

Abstract
The adhesion G protein-coupled receptors (aGPCRs) are a large yet poorly understood family of seven-transmembrane proteins. A defining characteristic of the aGPCR family is the conserved GAIN domain, which has autoproteolytic activity and can cleave the receptors near the first transmembrane domain. Several aGPCRs, including ADGRB1 (BAI1 or B1) and ADGRG1 (GPR56 or G1), have been found to exhibit significantly increased constitutive activity when truncated to mimic GAIN domain cleavage (ΔNT). Recent reports have suggested that the new N-terminal stalk, which is revealed by GAIN domain cleavage, can directly activate aGPCRs as a tethered agonist. We tested this hypothesis in studies on two distinct aGPCRs, B1 and G1, by engineering mutant receptors lacking the entire NT including the stalk (B1- and G1-SL, with “SL” indicating “stalkless”). These receptors were evaluated in a battery of signaling assays and compared with full-length wild-type and cleavage-mimicking (ΔNT) forms of the two receptors. We found that B1-SL, in multiple assays, exhibited robust signaling activity, suggesting that the membrane-proximal stalk region is not necessary for its activation. For G1, however, the results were mixed, with the SL mutant exhibiting robust activity in several signaling assays (including TGFα shedding, activation of NFAT luciferase, and β-arrestin recruitment) but reduced activity relative to ΔNT in a distinct assay (activation of SRF luciferase). These data support a model in which the activation of certain pathways downstream of aGPCRs is stalk-dependent, whereas signaling to other pathways is stalk-independent.
Keywords: arrestin, G protein-coupled receptor (GPCR), proteolysis, receptor structure-function, signal transduction, ubiquitylation (ubiquitination)
Introduction
The adhesion G protein-coupled receptors (aGPCRs)3 comprise a group of 33 seven-transmembrane-spanning (7TM) proteins that form the second largest family of GPCRs in humans (1). The aGPCRs are widely distributed and critical for many physiological processes, including cell adhesion, neural development, angiogenesis, and immune system function (2, 3). Despite their essential roles, the aGPCRs are poorly understood, with most members still considered orphan receptors with no known ligands. These receptors are characterized by large, multi-domain N termini that mediate cell-to-cell and cell-to-extracellular matrix interactions. Nearly all aGPCRs have an N-terminal juxtamembrane GPCR Autoproteolysis-Inducing (GAIN) domain, which can cleave the receptor into two non-covalently associated protomers (4). N-terminal cleavage is thought to be a critical activation step because a number of groups have reported that aGPCR truncated mutants that mimic post-cleavage receptors exhibit enhanced constitutive activity; these include BAI1/ADGRB1 (5), BAI2/ADGRB2 (6), GPR133/ADGRD1 (7), CD97/ADGRE5 (8), GPR110/ADGRF1 (9), GPR56/ADGRG1 (9, 10), GPR64/ADGRG2 (11, 12), GPR126/ADGRG6 (13) and VLGR1/ADGRV1 (14). These data prompted the proposal of a disinhibition model of aGPCR activation. In this model, the N-terminal fragment (NTF) inhibits the constitutive signaling ability of the 7TM protomer (also known as the C-terminal fragment or CTF) until the NTF is engaged by a large extracellular ligand, which results in a conformational change and/or removal of the NTF to relieve inhibition and unleash maximal receptor activity (15).
The disinhibition model is a general model that leaves open the mechanistic question of precisely how aGPCR NTF regions inhibit receptor signaling. At least two more mechanistically specific models have been discussed, one in which the NTF acts as a tethered antagonist to suppress signaling by the CTF and another model in which the NTF lacks antagonist activity per se but instead masks a cryptic agonist that becomes unveiled upon cleavage and removal of the NTF (Fig. 1) (2). Several recent reports have provided evidence in support of the cryptic agonist model (7, 9, 11). Liebscher and colleagues found that peptides mimicking the remaining post-cleavage NT stalk (also known as the “stachel”) can activate GPR126/ADGRG6, GPR133/ADGRD1 and GPR64/ADGRG2 (7, 11). Similarly, Stoveken et al. demonstrated that GPR110/ADGRF1 and GPR56/ADGRG1 can also be activated by stalk-mimetic peptides (9). These findings have raised the question of whether signaling by all aGPCRs is dependent upon agonistic sequences in the receptor N-terminal stalk regions.
FIGURE 1.
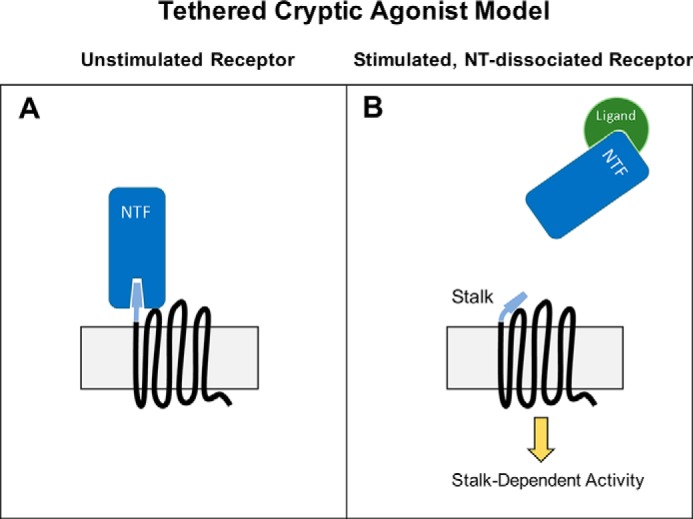
Tethered Cryptic Agonist Model of Adhesion GPCR Activation. A, according to this model, the unstimulated receptor is inactive due to the masking of an agonistic region of the stalk by the NTF. B, following ligand binding to the NTF, the NTF is released from the seven-transmembrane CTF to unveil a new N-terminal stalk, which then stimulates G protein-dependent signaling activity.
In the studies described here, we performed a series of tests of the cryptic agonist model for two distinct aGPCRs: GPR56/ADGRG1, hereafter referred to as “G1,” and BAI1/ADGRB1, hereafter referred to as “B1.” G1 is one of the most intensively-studied aGPCRs because mutations in this receptor cause a brain developmental disorder in humans (16). B1 is also a receptor of great physiological interest because it has been shown to play a critical role in regulating macrophage phagocytosis (17), muscle development (18) and synaptic plasticity in the brain (19–21).
