), patch clamp, receptor structure-function, structure-function, Cys-loop receptor superfamily, congenital myasthenic syndrome, invariant Cys-loop aspartate, invariant pre-M1 arginine, mutant cycle analysis
Introduction
Congenital myasthenic syndromes are heterogeneous disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Although no fewer than 20 congenital myasthenic syndrome disease genes have been identified, most congenital myasthenic syndromes arise from mutations in muscle acetylcholine receptor (AChR)2 subunits (1). The muscle AChR belongs to the cystine-loop (Cys-loop) superfamily of receptors and is a heteropentamer composed of homologous subunits with stoichiometry (α1)2β1δγ in fetal and (α1)2β1δϵ in adult muscle (2). Each subunit consists of an N-terminal extracellular domain composed of 10 β-strands that form inner and outer β-sheets, a transmembrane domain of four α-helices (M1–M4), a large cytoplasmic domain between M3 and M4, and an extracellular C-terminal tail.
The AChR Cys-loop divides the inner from the outer β-sheets of the extracellular domain and is situated at the interface between the extracellular and pore domains. Its name derives from a disulfide bond between a pair of cysteine residues separated by 13 residues, four of which are highly conserved. One of these conserved residues, Asp-138 in the muscle AChR α-subunit, is present at equivalent positions in all subtypes and species of AChR subunits (Fig. 1A) and also across all subunits of all species of the Cys-loop receptor superfamily. However, little is known about the contributions of the Cys-loop Asp of the different subunits to AChR expression and AChR activation.
FIGURE 1.

A, structural model of extracellular domains of human AChR viewed from the intracellular side indicating positions of invariant Cys-loop Asp and pre-M1 Arg residues (based on the crystal structure of the ACh binding protein (Protein Data Bank entry 1I9B) and mutational analyses of the human AChR binding domain (36)). B, side view of the α subunit showing that the pre-M1 Arg serves as a cationic hub linking the Cys-loop, β1-2 loop, and β8-9 loop in the AChR α subunit (Protein Data Bank entry 2BG9).
Recent crystal structures of eukaryotic Cys-loop receptors show that the residue equivalent to αAsp-138 localizes within the hydrophobic core of the subunit, where it establishes electrostatic contact with an invariant cationic residue equivalent to αArg-209 in the pre-M1 domain of the muscle AChR (3). In addition, anionic residues from the β1-2 and β8-9 loops interact with the residue equivalent to αArg-209 (4, 5), which may serve as a cationic hub linking multiple loops from the extracellular domain to the pre-M1 domain (Fig. 1B).
Studies of a variety of Cys-loop receptors suggest that residues equivalent to αAsp-138 interact functionally with cationic or polar residues from the pre-M1 domain, M2-M3 linker, and C-terminal tail. In the heteromeric GABAA receptor, analyses of charge reversal or disulfide trapping of residue pairs revealed that the residue equivalent to αAsp-138 interacted with cationic or polar residues from the pre-M1 (6) and M2-M3 regions (7). In the homomeric glycine receptor, studies of substituted cysteine pairs suggested that residues equivalent to αAsp-138 and αArg-209 form a salt bridge important for receptor function (8). Finally, in the muscle AChR, charge reversal experiments revealed functional interaction between the residue equivalent to αAsp-138 and a cationic residue in the C-terminal tail (9). All of these studies used macroscopic current measurements to assess function of the mutant receptors, and although functional interdependence of the residue pairs was demonstrated, functional consequences of the mutations at the level of individual receptor channels were not investigated.
Here we identify a mutation of the invariant Cys-loop Asp residue in the AChR δ-subunit in a patient with myasthenic symptoms since birth and analyze the consequences of the mutation at the level of individual receptor channels. Furthermore, we examine functional consequences of the Asp-to-Asn mutation at equivalent positions of the AChR α-, β-, and ϵ-subunits and use thermodynamic mutant cycle analyses to assess interactions between αAsp-138 and the invariant cationic residue αArg-209.
Experimental Procedures
Mutation Analysis
We directly sequenced muscle AChR α-, β-, δ-, and ϵ-subunit genes using the patient's genomic DNA. Using allele-specific PCR, we determined whether the patient's mutation was present in family members and 400 normal alleles of 200 unrelated controls.
Construction and Expression of Wild-type and Mutant AChR
Sources of human α-, β-, δ-, and ϵ-subunit cDNAs and subcloning of the cDNAs into the CMV-based mammalian expression vector pRBG4 were as previously described (10). The mutations were engineered into wild-type AChR subunit cDNAs in pRBG4 using the QuikChange site-directed mutagenesis kit (Agilent Technologies). The presence of each mutation and absence of unwanted mutations were confirmed by sequencing the entire inserts. HEK 293 cells were transfected with plasmid DNA encoding α-, β-, δ-, and ϵ-subunits and pEGFP-N1 at a ratio of 2:1:1:1:1, using the FuGENE 6 transfection reagent (Roche Applied Science) (11).
α-Bungarotoxin Binding Measurements
The total number of [125I]α-bungarotoxin ([125I]α-bgt) sites on the surface of transfected HEK cells was determined as described previously (10). Briefly, intact cells were harvested 3 days after transfection by gentle agitation in PBS with 5 mm EDTA. Cells were briefly centrifuged, resuspended in high potassium Ringer's solution, and divided into aliquots for measurements of α-bgt binding. The total number of α-bgt binding sites was determined by incubation for 1 h in the presence of 5 nm [125I]α-bgt. Unbound toxin was removed by washing with potassium Ringer's solution containing 300 μm d-tubocurarine, followed by filtration using a cell harvester (Brandel Inc.). Radioactivity retained by the glass fiber filters (Whatman GF-B, 1 μm cut-off) was measured with a γ counter. Nonspecific binding was determined in the presence of 300 μm d-tubocurarine. Binding properties of αδ dimers were determined in cells made permeable with saponin by modification of an established method (12). After removing the growth medium, cells were incubated for 5 min in PBS/EDTA plus 0.1% BSA and 0.5% saponin prior to incubation with [125I]α-bgt.
Patch Clamp Recordings and Single Channel Kinetic Analysis
Recordings were obtained in the cell-attached patch configuration at a membrane potential of −80 mV at 22 °C and with bath and pipette solutions containing 142 mm KCl, 5.4 mm NaCl, 1.8 mm CaCl2, 1.7 mm MgCl2, 10 mm HEPES, pH 7.4. Single channel currents were recorded using an Axopatch 200A amplifier (Axon Instruments) at a bandwidth of 50 kHz, digitized at 5-μs intervals using a Digidata 1322A (Axon Instruments), and recorded to a hard disk using the program Clampex version 8.2 (Axon Instruments). Recordings obtained with ACh concentrations of 1 μm or less were analyzed at a uniform bandwidth of 11.7 kHz with an imposed dead time of 15.3 μs. Recordings obtained with ACh concentrations of 10 μm or more were analyzed at a bandwidth of 10 kHz and an imposed dead time of 25 μs using TAC version 4.0.9 software (Bruxton). Dwell time histograms were plotted on a logarithmic abscissa and fitted with the sum of exponentials by maximum likelihood (13).