To explore the importance of the stalk regions of these two aGPCRs for receptor signaling, we took two approaches. First, since the cryptic agonist model is largely dependent on GAIN domain cleavage, we engineered a cleavage-deficient form of G1 by mutating the catalytic threonine (Thr-383) to alanine, and additionally assessed signaling by full-length B1, which is naturally cleavage-deficient in HEK cells (4). Second, we created mutant forms of G1 and B1 that lack almost the entire NT, including the stalk region. According to the cryptic agonist model, these deletions should render the receptors completely inactive due to a lack of the tethered agonist that is necessary for receptor activation. Since most if not all GPCRs can couple to multiple downstream pathways that may be differentially activated by distinct receptor active conformations (22), the signaling activities of the G1 and B1 stalkless mutants were assessed in a battery of different assays to provide a panoramic view of the importance of the stalk regions for receptor signaling.
Experimental Procedures
Constructs
Human B1ΔNT (927–1584) and B1-SL (944–1584) were subcloned into pcDNA3.1+ between 5′ EcoRI (B1ΔNT: AGA CCA GAA TTC ATG TCC ACC TTC GCC ATC TTA GCC CAG CTC) or HindIII (B1-SL: AGA CCA AAG CTT ATG GCG ACT CTG CCG TCG GTG ACG CTC) and 3′ XbaI (AGA CCA TCT AGA TCA GAC CTC GGT CTG GAG GTC GAT GAT GTC). Human G1ΔNT (383–693) and G1-SL (404–693) were subcloned into pcDNA3.1 between 5′ HindIII (G1ΔNT: GCA AAG AAG CTT ATG ACC TAC TTT GCA GTG CTG ATG; G1-SL: GCA AAG AAG CTT ATG AGC CTC CTC TCC TAC GTG GG) and 3′ XbaI (GCA AAG TCT AGA CTA GAT GCG GCT GGA CGA GGT).
FLAG-βarrestin2 was purchased from Addgene, the RGS domain of p115RhoGEF (RGSp115) was a gift from Tohru Kozasa (Univ. of Illinois Chicago), and HA-ubiquitin was kindly provided by Keqiang Ye (Emory University). These constructs have been described previously (5). Internal EE-tagged Gα13 was acquired from the cDNA Resource Center (cdna.org).
Cell Culture
HEK-293T/17 cells were acquired from ATCC (Manassas, VA) and maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humid, 5% CO2, 37 °C incubator. Cells were transfected using Mirus (Madison, WI) TransIT-LT1 according to the manufacturer's protocol.
Western Blot
Protein samples were reduced and denatured in Laemmli buffer, loaded into 4–20% Tris-Glycine gels (Bio-Rad) for SDS-PAGE, and then transferred to nitrocellulose membranes (Bio-Rad). Blots were blocked with 5% milk (in 50 mm NaCl, 10 mm HEPES, pH 7.3 with 1% Tween-20 (Sigma)) and incubated with primary antibodies for 1 h at room temperature or overnight at 4 °C. The B1 C-terminal antibody was a gift from Dr. Erwin Van Meir and has been described previously (5). The anti-GPR56 C-terminal antibody was developed by Orbigen, Inc. via injection of rabbits with a peptide (CSNSDSARLPISSGSTSSSRI) derived from the GPR56 C terminus, and has been characterized previously (10). The biotinylated anti-GPR56 N-terminal antibody was purchased from R&D Systems. Rat anti-HA (Roche), mouse HRP-conjugated anti-FLAG (Sigma), and mouse anti-Glu Glu (Abcam) antibodies were used to detect epitope-tagged proteins. HRP-conjugated secondary antibodies were purchased from GE Healthcare and antibody labeling of specific bands was visualized using Thermo Scientific SuperSignal West solutions.
Cell Surface Biotinylation
HEK-293T cells were transfected with 2 μg of DNA (empty vector or receptor). At 24-h post-transfection, cells were placed on ice and washed with ice-cold PBS+Ca2+ three times. Cells were then incubated with 10 mm Sulfo-NHS-Biotin (Thermo Scientific) in PBS+Ca2+ on ice for 30 min and then washed three more times with PBS+Ca2+ + 100 mm glycine. Cells were resuspended in 250 μl of lysis buffer (1% Triton X-100, 25 mm HEPES, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA, protease inhibitor mixture (Roche Diagnostics), and 2% glycerol) and lysed by slowly rotating on a spinning wheel for 30 min at 4 °C. Cell debris was cleared by centrifugation, and soluble cell lysates were incubated with 50 μl of streptavidin agarose beads (Thermo Scientific) for 1 h at 4 °C. Beads were washed three times with lysis buffer and resuspended in 60 μl of Laemmli buffer. Biotinylated proteins were detected via Western blot, as described above.
β-Arrestin Binding Assay
HEK-293T cells were transfected with a total of 6 μg of DNA (empty vector, receptor, FLAG-βArr2 or HA- βArr2). The next day, cells were washed with cold PBS+Ca2+ and lysed in harvest buffer (150 mm NaCl, 25 mm HEPES pH 7.3, 1 mm EDTA, 10 mm MgCl2, 1% Triton X-100, Roche EDTA-free complete protease inhibitor mixture tablet). Lysates were rotated at 4 °C for 45 min to solubilize integral membrane proteins and membranes were cleared by centrifugation (15 min at 17,000 × g, 4 °C). Solubilizates were added to magnetic anti-FLAG beads (Sigma) or anti-HA agarose beads (Sigma) and rotated at 4 °C for 1 h. Beads were washed 3× in harvest buffer and proteins were eluted in Laemmli buffer at 37 °C for 10–15 min and loaded in 4–20% Tris-glycine gels for SDS-PAGE and Western blotting. Western blot bands were quantified using Image Studio software (Licor, Lincoln, NE). For purposes of quantification, results from experiments using alternate B1ΔNT (929–1584) and B1-SL (941–1584) constructs were pooled as the results were indistinguishable.
G Protein Co-immunoprecipitation
HEK-293T cells were transfected with 1 μg of EE-tagged Gα13 and 1–4 μg of receptor DNA). EE-tagged G proteins were immunoprecipitated with anti-EE antibody (1:200, Abcam) and protein A/G beads (Thermo) as described above. Beads were washed 3×, and proteins were eluted in 2× Laemmli buffer.
Ubiquitination Assays
HEK-293T cells were plated and transfected as described above with 3 μg of receptor and 1 μg of HA-ubiquitin DNA. Four hours after transfection, cells were treated with 100 nm MG-132 (Tocris) to inhibit the proteasome overnight. The following day, cells were washed and harvested as described above. Cleared lysates were incubated with anti-HA agarose beads (Sigma) for 1 h, washed, and eluted in Laemmli buffer.
Luciferase Reporter Assays
HEK-293T cells were seeded in 96-well plates 20–24 h prior to transfection. Each well was transfected with 50 ng of firefly reporter, 1 ng of Renilla luciferase, and 10 ng of receptor or mock DNA. All reporter constructs (NFAT: pGL4.30, SRF: pGL4.34, Renilla pRLSV40) were acquired from Promega (Madison, WI). 24–48 h later Dual-Glo luciferase assays (Promega) were performed according to the manufacturer's protocol and plates were read on either a Biotek Synergy 3 or BMG Omega plate reader. Results were calculated for each assay by determining the luminescence ratio of firefly:Renilla luciferase counts, normalized to empty vector (EV) transfected wells. Error bars for all EV-transfected conditions were represented as the standard errors of the normalized raw value means.
AP-TGFα-shedding Assays
HEK-293T cells were seeded in 96-well plates 20–24 h prior to transfection. Each well was transfected with 50 ng of AP-TGFα plasmid (kindly provided by Shigeki Higashiyama, Ehime University) and 10 ng of receptor or mock DNA. Twenty-four hours later, the plate was incubated with p-nitrophenyl phosphate (New England BioLabs) and read on either a Biotek Synergy 3 or BMG Omega plate reader as per the protocol described by Inoue et al. (23).
Results
Cleavage-deficient Receptors Retain Signaling Activity
To test the cryptic agonist model of aGPCR activation, we performed parallel studies on two distinct aGPCRs: GPR56/ADGRG1 (“G1”) and BAI1/ADGRB1 (“B1”). According to the cryptic agonist model, signaling activity depends upon efficient GAIN domain cleavage followed by dissociation of the N-terminal fragment (NTF) to unveil the agonistic peptide found on the remaining N-terminal stalk of the 7TM protomer (the C-terminal fragment or CTF). Thus, we tested whether GAIN domain cleavage was indeed necessary for G1 and B1 basal constitutive activity.
As G1 is efficiently cleaved in transfected HEK-293T cells (10), we introduced a point mutation to the G1 GAIN domain at the site of cleavage (T383A) to create a cleavage-deficient version of the receptor (Fig. 2A). Human G1 mutations (C346S and W349S) that abrogate GAIN domain cleavage have been shown to result in the devastating neurological condition bilateral frontoparietal polymicrogyria, which led to speculation that autoproteolysis may be necessary for proper aGPCR function (24, 25). However, more recent crystallographic studies provided insights as to how GAIN domain cleavage can be abrogated without inducing misfolding of the GAIN domain (4). Using a cell surface biotinylation approach, we found that indeed G1-T383A (T383A) traffics to the plasma membrane, albeit at a somewhat reduced level compared with the wild-type receptor (Fig. 2B). To validate whether T383A is indeed cleavage-deficient, we probed for expression of the mutant receptor in Western blots using both CT- and NT-specific antibodies (Fig. 2C). The left panel of Fig. 2C displays the expression patterns of the wild-type and T383A receptors using a C-terminal specific antibody. For wild-type G1, there is a prominent band at ∼45 kDa which represents the monomeric, cleaved 7TM protomer. As expected, the T383A mutant lacks the ∼45 kDa band and instead displays a prominent band at ∼75 kDa which is the predicted molecular weight of full-length, uncleaved G1. Higher order bands for either the wild-type or T383A receptors are likely to be unresolved, oligomeric complexes. The righthand panel of Fig. 2C displays the expression patterns of both receptors as detected by an N-terminal specific antibody. Here bands are found at ∼70 kDa and ∼75 kDa for the wild-type receptor and T383A, respectively. The ∼75 kDa band of T383A is both C-terminally and N-terminally reactive, providing strong evidence that the point mutation does indeed abrogate cleavage to result in a single, uncleaved protein.
FIGURE 2.

GAIN domain cleavage is not necessary for G1 activity. A, schematic of T383A point mutation in G1. B, G1-T383A is expressed on HEK cell surface, albeit at a reduced level compared with the wild-type receptor. Molecular weight markers (in kDa) are shown on the left side of the blots. C, Western blots of G1 and G1-T383A reveal a ∼75 kDa band for G1-T383A that is both N-terminally and C-terminally reactive, suggesting non-cleavage of the mutant receptor. Equal amounts of protein (10–20 μg) were loaded in each lane for the blots shown in panels B and C, and these experiments were performed 3–4 times each. D and E, G1 and G1-T383A produce comparable activity in the AP-TGFα shedding and SRF-luciferase assays. Results for TGFα and SRF-luc are from 3–6 independent experiments (± S.E. shown, *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus cells transfected with empty vector, denoted by EV).
We next assessed the basal constitutive activity of the T383A mutant using two distinct downstream readouts: TGFα shedding and activation of SRF luciferase. G1 has previously been reported to couple to Gα12/13 (9, 10, 26), and GPCRs that activate Gαq- or Gα12/13-mediated pathways stimulate the ectodomain shedding of TGFα from the plasma membrane, with the amount of TGFα released into the conditioned media serving as a proxy for receptor activity (23). We observed that wild-type G1 and the T383A mutant displayed an equal level of activity in the TGFα shedding assay (Fig. 2D). Next, we compared the activities of wild-type G1 and T383A in a serum response factor (SRF)-luciferase reporter assay, another well-described readout for Gα12/13-coupled receptors that has previously been shown to be activated by G1 (27). In agreement with our TGFα shedding data, the T383A mutant mediated an approximately equal level of signaling activity to wild-type G1 in the SRF-luciferase assay (Fig. 2E). Regarding B1, this receptor is not efficiently cleaved in HEK-293T cells, as evidenced by >99% of the protein consistently appearing between 150–250 kDa in Western blots (5). Nonetheless, full-length, uncleaved B1 exhibits substantial constitutive G protein-dependent signaling activity, as shown in previously published studies (5) and also shown below. Taken together, these data provide evidence that GAIN domain cleavage is not necessary for activity, at least for G1 and B1.
Receptors Lacking the Entire N Terminus Retain Activity in a Battery of Signaling Assays
It is plausible that an agonistic peptide region of the stalk could still be important for activation of aGPCR signaling even in the absence of GAIN autoproteolysis, as cleavage-independent conformational changes that expose the cryptic agonist could conceivably be responsible for receptor activity. Therefore, to definitively answer the question of whether the stalk does indeed contain a requisite agonist for G1 and B1 signaling, we created mutant versions of the receptors that lack the entire NT (including the stalk) such that they begin very close to the start of the predicted first transmembrane domain (Fig. 3, A and C). These stalkless (“SL”) mutant receptors expressed and were trafficked to the plasma membrane at levels comparable to ΔNT versions of G1 and B1 (Fig. 3, B and D), revealing that removal of the stalk region did not result in impairments in receptor expression or trafficking.
FIGURE 3.