To estimate rate constants underlying AChR activation, we employed desensitizing concentrations of ACh that cause events from a single receptor channel to cluster into sequences of identifiable activation episodes (14). Clusters were identified as a series of closely spaced openings preceded and followed by closed intervals greater than a defined critical time. The critical time was determined by a method that misclassifies an equal number of events between two adjacent closed-time components (15). For each receptor, the critical time that provided the best fit for the closed-time histogram was chosen for the final analysis. Clusters with fewer than five openings were excluded from analysis. Individual clusters were examined for homogeneity based on the mean Po and open duration for each cluster, and clusters within 2 S.D. values of the mean were accepted for further analysis (16, 17). The resulting global set of open and closed dwell times from wild-type and mutant AChRs was analyzed using the program MIL (QuB suite), which uses an interval-based maximum likelihood method that also corrects for missed events (16) to yield fitted rate constants in a kinetic scheme for receptor activation.
For each wild-type or mutant AChR, single channel dwell times obtained over a range of ACh concentrations (10–1,000 μm) were fitted simultaneously. The final set of rate constants was checked by comparing probability density functions calculated from the rate constants and the experimental dwell time histograms and by the ability of the rate constants to predict the mean burst duration at limiting low concentrations of ACh (18, 19).
Simulation of Miniature Endplate Currents (MEPC)
MEPCs were simulated using MCell software (version 3.3) for a model of the rat neuromuscular junction (20, 21). The geometry of the synaptic cleft was defined as an open box with AChRs distributed on a 5 × 5-μm area that was 0.05 μm from the presynaptic membrane. For the wild-type AChR, the AChR density was set to 10,000/μm2 on the crests and 2,970/μm2 on deeper regions of the folds. For the mutant AChR from the patient, the density was reduced in proportion to the cell surface expression determined by the [125I]α-bgt binding measurements. A quantum of 1,000 ACh molecules, the maximum allowable, was released at time 0, and each molecule was followed with a time resolution of 0.5 μs. The diffusion constant of ACh in the cleft was 2.1 × 10−6 cm2/s. The association and dissociation rates for the formation of the complex between ACh and AChE were 2 × 108 m−1 s−1 and 1.4 × 104 s−1, respectively. The rate constants for the wild-type and mutant AChRs derived from kinetic analysis of open and closed dwell times were applied to the model. The amplitude of the simulated MEPC for the wild-type AChR was normalized to the mean amplitude of the MEPCs from control human EPs (22).
Thermodynamic Mutant Cycle Analysis
We performed mutant cycle analysis to determine whether the functional contributions of two residues, X and Y, are independent or interdependent. The functional consequences of mutating residue X, with and without the mutation of residue Y, are computed from the free energy change associated with a given reaction step for the wild-type AChR and the mutants AChRX, AChRY, and AChRXY. In the case of the AChR, the relevant reaction step was the apparent channel gating equilibrium constant of the diliganded receptor, θ, which has an associated free energy change (ΔG) = −RTlnθ. Although θ is a composite of closed-state priming and channel gating steps (23) and thus is an apparent equilibrium constant, it remains a highly sensitive measure of interresidue coupling. The changes in gating free energy due to the mutations X, Y, and XY relative to the wild-type are designated ΔΔGX, ΔΔGY, and ΔΔGXY. These terms are related to the free energy of interresidue interaction, ΔΔGint, as follows: ΔΔGXY = ΔΔGX + ΔΔGY + ΔΔGint (24, 25). Given this relationship and noting that ΔΔGs for the individual mutant AChR is ΔGX − ΔGW, ΔGY − ΔGW, ΔGXY − ΔGW, ΔΔGint can be calculated from −RTln((θW × θXY)/(θX × θY)), where θ is the apparent channel gating equilibrium constant for wild-type (W) and mutant (X, Y, and XY) AChRs (4, 26).
Statistics
Error estimates of the scheme rate constants for wild-type and mutant receptors in Tables 1 and 2 were computed by the MIL program from the curvature of the likelihood function at its maximum (16). To compute the S.E. for ΔΔG, the likelihood surfaces for each fitted β or α are assumed to be symmetrical about the maximum. The S.E. of ΔΔG is then computed from the S.E. for each rate constant (i.e. S.E. βN) as follows (5),
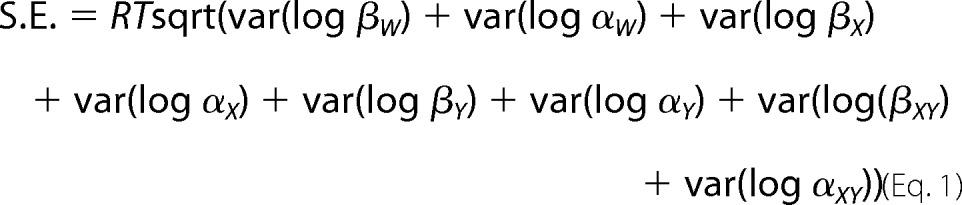 |
where var(log βN) = (S.E. βN/βN)2; var(log αN) = (S.E.αN/αN)2.
TABLE 1.
Burst durations of wild-type and mutant AChRs in HEK cells
Values indicate means ± S.E. with number of patches in parentheses. ACh concentration was 50 nm for all except 1 μm for αD138N and δD140N/ϵD138N. Temperature was 22 ± 0.5 °C, bandwidth was 11.7 kHz, and membrane potential was −80 mV.
| Bursts (measured) |
|||
|---|---|---|---|
| τ1 (area) | τ2 (area) | τ3 (area) | |
| ms (no. of patches) | |||
| Wild type (n = 21) | 0.036 ± 0.002a (0.24 ± 0.02) | 0.47 ± 0.06 (0.21 ± 0.03) | 3.31 ± 0.12 (0.58 ± 0.04) |
| δD140N (n = 4) | 0.32 ± 0.060 (0.41 ± 0.019) | 1.81 ± 0.18 (0.59 ± 0.023) | |
| αD138N (n = 4) | 0.22 ± 0.015 (1.00 ± 0.00) | ||
| βD138N (n = 3) | 0.049b (0.19) | 0.60 ± 0.050 (0.94 ± 0.060) | |
| ϵD138N (n = 4) | 0.040 ± 0.018 (0.21 ± 0.022) | 1.67 ± 0.14 (0.84 ± 0.055) | |
| δD140N/ ϵD138N (n = 5) | 1.70 ± 0.082 (1.00 ± 0.00) | ||
a Not detected at three patches.
b Not detected at two patches.
TABLE 2.