Generation of G1 and B1 SL receptors. A and C, schematic of G1-SL and B1-SL alongside their ΔNT counterparts. B and D, SL mutants exhibit comparable surface expression in HEK cells to their ΔNT counterparts. Molecular weight markers (in kDa) are shown on the left side of the blots. For G1, prominent C-terminally reactive bands between ∼40–45 kDa correspond to monomeric 7TM regions. Lower molecular weight bands at ∼25 kDa (for full-length G1) and ∼20 kDa (for G1-ΔNT or -SL) likely represent further cleaved forms of the proteins and/or differential conformations. Equal amounts of protein (10–20 μg) were loaded in each lane for the blots shown here, and the data shown in this figure are representative of 3–4 experiments for each pair of mutants.
It has previously been reported that deletion of the NT up to the site of GAIN cleavage results in significant increases in the constitutive activity of aGPCRs, including G1 and B1 (5, 9, 10). According to the cryptic agonist model, further NT deletions that remove some or all of the stalk region should render the receptors inactive. To assess the activities of G1-SL and B1-SL, we utilized a battery of signaling assays. In the TGFα shedding assay, G1-SL mediated significant signaling compared with mock-transfected cells (Fig. 4A: main effect of receptor F (3,20) = 19.98, p < 0.0001, post hoc EV versus G1-SL p < 0.0001), with the extent of signaling not significantly different from that mediated by G1ΔNT (p > 0.05). The results were different, though, in the SRF-luciferase assay, in which G1ΔNT exhibited robust activity but G1-SL did not (Fig. 4B: main effect of receptor F (4, 25) = 16.97, p < 0.0001, post hoc EV versus G1-SL p > 0.05). Given these contrasting results, we measured receptor activity using a third signaling output: transcription of the nuclear factor of activated T-cells (NFAT), a readout that has been shown to be downstream of some Gα12/13-coupled receptors (27, 28) including G1 (27). We found that G1ΔNT signals strongly in the NFAT-luciferase assay but the full-length receptor does not (Fig. 4C: one-way ANOVA, main effect of receptor F (3, 12) = 20.52, p < 0.0001). Interestingly, we found that G1-SL also strongly activated the NFAT pathway, achieving an extent of activation comparable to G1ΔNT (Fig. 4C: G1ΔNT p < 0.001, G1-SL p < 0.01). This signaling was sensitive to inhibition by the RGS domain of p115RhoGEF (29) (Fig. 5A, one-sample t test, G1ΔNT p = 0.0063, G1-SL p = 0.0080 compared with 0.0) and the broad-spectrum Gβγ inhibitor gallein (30) (Fig. 5B: one-sample t test, G1ΔNT p = 0.0034, G1-SL p = 0.0017 compared with 0.0), demonstrating that it is mediated predominantly by heterotrimeric G proteins.
FIGURE 4.
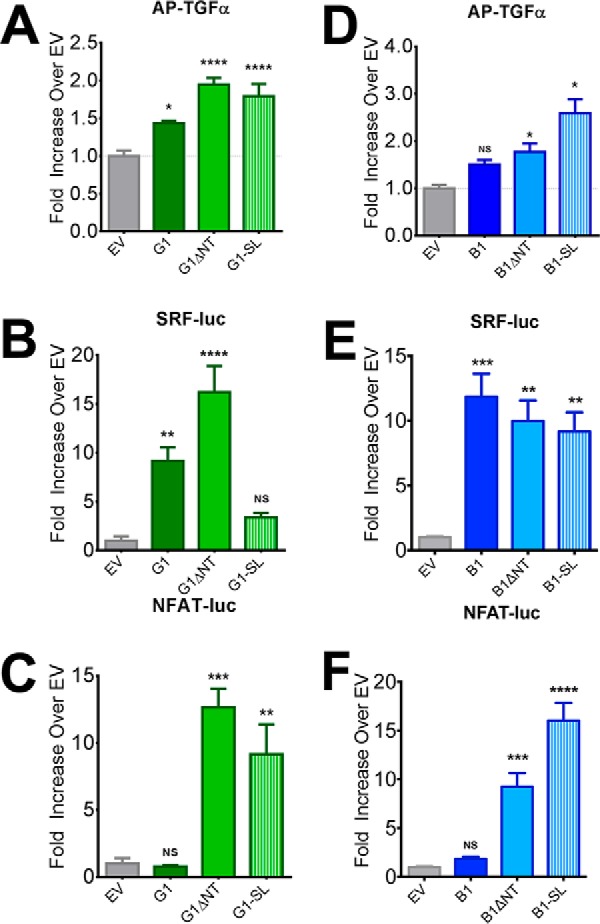
G1-SL exhibits differential levels of signaling activity in distinct assays whereas B1-SL is consistently active. G1-SL exhibited significant signaling activity in the TGFα-shedding (A) and NFAT luciferase (C) assays but was found to not be significantly active in the SRF-luciferase assay (B). However, B1-SL was significantly active at comparable levels to B1ΔNT in all three assays (D: TGFα-shedding, E: SRF-luc, F: NFAT-luc) demonstrating the dispensability of the B1 post-cleavage stalk. All experiments performed in HEK cells. TGFα, SRF-luc, and NFAT-luc results are from 4–6 independent experiments (± S.E. shown, *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 versus cells transfected with empty vector, denoted by EV).
FIGURE 5.
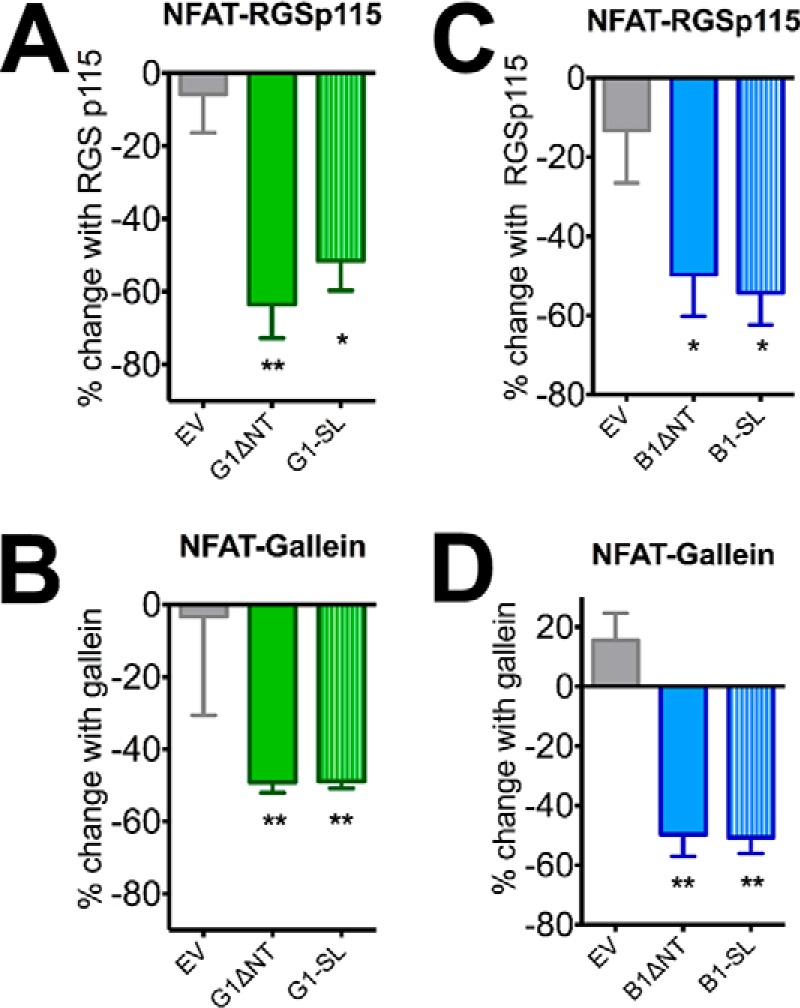
Inhibitors of Gα12/13 and Gβ/γ block ΔNT and -SL signaling activity. The RGS domain of p115RhoGEF, a Gα12/13 inhibitor, as well as the Gβ/γ inhibitor gallein, significantly block ΔNT and -SL signaling to NFAT for both receptors (5A-B: G1, 5C-D: B1). Results are from 3–6 independent experiments (± S.E. shown, *, p < 0.05; **, p < 0.01 versus G1/B1).