Kinetic analysis of wild-type and mutant AChRs
Kinetic parameters and error estimates are derived from global fitting of a kinetic scheme to data obtained over a wide range of ACh concentrations (see “Experimental Procedures”). Rate constants are in μm−1 ·s−1 for association rate constants and s−1 for all others. The dissociation constants, K1 and K2, are the ratios of k−/k+. ΔG0 indicates free energy change compared with that in wild type in kcal/mol. The apparent channel gating equilibrium constant θ is the ratio of the apparent opening rate (β) to apparent closing rate (α) constants. Free energy change ΔG0 = −RTln(θmutant/θwild type), where R is the gas constant (1.987 cal/K/mol) and T is the absolute temperature (295 K).
| AChR | k+1 | k−1 | K1 | k+2 | k−2 | K2 | β | α | θ | k+b | k−b | KB | ΔG0 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μm | μm | mm | kcal/mol | ||||||||||
| Wild type | 210 ± 2 | 5,300 ± 81 | 25 | 100 ± 1 | 11,000 ± 160 | 110 | 52,000 ± 1,200 | 2,200 ± 63 | 24 ± 0.44 | 29 ± 2 | 110,000 ± 2,200 | 3.8 | |
| δD140N | 430 ± 12 | 7,400 ± 260 | 17 | 220 ± 5 | 15,000 ± 520 | 68 | 18,000 ± 350 | 1,000 ± 14 | 18 ± 0.22 | 44 ± 2 | 150,000 ± 1,828 | 3.4 | 0.17 ± 0.013 |
| αD138N | 140 ± 13 | 2,000 ± 220 | 14 | 70 ± 6 | 4,000 ± 450 | 57 | 2,200 ± 64 | 8,000 ± 140 | 0.28 ± 0.0048 | 59 ± 5 | 97,000 ± 3,900 | 1.6 | 2.61 ± 0.024 |
| βD138N | 400 ± 13 | 11,000 ± 479 | 28 | 200 ± 6 | 23,000 ± 1,200 | 120 | 16,000 ± 400 | 3,400 ± 59 | 4.7 ± 0.073 | 12 ± 1 | 71,000 ± 3,600 | 5.9 | 0.24 ± 0.014 |
| ϵD138N | 410 ± 10 | 8,300 ± 270 | 20 | 210 ± 5 | 17,000 ± 530 | 81 | 15,000 ± 390 | 1,200 ± 19 | 13 ± 0.19 | 23 ± 4 | 110,000 ± 6,800 | 4.8 | 0.36 ± 0.014 |
| δD140N/ϵD138N | 310 ± 11 | 5,700 ± 280 | 18 | 160 ± 5 | 12,000 ± 570 | 75 | 19,000 ± 610 | 1,400 ± 14 | 14 ± 0.23 | 14 ± 2 | 84,000 ± 4,400 | 6.0 | 0.32 ± 0.015 |
| αD138E | 320 ± 11 | 4,200 ± 180 | 13 | 160 ± 5 | 8,400 ± 370 | 55 | 25,000 ± 550 | 1,400 ± 36 | 18 ± 0.31 | 12 ± 2 | 90,000 ± 7,000 | 7.5 | 0.17 ± 0.015 |
| αR209K | 400 ± 22 | 1,800 ± 120 | 5 | 200 ± 11 | 3,600 ± 250 | 18 | 19,000 ± 570 | 2,100 ± 52 | 9.1 ± 0.18 | 12 ± 3 | 100,000 ± 6,400 | 8.3 | 0.54 ± 0.016 |
| αD138E/αR209K | 52 ± 6 | 700 ± 90 | 14 | 26 ± 3 | 1,400 ± 180 | 54 | 3,800 ± 180 | 4,100 ± 130 | 0.93 ± 0.027 | 8 ± 1 | 46,000 ± 5,700 | 5.8 | 1.74 ± 0.021 |
Study Approval
All human studies were in accordance with the guidelines of and were approved by the institutional review board of the Mayo Clinic. The parents of the patient reported in this study provided informed consent to participate.
Results
Characteristics of the Congenital Myasthenic Syndrome Patient
An 11-year-old girl was hypotonic and apneic at birth. During infancy and early childhood, she had severe weakness of all muscles. Because she could not swallow, she was fed through a gastrostomy tube, and she had recurrent apneic episodes requiring ventilatory support. At 1 year of age, electromyographic studies revealed a defect in neuromuscular transmission. She improved with high doses of the cholinesterase inhibitor pyridostigmine, which prolongs the lifetime of ACh in the synaptic cleft, and improved further with 3,4-diaminopyridine, which enhances the number of acetylcholine quanta released by nerve impulse. At age 11, she has no further apneic episodes, is able to walk with support, can speak a few words with a palatal support, and attends a school for children with special needs. There are no similarly affected family members.
Mutation Analysis
To determine the molecular basis of the patient's illness, we sequenced each AChR subunit gene. This revealed two mutations in the δ-subunit of the receptor (CHRND): a paternally inherited c.481G→A/p.δD140N mutation located in the signature Cys-loop (Fig. 2) and a maternally inherited splice site mutation within intron 7, c.821–2A→C. Because the parents are asymptomatic, each mutation in the AChR δ subunit is recessive. Neither mutation was detected in 400 normal control alleles. Neither variant is listed in the Exome Variant Database (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA), and δD140N is predicted to be disease-causing by both Mutation Taster and Polyphen-2. The splice site mutation disrupts splicing of exon 8 in one δ-subunit allele, so the second allele harboring the δD140N mutation determines the phenotype. Nucleotide numbers start from the translational start site, with +1 corresponding to the A of the ATG translation initiation codon (NM_000751.2). Codon numbers start from the first codon of the mature peptide (NP_000742.1). The mutated δAsp-140 is conserved at equivalent positions of all subunits of the Cys-loop superfamily of receptors of all species. To date, mutations of equivalent residues in non-δ-subunits of AChR have not been reported.
FIGURE 2.
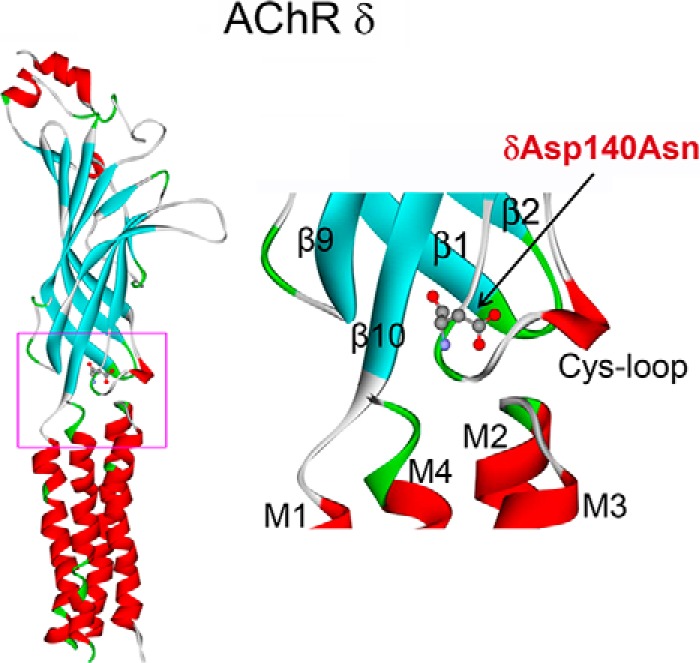
Position of the δD140N mutation in the δ subunit of AChR (Protein Data Bank entry 2BG9). Left, extracellular and transmembrane domains of the δ subunit. Right, structure of the interface between extracellular and transmembrane domain demarcated by a rectangle in the left panel.
Expression of δD140N in HEK Cells
To assess the pathogenic effects of the δD140N mutation, we engineered the mutation into the wild-type human δ-subunit and co-transfected cDNAs encoding mutant or wild-type δ-subunits with complementary wild-type α-, β-, and ϵ-subunits into HEK cells. Expression of the mutant AChR on the surface of intact cells, measured from the binding of [125I]α-bgt, was reduced to 20% of wild-type (Fig. 3A). Furthermore, substitution of Glu for δAsp-140, which maintains the negative charge, reduced expression to ∼40% of wild type, and substitution of positively charged, aromatic, or hydrophobic residues reduced expression to 10% or less (Fig. 3A). Substituting Asn for the equivalent Asp in the different AChR subunits decreased cell surface expression in the following subunit-dependent manner: wild type = ϵm > βm > αm > δm, where the subscript specifies the mutant subunit (Fig. 3B). Note that the α-subunit mutation introduces two mutations per AChR but has a smaller effect than the single mutation in the δ-subunit. Thus, the contribution of the invariant Cys-loop Asp residue to cell surface expression depends on the electrostatic charge and size of the side chain and also on the subunit harboring the Asp residue. To gain insight into why the δD140N mutant reduces AChR surface expression, we determined the ability of the mutant subunit to form a stable complex with the wild-type α-subunit. Compared with cells expressing wild-type α- and δ-subunits, which form intracellular α-δ dimers, cells expressing wild-type α- and mutant δ-subunits exhibited a 60% decrease in [125I]α-bgt binding (Fig. 3C). Thus, the δD140N mutation impairs dimerization of the α- and δ-subunits, which is an early step in AChR assembly (27).
FIGURE 3.
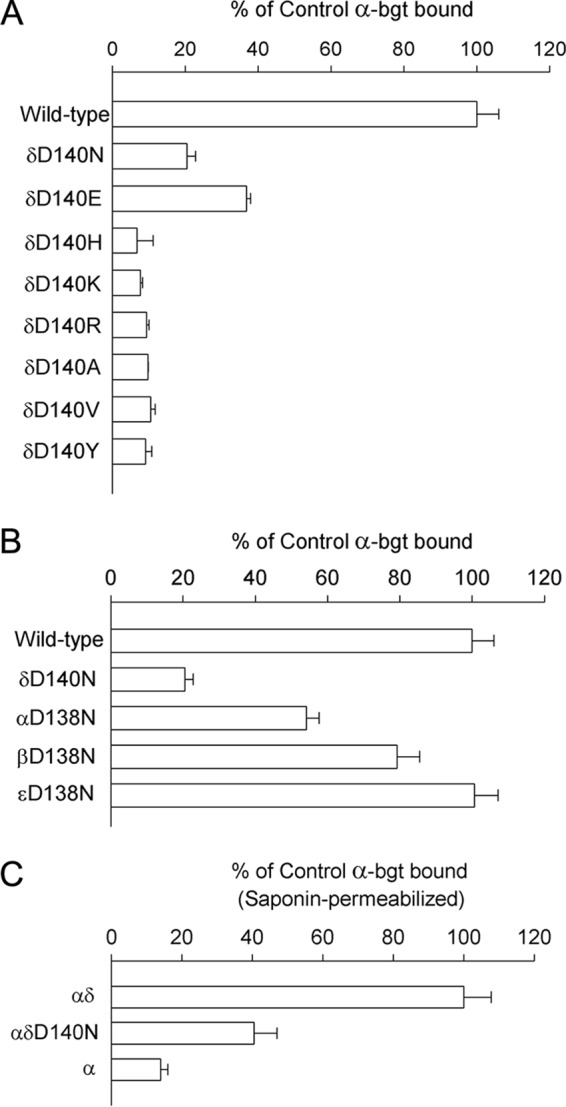
Specific α-bgt binding to intact (A and B) and saponin-permeabilized (C) HEK cells transfected with the indicated AChR subunit cDNAs (see “Experimental Procedures”). The results are normalized for α-bgt binding to wild-type AChR (A and B) or wild-type αδ dimers (C) and represent mean ± S.D. (error bars) of at least three experiments.
Single Channel Currents from the δD140N Mutant Elicited by Low ACh Concentrations
To gain insight into the effect of the δD140N mutation on AChR function, we recorded single channel currents in the presence of a limiting low concentration of ACh (50 nm). At a low concentration of ACh, channel openings appear as bursts of several openings in quick succession, with each burst arising from a different channel. The mean burst duration equals the time constant for decay of the synaptic current (18, 28) and thus provides insight into the consequences for neuromuscular transmission. Compared with bursts recorded from the wild-type AChR, bursts from the mutant receptor are noticeably briefer (Fig. 4). Histograms of burst durations reveal several exponential components, the longest of which has a mean duration of 3.3 ms for the wild-type AChR and 1.8 ms for the mutant (Table 1). The decrease in mean burst duration predicts a more rapid decay of the synaptic response at mutant compared with normal endplates, reducing the safety margin of neuromuscular transmission.
FIGURE 4.

Single channel currents elicited by ACh from HEK cells expressing wild-type, δD140N, and αD138N AChRs. Left, channel openings are shown as upward deflections. Right, logarithmically binned burst duration histograms fitted to the sum of exponentials. Arrows, peaks of burst components. The values for major components are indicated for each AChR.
To investigate the subunit dependence of the invariant Cys-loop Asp on mean burst duration, we made analogous recordings from mutant AChRs in the presence of low concentrations of ACh. The mean burst duration decreased in the following subunit-dependent manner: wild-type > δm > ϵm > βm > αm (Table 1 and Fig. 4). Notably, the mutation in the α-subunit reduced the mean burst duration by 15-fold compared with that of the wild-type AChR. Thus, as observed for cell surface expression, the Asp-to-Asn substitution affects the mean burst duration in a subunit-dependent manner.
Patch Clamp Recordings from the δD140N Mutant at Increased ACh Concentrations
To gain further insight into the functional consequences of the δD140N mutation, we recorded single channel currents over a range of increased ACh concentrations. Under these conditions, individual AChR channels activate in clusters of many channel openings in quick succession, which terminate by desensitization. The durations of openings and closings within clusters give information on the kinetics of state transitions within a mechanism of receptor activation, including rate constants for agonist association and dissociation and channel opening and closing. The rate constants are estimated by fitting a kinetic scheme to the sequences of intracluster open and closed dwell times.