In contrast to G1-SL, which displayed activity in two out of three signaling assays utilized, B1-SL displayed significant signaling activity over mock-transfected cells in all three assays (Fig. 4, D–F). First, we found that B1ΔNT and B1-SL induced significantly more TGFα shedding than mock-transfected cells (Fig. 4D: main effect of receptor transfection, one-way ANOVA, F (3, 19) = 15.17, post-hoc B1: p > 0.05, B1ΔNT: p < 0.01, B1-SL: p < 0.0001). In the SRF-luciferase assay (4E), one-way ANOVA found a main effect of receptor transfection (F (3, 16) = 11.67, p = 0.0003). Post-hoc comparisons revealed that B1 (p < 0.001), B1ΔNT (p < 0.01) and B1-SL (p < 0.01) all displayed significant activity over empty vector (EV) transfected controls but were not significantly different from each other. However, in measuring NFAT luciferase activity (4F), we again found a main effect of receptor transfection in one-way ANOVA (F (3, 12) = 37.46, p < 0.0001) but found that full-length B1 showed no significant activity over empty vector whereas B1ΔNT (p < 0.001) and B1-SL (p < 0.0001) exhibited robust activity that was highly significant over baseline. In fact, B1-SL was significantly more active than B1ΔNT (p < 0.01).
Similar to our findings with G1, this signaling activity from B1 was significantly inhibited by co-transfection with the RGS domain of p115RhoGEF (Fig. 5C: one-sample t test, B1ΔNT p = 0.0032, B1-SL p = 0.0005 compared with 0.0) and also inhibited by overnight treatment (50 μm) with gallein (Fig. 5D: one-sample t test, B1ΔNT p = 0.0064, B1-SL p = 0.0025 compared with 0.0) demonstrating that this signaling activity is dependent on heterotrimeric G proteins.
Finally, to further assess the abilities of the SL mutants to couple to heterotrimeric G proteins, we performed co-immunoprecipitation experiments with each receptor and co-transfected EE-tagged Gα13. In these experiments, the tagged G protein was immunoprecipitated and Western blots were performed to detect any co-immunoprecipitated receptor. For both G1 and B1, we found that the truncated ΔNT and SL forms strongly and significantly co-immunoprecipitated with Gα13 but the full-length forms of the receptors did not (Fig. 6A: G1 one-way ANOVA F (2, 6) = 10.49, p < 0.05, post-hoc test versus full-length G1, G1ΔNT p < 0.05; G1-SL p < 0.05. Fig. 6B, B1: one way ANOVA main effect of receptor, Holm-Sidak multiple comparisons B1ΔNT p < 0.05, B1-SL p < 0.01 versus B1).
FIGURE 6.

G1-SL and B1-SL couple to G proteins. Western blots of co-immunoprecipitation experiments in HEK cells demonstrating that ΔNT and -SL receptors (6A: G1, 6B: B1) robustly associate with Gα13 whereas the full-length versions of the receptors do not. Equal amounts of protein (10–20 μg) were loaded in each lane for the blots shown here, and the results shown are from 3–4 independent experiments (± S.E. shown, *, p < 0.05; **, p < 0.01 versus G1/B1).
Stalk-deficient Receptors Exhibit Robust β-Arrestin Association and Ubiquitination, Two Correlates of Enhanced GPCR Activity
To further assess the activity of the stalk-deficient receptors, we employed a β-arrestin2 recruitment assay. β-Arrestin recruitment is a classical hallmark of highly active GPCRs (31). In support of their constitutively-active nature, both B1ΔNT and G1ΔNT have been reported to strongly bind β-arrestin2 whereas their full-length counterparts do not (5, 10). In the present study, we confirmed that B1ΔNT and G1ΔNT robustly co-immunoprecipitate with β-arrestin2, and additionally found that both G1-SL and B1-SL bind β-arrestin2 to a comparably robust extent as the ΔNT mutants (Fig. 7). Multiple iterations of this experiment were quantified and revealed a main effect of receptor transfected with β-arrestin2 relative to β-arrestin2 transfected alone (B1: one-way ANOVA F (2, 6) = 11.58, p = 0.0087, post-hoc: B1 p > 0.05, B1ΔNT p < 0.05, B1-SL p < 0.01; G1: F (2,6) = 31.93, p = 0.0006, G1 p > 0.05, G1ΔNT p < 0.001, G1-SL p < 0.01). Similar to our reporter assay results shown earlier, G1-SL binding to β-arrestin2 was found to be significantly lower (One-way ANOVA, post-hoc p < 0.05, n = 3) than that of G1ΔNT whereas there was no significant difference between B1ΔNT and B1-SL.
FIGURE 7.

G1-SL and B1-SL bind robustly to βArrestin2. Western blots of co-immunoprecipitation experiments in HEK cells with HA or FLAG-tagged β-arrestin2 revealed that ΔNT and -SL receptors bound to β-arrestin2 significantly more than full-length receptors. Molecular weight markers (in kDa) are shown on the left side of the blots. Equal amounts of protein (10–20 μg) were loaded in each lane for the blots shown here, and the results shown are from three independent experiments (± S.E. shown, *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus G1/B1).
In addition to binding to β-arrestins, highly active GPCRs are often ubiquitinated prior to down-regulation and degradation (32). As with β-arrestin association, B1ΔNT and G1ΔNT have previously been found to be heavily ubiquitinated whereas their full-length counterparts are not (5, 10). As shown in Fig. 8, we found that B1-SL and G1-SL are ubiquitinated to a similar extent as their ΔNT counterparts (B1: one-way ANOVA F (2, 6) = 132.9, p < 0.0001, post-hoc test versus full-length B1, B1ΔNT p < 0.0001, B1-SL p < 0.0001; G1 F (2,6) = 27.50, p = 0.001, G1 p > 0.05, G1ΔNT p < 0.001, G1-SL p < 0.01), which provides further support for the idea that the SL mutants are highly active receptors.
FIGURE 8.

G1-SL and B1-SL are heavily ubiquitinated. Western blots of co-immunoprecipitation experiments with HA-ubiquitin demonstrated that ΔNT and -SL receptors were significantly more ubiquitinated than full-length receptors. Molecular weight markers (in kDa) are shown on the left side of the blots. Equal amounts of protein (10–20 μg) were loaded in each lane for the blots shown here, and the results are from three independent experiments (± S.E. shown, *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 versus G1/B1).
Discussion
Substantial recent progress has been made in understanding the mechanisms of aGPCR activation. These mechanisms are important to understand, given the association of these receptors with several human diseases and the potential value of these receptors as drug targets (1). Specifically, it has been found for a number of aGPCRs that truncation of the receptor N termini up to the point of predicted GAIN domain cleavage leads to increased constitutive activity (5–14). These observations led to the proposal of the disinhibition model of aGPCR activation, which posits that the NTF exerts an inhibitory constraint on signaling by the CTF, with this inhibitory constraint being removed following engagement of the NTF with a large extracellular ligand that results in either dissociation of the NTF from the CTF and/or a conformational change that reduces NTF-mediated inhibition (15). Subsequently, more mechanistically specific variations of the disinhibition model have been proposed, including the cryptic agonist model (7, 9, 11), wherein GAIN domain cleavage and NTF dissociation result in the unveiling of a cryptic agonist peptide on the post-cleavage stalk of the CTF in a manner analogous to protease-activated receptors (33).
In the studies described above, we tested the cryptic agonist model directly in parallel experiments on two distinct aGPCRs by deleting the stalk regions and broadly assessing receptor activity using a variety of downstream outputs. Our results provide evidence that the stalk region is not a requisite agonist for these two aGPCRs, as we observed that deleting the stalk does not affect signaling to most pathways measured. In the case of ADGRB1/BAI1 (“B1”), removal of the stalk region made no difference whatsoever for any assessment of receptor activity. In fact, we found B1-SL to be significantly more active in the NFAT luciferase assay than B1ΔNT, which suggests that the stalk may actually attenuate the full signaling ability of this receptor. In contrast, studies on ADGRG1/GPR56 (“G1”) resulted in mixed findings, with removal of the stalk region largely abrogating the receptor's ability to stimulate SRF luciferase but having little effect on the other readouts examined. Additionally, both B1- and G1-SL retained the ability to robustly co-immunoprecipitate with Gα13. Based on these findings, we propose that aGPCRs can mediate both stalk-dependent and stalk-independent signaling, with the relative contribution of the stalk to receptor activity varying substantially between different receptors as well as between different outputs examined.
A recent study by Stoveken et al. (2015) suggested that the NT stalk of G1 is necessary for signaling activity (9). This study reported signaling data from SRE luciferase experiments in transfected cells and GTP loading experiments in a reconstitution system. Our data reported here are in agreement with the findings of Stoveken et al., as we found that the activity of G1-SL was sharply reduced compared with G1ΔNT in the SRF luciferase assay, which is very similar to the SRE luciferase assay. However, in other assays in which we assessed G1 activity (TGFα shedding, NFAT luciferase, β-arrestin recruitment and receptor ubiquitination), we found G1-SL to be in an active conformation and capable of mediating receptor signaling to a similar extent as G1ΔNT. These results suggest that the stalk region of G1 is necessary for certain aspects of receptor signaling activity but dispensable for others.
There have been prior indications that the cryptic agonist model may represent an incomplete description of aGPCR activation. For example, studies on the Caenorhabditis elegans aGPCR lat-1 demonstrated that mutations blocking cleavage of the receptor's GAIN domain exerted no effect on the in vivo function of the receptor (34). These in vivo data find a parallel in the in vitro findings reported here regarding the non-cleaving G1-T383A mutant, which we found to exhibit no change in signaling activity relative to wild-type G1. Similarly, a recent report from Peeters et al. demonstrated that non-cleavable versions of GPR64/ADGRG2 can still activate downstream signaling (12). According to the cryptic agonist model, GAIN-mediated cleavage should be essential for exposure of the agonistic peptide sequence on the stalk region. Thus, observations that the activity of at least some aGPCRs is not modulated by GAIN cleavage obviously run counter to this model. Moreover, there is convincing evidence that some aGPCRs do not undergo GAIN-mediated cleavage (35), an observation that needs to be taken into account in general models of aGPCR activation.
Another challenge faced by the cryptic agonist model is the uncertainty surrounding how aGPCR NTF regions become dissociated from their cognate 7TM regions. In the cryptic agonist model, it is envisioned that the N-terminal portion of a cleaved GAIN domain can be released from the receptor's stalk region in a regulated manner, thereby exposing the agonistic stalk peptide sequence (7, 9, 11). However, the first crystal structures of GAIN domains reported by Arac et al. have cast doubt on whether GAIN domains can actually exist as stable folded protein units in the absence of the hydrophobic stalk peptides (4). Thus, while it is clear that aGPCR NTF regions can become dissociated from their cognate CTF regions (2), it is uncertain whether dissociated GAIN domains leave the stalk behind or take the stalk with them. Interestingly, studies on ADGRL1/CIRL/latrophilin-1 provided evidence that the release of this receptor NTF region is dependent on two proteolytic steps, with GAIN domain cleavage followed by a second cleavage event that cleaves the receptor's stalk region to release the GAIN domain and stalk together (36). According to the cryptic agonist model, the resultant 7TM region of such a twice-cleaved aGPCR would be devoid of signaling activity, as the stalk region containing the agonistic peptide would have been lost with the second cleavage event. However, our studies presented here on stalkless versions of G1 and B1 demonstrate that stalkless receptors can still exert significant downstream signaling, albeit signaling that is altered in some cases (as in the case of G1) relative to receptors that possess an intact stalk.