For both the wild-type and mutant AChR, closed dwell times within clusters become progressively briefer as the ACh concentration increases, and the closed duration histograms mirror the concentration-dependent change from long to brief intracluster closings (Fig. 5). Notably, the change from long to brief intracluster closings occurs at a higher concentration of ACh for the mutant compared with the wild-type AChR. In addition, at intermediate ACh concentrations, the ratio of brief to long closings is greater for the wild-type than for the mutant AChR. These qualitative changes in intracluster closings suggest that the mutation affects one or more kinetic transitions that precede the channel-opening step.
FIGURE 5.

Activation kinetics of wild-type (A), δD140N (B), and αD138N AChRs (C). The left column shows representative single channel currents at the indicated AChR concentrations recorded from HEK cells. Bandwidth was 10 kHz. The center and right columns show corresponding histograms of closed and open duration dwell times with superimposed global fits for Scheme 1. Fitted rate constants are shown in Table 2.
To identify rate constants altered by the mutation, we fitted the following minimal kinetic scheme to sequences of open and closed dwell times obtained over a range of increased ACh concentrations.
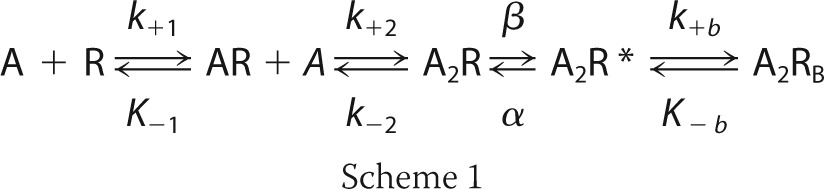 |
In this scheme, two agonists (A) bind to the receptor (R) with association rate constants k+1 and k+2 and dissociate with rate constants k−1 and k−2. The doubly occupied receptor opens with rate constant β and closes with rate constant α, and ACh blocks the open channel with rate constant k+b, and it dissociates from the block state with rate constant k−b. Due to bandwidth limitations, the previously identified closed state between A2R and A2R*, known as flip or primed (23, 29), is not included in this scheme; thus, the fitted rate constants β, α, and k−2 are apparent rate constants. Also, a desensitized state is not included because each cluster begins following recovery from desensitization and ends upon return to the desensitized state, so that closings within clusters represent transitions between activatable states.
For both the wild-type and δD140N AChRs, the above kinetic scheme describes the closed and open dwell time distributions over the range of ACh concentrations, as indicated by the close correspondence between the computed probability density functions and the dwell time histograms (Fig. 5). The fitting analysis reveals that for the mutant relative to the wild-type AChR, the apparent channel opening rate constant β decreases, the apparent ACh dissociation rate constant k−2 increases, and the apparent channel closing rate constant α decreases (Table 2). These changes in rate constants are expected to alter the synaptic response to nerve-released ACh. First, the probability that a doubly occupied AChR will transition to the open state, given by the ratio β/(β + k−2), is 0.55 for the mutant and 0.83 for the wild-type AChR. Second, the channel opening equilibrium constant, given by the ratio β/α, is 18 for the mutant and 24 for the wild-type AChR. Third, the mean burst duration, given by (β/k−2 + 1)/α, is 2.2 ms for the mutant and 2.6 ms for the wild-type AChR; these values are similar to the mean burst durations obtained from recordings at limiting low ACh concentrations (Table 1). Thus, the δD140N mutation reduces the probability that a doubly occupied receptor channel will open, reduces the opening equilibrium constant, and shortens the burst duration.
Simulation of MEPCs
The preceding kinetic analysis shows that the δD140N mutation alters multiple steps in the activation process, whereas the [125I]α-bgt binding measurements show that the mutation reduces cell surface expression. To assess the overall consequences for neuromuscular transmission, we simulated MEPCs for the wild type and mutant AChRs based on the kinetic scheme, the fitted rate constants, and the AChR density inferred from measurements of cell surface expression (see “Experimental Procedures”). The simulations show that the mutation attenuates the peak of the MEPC to 58% of normal (Fig. 6A). However, this attenuation is not as great as the decrease in cell surface expression alone, which was 20% of normal. To reconcile the apparent disparity between the peak of the MEPC and AChR density, we repeated the simulations for a range of AChR densities. The simulations reveal a non-linear relationship between the peak of the MEPC and AChR density, so that reducing the AChR density to 20% reduces the peak response to only 50% (Fig. 6B). This non-linear relationship is strikingly similar to that from a landmark study of MEPC amplitude following neuromuscular blockade (30). In that study, the authors noted that the number of AChRs at the EP is much greater than the number of ACh molecules in a quantum, so that the number of AChRs can be reduced substantially before the number of bound ACh molecules is reduced.
FIGURE 6.
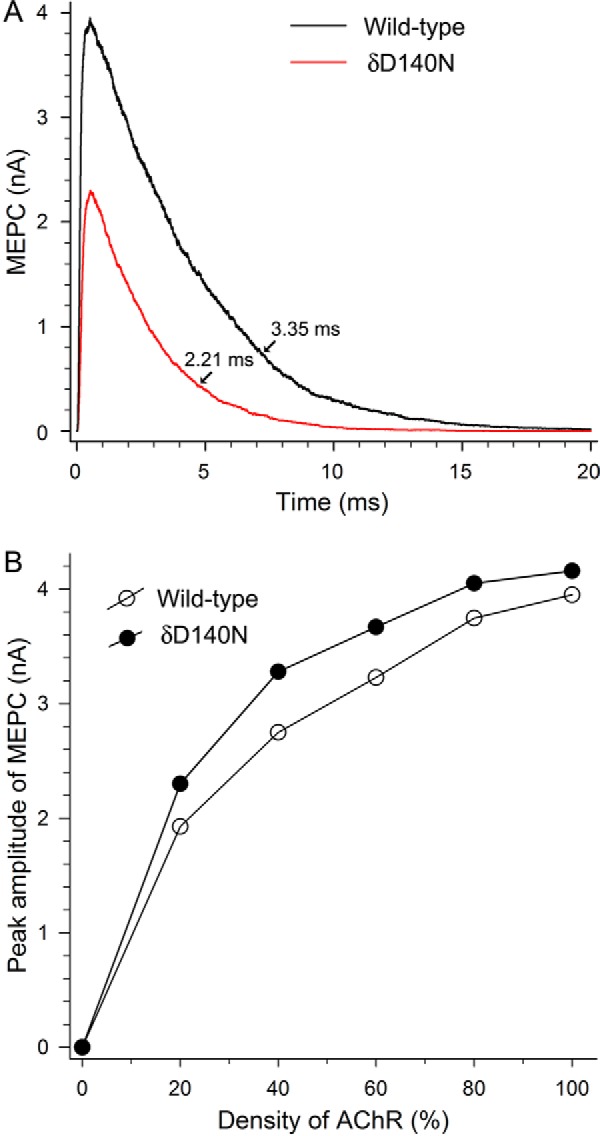
A, simulated MEPC at the control EP for wild-type AChR and at the patient EP for δD140N AChR. The rate constants for wild-type and mutant AChRs shown in Table 2 were used in the simulations. Other parameters for the simulation are indicated under “Experimental Procedures.” MEPC decay times for wild-type and δD140N AChR are obtained by exponential fitting of the curves. B, nonlinear relations between the peak MEPC amplitude and the AChR density at the control and the patient's EPs.