Further work will be needed to elucidate the differences in signaling intermediates that presumably exist between the stalk-dependent versus stalk-independent signaling activities observed in our studies. As shown above, the activation of NFAT luciferase by G1 and B1 was significantly blocked by the Gβγ inhibitor gallein and also blocked to a similar extent by the RGS domain of p115RhoGEF, which would be expected to attenuate signaling by Gα12/13 as well as any Gβγ subunits released from activated Gα12/13. Insofar as the SRF luciferase assay represents a more pure readout of Gα12/13 action, the differential change in activity observed for G1-SL in the SRF versus NFAT luciferase assays may represent a difference in the relative importance of Gα12/13- versus Gβγ-dependent pathways (37). Yet another possibility is that β-arrestins may contribute to G1 and B1 signaling, with the presence of the stalk having little effect on β-arrestin-mediated signaling. Indeed, we found that the ΔNT and SL versions of G1 and B1 both exhibited strong interactions with β-arrestin2. However, the specific contributions of the various G protein subunits and β-arrestins to signaling by G1, B1, and other adhesion GPCRs will require further investigation to truly assess whether the stalk regions might confer bias toward certain receptor-initiated pathways and away from others (38).
Understanding the natural mechanism(s) of aGPCR activation is a critical step toward facilitating drug development efforts aimed at these receptors. For example, the elucidation of agonistic peptide sequences on the N-terminal stalks of certain aGPCRs (7, 9, 11) has provided insights that may lead to the development of peptidomimetic small molecules with agonistic activity at these receptors. Similarly, the findings reported here that cryptic agonist sequences on aGPCR stalks do not account for the entirety of aGPCR signaling are important because these observations suggest an additional antagonistic effect of tethered GAIN domains on aGPCR activity beyond the simple masking of the stalk region. Therefore, we propose an allosteric antagonist model of aGPCR activation (Fig. 9), in which the NTF can antagonize receptor activity in two distinct ways: (i) by masking the stalk region and (ii) by directly antagonizing the inherent stalk-independent constitutive activity of the 7TM region. The word “allosteric” in this context is meant to convey that the NTF presumably does not block agonist binding in the manner of a competitive antagonist, but rather constrains receptor activity in an allosteric fashion. This model is consistent with the data presented here as well as in previous studies (7, 9, 11) and furthermore is consistent with the possibility that aGPCRs may still signal even if they are not cleaved at the GAIN domain or lose their stalk following GAIN cleavage.
FIGURE 9.

Allosteric Antagonist Model of aGPCR Activation. A, in this model, the NTF behaves as an allosteric antagonist in two ways: (i) masking the stalk region and (ii) directly antagonizing the constitutive stalk-independent activity possessed by the 7TM region. B, conformational change of the NTF induced by ligand binding is sufficient to allow for enhanced stalk-dependent activity. C, ligand binding can also result in either NTF dissociation or a conformational change that relieves the inhibitory constraint of the NTF upon the 7TM region, such that both stalk-dependent and stalk-independent pathways are activated.
Further insights into the structural determinants of the antagonistic relationship between aGPCR NTF and 7TM regions may help to facilitate discovery of distinct classes of small-molecule aGPCR modulators that either block or potentiate NTF-mediated suppression of aGPCR 7TM signaling. Additionally, a model in which aGPCRs can mediate both stalk-dependent and stalk-independent signaling has clear implications for the future development of biased agonists targeting these receptors (38). In many cases, it is therapeutically desirable to target some but not all pathways downstream of a given receptor. Thus, it will be of interest going forward to study the various members of the aGPCR family on a receptor-by-receptor basis in order to understand the structural determinants of receptor coupling to different downstream signaling pathways to facilitate the discovery of biased ligands possessing therapeutic potential.
Author Contributions
A. K., R. H. P., and R. A. H. designed the experiments and wrote the manuscript. AK performed the experiments focused on ADGRG1 as well as the TGFα shedding assays focused on ADGRB1. R. H. P. and Z. N. T. performed all other experiments focused on ADGRB1. A. K., R. H. P., and R. A. H. analyzed the data and created the figures. All authors made intellectual contributions to the paper and take responsibility for the data.
Acknowledgments
We thank Michelle Giddens (Emory University) for helpful comments about this work and Tohru Kozasa (University of Illinois, Chicago), Keqiang Ye (Emory University) and Shigeki Higashiyama (Ehime University) for kindly providing constructs to facilitate these studies.
This work was supported by Grant R01 NS072394 from the National Institutes of Health (to R. A. H.), NIH Training Grant T32 GM008602 (to A. K.), and NIH Training Grant T32 GM008605 (to R. H. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- aGPCR
- adhesion G protein-coupled receptor
- SL
- stalkless
- NTF
- N-terminal fragment
- CTF
- C-terminal fragment
- 7TM
- seven-transmembrane domain
- GAIN
- GPCR autoproteolysis-inducing.
References
- 1. Langenhan T., Aust G., and Hamann J. (2013) Sticky signaling–adhesion class G protein-coupled receptors take the stage. Sci. Signal. 6, re3. [DOI] [PubMed] [Google Scholar]
- 2. Hamann J., Aust G., Araç D., Engel F. B., Formstone C., Fredriksson R., Hall R. A., Harty B. L., Kirchhoff C., Knapp B., Krishnan A., Liebscher I., Lin H. H., Martinelli D. C., Monk K. R., Peeters M. C., Piao X., Prömel S., Schöneberg T., Schwartz T. W., Singer K., Stacey M., Ushkaryov Y. A., Vallon M., Wolfrum U., Wright M. W., Xu L., Langenhan T., and Schiöth H. B. (2015) International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 67, 338–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prömel S., Langenhan T., and Araç D. (2013) Matching structure with function: the GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 34, 470–478 [DOI] [PubMed] [Google Scholar]
- 4. Araç D., Boucard A. A., Bolliger M. F., Nguyen J., Soltis S. M., Südhof T. C., and Brunger A. T. (2012) A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31, 1364–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stephenson J. R., Paavola K. J., Schaefer S. A., Kaur B., Van Meir E. G., and Hall R. A. (2013) Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J. Biol. Chem. 288, 22248–22256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okajima D., Kudo G., and Yokota H. (2010) Brain-specific angiogenesis inhibitor 2 (BAI2) may be activated by proteolytic processing. J. Recept Signal Transduct. Res. 30, 143–153 [DOI] [PubMed] [Google Scholar]
- 7. Liebscher I., Schön J., Petersen S. C., Fischer L., Auerbach N., Demberg L. M., Mogha A., Cöster M., Simon K. U., Rothemund S., Monk K. R., and Schöneberg T. (2014) A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 9, 2018–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ward Y., Lake R., Yin J. J., Heger C. D., Raffeld M., Goldsmith P. K., Merino M., and Kelly K. (2011) LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 71, 7301–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stoveken H. M., Hajduczok A. G., Xu L., and Tall G. G. (2015) Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. U.S.A. 112, 6194–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paavola K. J., Stephenson J. R., Ritter S. L., Alter S. P., and Hall R. A. (2011) The N terminus of the adhesion G protein-coupled receptor GPR56 controls receptor signaling activity. J. Biol. Chem. 286, 28914–28921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Demberg L. M., Rothemund S., Schöneberg T., and Liebscher I. (2015) Identification of the tethered peptide agonist of the adhesion G protein-coupled receptor GPR64/ADGRG2. Biochem. Biophys. Res. Commun. 464, 743–747 [DOI] [PubMed] [Google Scholar]
- 12. Peeters M. C., Fokkelman M., Boogaard B., Egerod K. L., van de Water B., IJzerman A. P., and Schwartz T. W. (2015) The adhesion G protein-coupled receptor G2 (ADGRG2/GPR64) constitutively activates SRE and NFκB and is involved in cell adhesion and migration. Cell. Signal. 27, 2579–2588 [DOI] [PubMed] [Google Scholar]
- 13. Paavola K. J., Sidik H., Zuchero J. B., Eckart M., and Talbot W. S. (2014) Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 7, ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Q. X., Dong J. H., Du H. B., Zhang D. L., Ren H. Z., Ma M. L., Cai Y., Zhao T. C., Yin X. L., Yu X., Xue T., Xu Z. G., and Sun J. P. (2014) Constitutive Gαi coupling activity of very large G protein-coupled receptor 1 (VLGR1) and its regulation by PDZD7 protein. J. Biol. Chem. 289, 24215–24225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paavola K. J., and Hall R. A. (2012) Adhesion G protein-coupled receptors: signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 82, 777–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piao X., Hill R. S., Bodell A., Chang B. S., Basel-Vanagaite L., Straussberg R., Dobyns W. B., Qasrawi B., Winter R. M., Innes A. M., Voit T., Ross M. E., Michaud J. L., Déscarie J. C., Barkovich A. J., and Walsh C. A. (2004) G protein-coupled receptor-dependent development of human frontal cortex. Science 303, 2033–2036 [DOI] [PubMed] [Google Scholar]
- 17. Park D., Tosello-Trampont A. C., Elliott M. R., Lu M., Haney L. B., Ma Z., Klibanov A. L., Mandell J. W., and Ravichandran K. S. (2007) BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434 [DOI] [PubMed] [Google Scholar]
- 18. Hochreiter-Hufford A. E., Lee C. S., Kinchen J. M., Sokolowski J. D., Arandjelovic S., Call J. A., Klibanov A. L., Yan Z., Mandell J. W., and Ravichandran K. S. (2013) Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497, 263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duman J. G., Tzeng C. P., Tu Y. K., Munjal T., Schwechter B., Ho T. S., and Tolias K. F. (2013) The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J. Neurosci. 33, 6964–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu D., Li C., Swanson A. M., Villalba R. M., Guo J., Zhang Z., Matheny S., Murakami T., Stephenson J. R., Daniel S., Fukata M., Hall R. A., Olson J. J., Neigh G. N., Smith Y., Rainnie D. G., and Van Meir E. G. (2015) BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J. Clin. Invest. 125, 1497–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stephenson J. R., Purcell R. H., and Hall R. A. (2014) The BAI subfamily of adhesion GPCRs: synaptic regulation and beyond. Trends Pharmacol. Sci. 35, 208–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kenakin T. (2011) Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336, 296–302 [DOI] [PubMed] [Google Scholar]
- 23. Inoue A., Ishiguro J., Kitamura H., Arima N., Okutani M., Shuto A., Higashiyama S., Ohwada T., Arai H., Makide K., and Aoki J. (2012) TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat. Methods 9, 1021–1029 [DOI] [PubMed] [Google Scholar]
- 24. Jin Z., Tietjen I., Bu L., Liu-Yesucevitz L., Gaur S. K., Walsh C. A., and Piao X. (2007) Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum. Mol. Genet. 16, 1972–198517576745 [Google Scholar]
- 25. Chiang N. Y., Hsiao C. C., Huang Y. S., Chen H. Y., Hsieh I. J., Chang G. W., and Lin H. H. (2011) Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J. Biol. Chem. 286, 14215–14225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iguchi T., Sakata K., Yoshizaki K., Tago K., Mizuno N., and Itoh H. (2008) Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a Gα12/13 and Rho pathway. J. Biol. Chem. 283, 14469–14478 [DOI] [PubMed] [Google Scholar]
- 27. Wu M. P., Doyle J. R., Barry B., Beauvais A., Rozkalne A., Piao X., Lawlor M. W., Kopin A. S., Walsh C. A., and Gussoni E. (2013) G-protein coupled receptor 56 promotes myoblast fusion through serum response factor- and nuclear factor of activated T-cell-mediated signalling but is not essential for muscle development in vivo. FEBS J. 280, 6097–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishida M., Onohara N., Sato Y., Suda R., Ogushi M., Tanabe S., Inoue R., Mori Y., and Kurose H. (2007) Gα12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J. Biol. Chem. 282, 23117–23128 [DOI] [PubMed] [Google Scholar]
- 29. Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., and Sternweis P. C. (1998) p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science 280, 2109–2111 [DOI] [PubMed] [Google Scholar]
- 30. Lehmann D. M., Seneviratne A. M., and Smrcka A. V. (2008) Small molecule disruption of G protein βγ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol. Pharmacol. 73, 410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reiter E., and Lefkowitz R. J. (2006) GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 32. Marchese A., and Trejo J. (2013) Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell. Signal. 25, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 34. Prömel S., Frickenhaus M., Hughes S., Mestek L., Staunton D., Woollard A., Vakonakis I., Schöneberg T., Schnabel R., Russ A. P., and Langenhan T. (2012) The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep. 2, 321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prömel S., Waller-Evans H., Dixon J., Zahn D., Colledge W. H., Doran J., Carlton M. B., Grosse J., Schöneberg T., Russ A. P., and Langenhan T. (2012) Characterization and functional study of a cluster of four highly conserved orphan adhesion-GPCR in mouse. Dev. Dyn. 241, 1591–1602 [DOI] [PubMed] [Google Scholar]
- 36. Krasnoperov V., Deyev I. E., Serova O. V., Xu C., Lu Y., Buryanovsky L., Gabibov A. G., Neubert T. A., and Petrenko A. G. (2009) Dissociation of the subunits of the calcium-independent receptor of α-latrotoxin as a result of two-step proteolysis. Biochemistry 48, 3230–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smrcka A. V. (2008) G protein βγ subunits: central mediators of G protein-coupled receptor signaling. Cell Mol. Life Sci. 65, 2191–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luttrell L. M., Maudsley S., and Bohn L. M. (2015) Fulfilling the promise of ‘biased’ GPCR agonism. Mol. Pharmacol. 88, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]