The simulations also show that the mutation accelerates the decay of the MEPC to 66% of normal. For the wild-type AChR, the simulated MEPC decay time constant of 3.35 ms is very similar to that measured at voltage-clamped human endplates (22). For the mutant AChR, the simulated MEPC decay time constant of 2.2 ms is slightly longer than the mean burst duration of 1.8 ms obtained from the mean burst duration at limiting low ACh concentrations (Table 1) but coincides with that computed from the formula (β/k−2 + 1)/α and the rate constants in Table 2. Thus, at the mutant endplate, the attenuated peak and the accelerated decay reduce the net current during an MEPC to 38% of normal.
Mutations of Equivalent Asp Residues in Non-δ-subunits
To determine the kinetic consequences of mutations of the equivalent Cys-loop Asp residues in the non-δ-subunits, we again recorded single channel currents over a range of increased ACh concentrations and analyzed the sequences of single channel dwell times as described for the mutation δD140N. The analyses reveal that the mutants, βD138N and ϵD138N, attenuate the apparent equilibrium constant for channel gating to an extent similar to that by the δD140N mutant (Table 2). However, as anticipated from measurements of mean burst duration (Table 1), the αD138N mutation attenuates the apparent channel gating equilibrium constant by 2 orders of magnitude, due to a decrease in the apparent channel opening and an increase in the apparent channel closing rate constants.
Functional Consequences of the Number of Subunits with Mutated Asp Residues
The δD140N mutant is present in one copy per AChR, whereas the αD138N mutation is present in two copies per AChR. To determine whether the severe consequences of the αD138N mutation arise from the presence of two mutant subunits, we combined δD140N and ϵD138N into a single AChR and determined the functional consequences. Recordings in the presence of a limiting low concentration of ACh show that the mean burst duration of the double mutant AChR (1.7 ms) is very close to that of either single mutant (1.8 and 1.7 ms), yet it is much longer than that for the αD138N mutant (0.22 ms) (Table 1). Analysis of recordings in the presence of increased ACh concentrations shows that the apparent channel gating equilibrium constant for the double mutant AChR is essentially the same as that for either single mutant AChR, yet it is much greater than that for the αD138N AChR (Table 2).
Interaction of αAsp-138 with Surrounding Residues
Recent x-ray crystal structures of Cys-loop receptors show that within the structural transition zone between the extracellular and transmembrane domains, electron-rich residues from three structural regions converge upon an invariant arginine residue from the pre-M1 domain (3, 31–33) (Fig. 7A). The conserved αGlu-45 at the apex of the β1-2 loop and the invariant αArg-209 from the pre-M1 domain were the first pairwise contacts in this region shown to be functionally interdependent (4). Subsequently, functional interdependence was demonstrated between the conserved αGlu-175 and αTrp-176 from the β8-9 loop and αArg-209 (5). However, it remained unknown whether the anionic αAsp-138 from the Cys-loop, and αArg-209 were functionally interdependent. To determine whether αAsp-138 and αArg-209 are functionally interdependent, we generated a series of pairwise mutations of αAsp-138 and αArg-209 and used single channel kinetic analysis to determine the functional consequences of the single and double mutants.
FIGURE 7.

A, stereo view of the interface between extracellular domains and transmembrane domains shows spatial disposition of interacting charged residues in Cys-loop, pre-M1, β1-2 loop, and β8-9 loop (based on the crystal structure of 5-HT3A (Protein Data Bank entry 4PIR)). B, a mutant cycle of energetic interaction among αAsp-138 and αArg-209. Single channel currents correspond to each species of AChR elicited by 300 μm ACh. Numbers over the arrows indicate the difference in the apparent gating free energy between the two different AChRs in kcal/mol. S.E. values were computed as described under “Experimental Procedures.” The 95% confidence limit, or twice the S.E., indicates a coupling energy significantly different from zero. The indicated diagonal arrow shows the coupling free energy (ΔΔGint) for αD138E/αR209K.
Initially, we tested the following pairwise mutations designed to maintain a strong interaction between the pair of residues: αD138E/αR209H, αD138E/αR209Q, αD138N/αR209S, αD138N/αR209G, αD138N/αR209Q and αD138N/αR209H, and αD138C/αR209C. However, none of these pairwise mutants yielded detectable single channel currents, suggesting a delicate structural interplay between αAsp-138 and αArg-209 or between this pair and the surrounding residues. We therefore made more conservative mutations of αAsp-138 and αArg-209 that maintained the charge on each residue but altered either the size or branching pattern of the side chains (Fig. 7B). The more conservative pairwise mutation, αD138E/αR209K, yielded readily detectable single channel currents marked by notably brief channel openings flanked by prolonged closings, suggesting that the apparent channel gating step was impaired. Kinetic analysis of the single channel dwell times reveals a 14-fold decrease of the apparent channel opening rate constant and a 2-fold increase of the apparent channel closing rate constant, resulting in a 26-fold decrease of the apparent channel gating equilibrium constant (Table 2). By contrast, analyses of the single residue mutations, αD138E and αR209K, reveal only modest attenuation of the apparent channel gating equilibrium constant (Table 2). Thus, the functional consequences of the double mutation are much greater than those of either individual mutation. Mutant cycle analysis (24), based on the apparent channel gating equilibrium constants, reveals an interresidue coupling free energy of 1.2 kcal/mol (Fig. 7B). The magnitude of this coupling free energy, although less than expected for charge reversal mutations of both residues, could be rationalized based on the conservative structural changes of the substituted side chains. Thus, αAsp-138 and αArg-209, which are proximal in crystal structures of multiple Cys-loop receptors, contribute to the apparent channel gating step in a functionally interdependent manner. Together with previous results showing functional interdependence between αArg-209 and surrounding anionic and electron-rich residues (4, 5), the present results further show that the apparent channel gating step depends on multiple charge-charge interactions within the region between the extracellular and transmembrane domains.
Discussion
We identify the cause of a congenital myasthenic syndrome and, in the course of investigating the underlying mechanism, reveal new and unexpected insights into AChR structure-function relationships. To our knowledge, the δD140N mutation is the first naturally occurring mutation of the invariant Cys-loop Asp in the eponymous receptor superfamily. The mutation reduces cell surface expression of AChR to 20% of wild type and impairs the speed and efficiency of the apparent channel gating step. The effects of the mutation on AChR expression and function are expected to reduce the amplitude and accelerate the decay of the endplate current, reducing the patient's safety margin for neuromuscular transmission. Furthermore, by analyzing the kinetics of single channel open and closed dwell times, we find that the invariant Cys-loop Asp is essential for the proper kinetics of AChR activation, that its contribution to the kinetics is subunit-dependent, and that in the α-subunit, it is one of three conserved anionic residues that interact with an invariant Arg to promote rapid and efficient activation.
The δD140N mutation reduces AChR expression on the cell surface through impaired association with the α-subunit, an early step in the assembly process (27). The ability of the mutant δ-subunit to associate with the α-subunit depends exquisitely on both the negative charge and size of the side chain at position δAsp-140 because both charge neutralization (Asp to Asn) and increasing the size (Asp to Glu) markedly reduce expression. On the other hand, mutations in the β- and α-subunits reduce expression to a lesser extent, whereas mutation of the ϵ-subunit does not alter expression. Because AChR expression is a multistep process, our studies do not exclude possible effects of the mutation on subunit biosynthesis, protein folding, post-translational modification, and targeting for proteasome degradation. In addition, given the unavailability of a muscle biopsy, a decrease in AChR density at the patient's endplates could not be confirmed. However, because the kinetic consequences of the mutation are moderate and the patient's symptoms are severe, a decrease in AChR density at the patient's endplates seems likely.
Our single channel kinetic analyses reveal that the δD140N mutation alters kinetic steps essential to rapid and efficient neuromuscular transmission. The apparent rate for channel opening is reduced, whereas that for ACh dissociation is increased (Table 2). As a consequence, the probability that a doubly occupied AChR will open decreases from 0.83 for the wild-type AChR to 0.55 for the mutant, an overall decrease to 66% of normal. In addition, the mean burst duration decreases from between 3.3 and 2.6 ms for the wild type to between 2.2 and 1.8 for the mutant (Tables 1 and 2).
To assess the functional consequences of the reduced expression and altered activation kinetics, we simulated MEPCs for a model neuromuscular junction. The simulations incorporated the fitted activation rate constants and an AChR density proportional to the measurements of cell surface expression. The simulations confirm that the mutation reduces the peak of the MEPC and accelerates the decay. Due to the non-linear relationship between the peak of the MEPC and AChR density (30), the reduction of the peak to 58% of normal is close to that expected from the reduction in AChR density alone (Fig. 6). The accelerated decay of the simulated MEPC is very close (2.2 ms) to the mean burst duration computed from the formula (β/k−2 + 1)/α and the rate constants in Table 2. A further reduction of the ionic current is expected from the reduced probability that a doubly occupied AChR will open, given by the formula, β/(β + k−2), to 66% of normal. This reduction is not evident in the simulated MEPC because the number of ACh molecules in a quantum is limited to a maximum of 1,000, or about 10-fold less than in a quantum of ACh at the motor synapse (34), and the assumption of instantaneous ACh occupancy in the above formula does not hold. The decrease in peak current (58%), accelerated decay (66%), and reduced opening probability (66%) predict that the mutation reduces the total ionic current to 25% of normal and thus compromises the safety margin for neuromuscular transmission.
In the course of defining the structural mechanism of the pathogenic mutation δD140N, we found that the equivalent Asp in the α-subunit is even more important for rapid and efficient channel gating. The greater contribution of the α-subunit is not due to two rather than one mutant subunit, because combining mutations of the δ- and ϵ-subunits into a single AChR results in apparent channel gating equivalent to that of either mutant subunit alone. Furthermore, we find that αAsp-138 couples energetically to the invariant cationic residue αArg-209 in the pre-M1 domain. These are the first single channel studies of the mutation αD138N from the muscle AChR, although previous macroscopic current measurements from the same mutant did not detect functional AChR channels (9). Our study complements previous work on the homomeric glycine receptor, where the equivalent Cys-loop Asp residue was found to be required for efficient activation (35), and, further, it was found that this residue interacted with the Arg residue in the pre-M1 domain equivalent to αArg-209 (8). In the latter study, cysteine substitution of either the Asp or the Arg prevented agonist-elicited macroscopic currents, whereas the double cysteine mutant afforded agonist-elicited currents with reduced magnitude. However, in the AChR, we found that the double cysteine mutant, αD138C/αR209C, did not exhibit agonist-elicited single channel currents. Although the two studies suggest differences in local structure between the AChR and glycine receptor, the muscle AChR is a heteromer, and the glycine receptor is a homomer, potentially voiding comparison.
In crystal structures from different Cys-loop receptors (3, 31–33), the equivalent Cys-loop Asp residue is one of three anionic residues engaged in electrostatic interactions with the invariant Arg from the pre-M1 domain. The strength of these electrostatic interactions is enhanced due to their location within the hydrophobic core of the subunit, suggesting that in joining multiple regions of the protein main chain, these contacts are particularly stable. We find that the mutation αD138N markedly suppresses apparent channel gating, and analysis of pairwise mutants of αAsp-138 and αArg-209 reveals interresidue energetic coupling. In addition, two more proximal anionic residues, αGlu-45 and αGlu-175, energetically couple to αArg-209 (4, 5). Thus, the collective structural and functional data show that within the protein core between the ligand binding and pore domains, multiple charge-charge interactions are crucial for rapid and efficient channel gating.
Although the kinetic consequences of the δD140N mutation are severe enough to be pathogenic, mutation of the equivalent Asp residue in the α-subunit has an even greater effect. Thus, although the five Cys-loop Asp residues are positioned symmetrically around the pentameric ring, their contributions to AChR function are asymmetric. This asymmetry probably arose during the homomer to heteromer transition, where the non-α-subunits diverged and the α-subunits retained attributes of the homomeric ancestor. Two of the retained attributes, formation of the principal face of the ACh binding site and tight coupling between the ligand binding and pore domain, thus appear inextricably linked. Furthermore, our findings show that deciphering causes of a pathogenic mutation can provide unanticipated insights into AChR structure-function relationships.
Author Contributions
X. M. S. designed, performed, analyzed, and interpreted the experiments shown in Tables 1 and 2 and Figs. 3–7 and wrote the paper; J. B. contributed to the detection of mutations and performed the experiments shown in Fig. 3; D. N. recognized the research subject and contributed to the detection of the mutations; S. M. S. designed, analyzed, and interpreted the experiments shown in Figs. 5 and 7 and Table 2 and wrote the paper; A. G. E. designed and interpreted the experiments and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Hai-Long Wang for help using the software for the MEPC simulation.
This work was supported by National Institutes of Health Grants NS6277 (to A. G. E.) and NS31744 (to S. M. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- AChR
- acetylcholine receptor
- ACh
- acetylcholine
- M1
- M2, M3, and M4, a transmembrane domain of the first, second, third, and fourth α-helix of AChR, respectively
- HEK
- human embryonic kidney
- MEPC
- miniature endplate current
- EP
- endplate
- α-bgt
- bungarotoxin.
References
- 1. Engel A. G., Shen X. M., Selcen D., and Sine S. M. (2015) Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 14, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karlin A., Holtzman E., Yodh N., Lobel P., Wall J., and Hainfeld J. (1983) The arrangement of the subunits of the acetylcholine receptor of Torpedo californica. J. Biol. Chem. 258, 6678–6681 [PubMed] [Google Scholar]
- 3. Hibbs R. E., and Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee W. Y., and Sine S. M. (2005) Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature 438, 243–247 [DOI] [PubMed] [Google Scholar]
- 5. Mukhtasimova N., and Sine S. M. (2013) Nicotinic receptor transduction zone: invariant arginine couples to multiple electron-rich residues. Biophys. J. 104, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kash T. L., Dizon M. J., Trudell J. R., and Harrison N. L. (2004) Charged residues in the β2 subunit involved in GABAA receptor activation. J. Biol. Chem. 279, 4887–4893 [DOI] [PubMed] [Google Scholar]
- 7. Kash T. L., Jenkins A., Kelley J. C., Trudell J. R., and Harrison N. L. (2003) Coupling of agonist binding to channel gating in the GABA(A) receptor. Nature 421, 272–275 [DOI] [PubMed] [Google Scholar]
- 8. Pless S. A., Leung A. W., Galpin J. D., and Ahern C. A. (2011) Contributions of conserved residues at the gating interface of glycine receptors. J. Biol. Chem. 286, 35129–35136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiu X., Hanek A. P., Wang J., Lester H. A., and Dougherty D. A. (2005) A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J. Biol. Chem. 280, 41655–41666 [DOI] [PubMed] [Google Scholar]
- 10. Ohno K., Wang H. L., Milone M., Bren N., Brengman J. M., Nakano S., Quiram P., Pruitt J. N., Sine S. M., and Engel A. G. (1996) Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ϵ subunit. Neuron 17, 157–170 [DOI] [PubMed] [Google Scholar]
- 11. Shen X. M., Ohno K., Sine S. M., and Engel A. G. (2005) Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3-M4 linker. Brain 128, 345–355 [DOI] [PubMed] [Google Scholar]
- 12. Blount P., and Merlie J. P. (1989) Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron 3, 349–357 [DOI] [PubMed] [Google Scholar]
- 13. Sigworth F. J., and Sine S. M. (1987) Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J. 52, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakmann B., Patlak J., and Neher E. (1980) Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature 286, 71–73 [DOI] [PubMed] [Google Scholar]
- 15. Colquhoun D., and Sakmann B. (1985) Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J. Physiol. 369, 501–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin F., Auerbach A., and Sachs F. (1997) Maximum likelihood estimation of aggregated Markov processes. Proc. Biol. Sci. 264, 375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen X. M., Ohno K., Tsujino A., Brengman J. M., Gingold M., Sine S. M., and Engel A. G. (2003) Mutation causing severe myasthenia reveals functional asymmetry of AChR signature cystine loops in agonist binding and gating. J. Clin. Invest. 111, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colquhoun D., and Hawkes A. G. (1995) in Single-channel Recording (Sakmann B., and Neher E., eds) pp. 397–482, Plenum, New York [Google Scholar]
- 19. Colquhoun D., and Hawkes A. G. (1995) in Single-channel Recording (Sakmann B., and Neher E., eds) pp. 589–633, Plenum, New York [Google Scholar]
- 20. Stiles J. R., Van Helden D., Bartol T. M. Jr., Salpeter E. E., and Salpeter M. M. (1996) Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle. Proc. Natl. Acad. Sci. U.S.A. 93, 5747–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou M., Engel A. G., and Auerbach A. (1999) Serum choline activates mutant acetylcholine receptors that cause slow channel congenital myasthenic syndromes. Proc. Natl. Acad. Sci. U.S.A. 96, 10466–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen X. M., Ohno K., Fukudome T., Tsujino A., Brengman J. M., De Vivo D. C., Packer R. J., and Engel A. G. (2002) Congenital myasthenic syndrome caused by low-expressor fast-channel AChR δ subunit mutation. Neurology 59, 1881–1888 [DOI] [PubMed] [Google Scholar]
- 23. Mukhtasimova N., Lee W. Y., Wang H. L., and Sine S. M. (2009) Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature 459, 451–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horovitz A., and Fersht A. R. (1990) Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 214, 613–617 [DOI] [PubMed] [Google Scholar]
- 25. Wells J. A. (1990) Additivity of mutational effects in proteins. Biochemistry 29, 8509–8517 [DOI] [PubMed] [Google Scholar]
- 26. Shen X. M., Fukuda T., Ohno K., Sine S. M., and Engel A. G. (2008) Congenital myasthenia-related AChR δ subunit mutation interferes with intersubunit communication essential for channel gating. J. Clin. Invest. 118, 1867–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu Y., Forsayeth J. R., Verrall S., Yu X. M., and Hall Z. W. (1991) Assembly of the mammalian muscle acetylcholine receptor in transfected COS cells. J. Cell Biol. 114, 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mathie A., Cull-Candy S. G., and Colquhoun D. (1987) Single-channel and whole-cell currents evoked by acetylcholine in dissociated sympathetic neurons of the rat. Proc. R. Soc. Lond. B Biol. Sci. 232, 239–248 [DOI] [PubMed] [Google Scholar]
- 29. Lape R., Colquhoun D., and Sivilotti L. G. (2008) On the nature of partial agonism in the nicotinic receptor superfamily. Nature 454, 722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pennefather P., and Quastel D. M. (1981) Relation between subsynaptic receptor blockade and response to quantal transmitter at the mouse neuromuscular junction. J. Gen. Physiol. 78, 313–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller P. S., and Aricescu A. R. (2014) Crystal structure of a human GABA receptor. Nature 512, 270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hassaine G., Deluz C., Grasso L., Wyss R., Tol M. B., Hovius R., Graff A., Stahlberg H., Tomizaki T., Desmyter A., Moreau C., Li X. D., Poitevin F., Vogel H., and Nury H. (2014) X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 512, 276–281 [DOI] [PubMed] [Google Scholar]
- 33. Zouridakis M., Giastas P., Zarkadas E., Chroni-Tzartou D., Bregestovski P., and Tzartos S. J. (2014) Crystal structures of free and antagonist-bound states of human alpha9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 21, 976–980 [DOI] [PubMed] [Google Scholar]
- 34. Kuffler S. W., and Yoshikami D. (1975) The number of transmitter molecules in a quantum: an estimate from iontophoretic application of acetylcholine at the neuromuscular synapse. J. Physiol 251, 465–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schofield C. M., Jenkins A., and Harrison N. L. (2003) A highly conserved aspartic acid residue in the signature disulfide loop of the α1 subunit is a determinant of gating in the glycine receptor. J. Biol. Chem. 278, 34079–34083 [DOI] [PubMed] [Google Scholar]
- 36. Sine S. M., Wang H. L., and Bren N. (2002) Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J. Biol. Chem. 277, 29210–29223 [DOI] [PubMed] [Google Scholar]