

Abstract
TnrA is a master regulator of nitrogen assimilation in Bacillus subtilis. This study focuses on the mechanism of how glutamine synthetase (GS) inhibits TnrA function in response to key metabolites ATP, AMP, glutamine, and glutamate. We suggest a model of two mutually exclusive GS conformations governing the interaction with TnrA. In the ATP-bound state (A-state), GS is catalytically active but unable to interact with TnrA. This conformation was stabilized by phosphorylated l-methionine sulfoximine (MSX), fixing the enzyme in the transition state. When occupied by glutamine (or its analogue MSX), GS resides in a conformation that has high affinity for TnrA (Q-state). The A- and Q-state are mutually exclusive, and in agreement, ATP and glutamine bind to GS in a competitive manner. At elevated concentrations of glutamine, ATP is no longer able to bind GS and to bring it into the A-state. AMP efficiently competes with ATP and prevents formation of the A-state, thereby favoring GS-TnrA interaction. Surface plasmon resonance analysis shows that TnrA bound to a positively regulated promoter fragment binds GS in the Q-state, whereas it rapidly dissociates from a negatively regulated promoter fragment. These data imply that GS controls TnrA activity at positively controlled promoters by shielding the transcription factor in the DNA-bound state. According to size exclusion and multiangle light scattering analysis, the dodecameric GS can bind three TnrA dimers. The highly interdependent ligand binding properties of GS reveal this enzyme as a sophisticated sensor of the nitrogen and energy state of the cell to control the activity of DNA-bound TnrA.
Keywords: AMP, ATP, glutamate, glutamine, transcription regulation, transcription factor TnrA, glutamine synthetase, Bacillus subtilis
Introduction
The Gram-positive soil bacterium Bacillus subtilis is able to utilize nitrate, nitrite, and urea in the absence of its preferred nitrogen sources like ammonium ions or glutamine (1–3). The metabolism of such compounds is tightly regulated and requires a large energy investment. Under conditions of nitrogen limitation, the global transcription regulator TnrA activates genes and operons of nitrate and nitrite reduction(nasABCDEF), urea (ureABC) and nucleotide assimilation, ammonia transport (nrgAB), and its own gene. Meanwhile, it represses operons required for ammonium assimilation like the glnRA and gltAB operons (1, 4–6). Furthermore, TnrA was suggested to be involved in the control of amino acid and purine utilization and might even be involved in oxidative stress response (7).
Under nitrogen-poor conditions, TnrA interacts with the PII-like protein GlnK, which itself is membrane-associated via the ammonium transporter AmtB (3, 8). After sudden exposure to conditions of nitrogen excess, TnrA is released from GlnK (8), and its transcriptional activity is repressed by interaction with GS.2 Previous biochemical studies demonstrated that GS can only interact with TnrA in the presence of GS feedback inhibitors glutamine or AMP, and this interaction would prevent DNA binding activity of TnrA (9). Furthermore, feedback-inhibited GS stabilizes DNA binding activity of GlnR, a repressor for glnRA and ureABC operons as well as the tnrA gene (10). Therefore, GS in B. subtilis is regarded as a trigger enzyme, which participates in primary metabolism and controls gene expression indirectly through TnrA and GlnR (9, 11, 12).
Both transcription factors (TnrA and GlnR) have a high sequence similarity at the N terminus and bind the same DNA consensus sequence (TGTNAN7TNACA), in which only 4 nucleotides in each operator half-site are required for their specific DNA binding (1, 5, 6, 13, 14). Three conserved residues of the second α-helix (Tyr-32, Arg-28, and Arg-31 of TnrA and Tyr-30, Arg-26, and Arg-29 of GlnR) recognize the consensus sequence (14). TnrA serves in most cases as an activator, whereas in a few cases, it acts like GlnR as a repressor (1, 5, 6). The C terminus of these proteins differs completely and is considered to be a signal transduction domain (10, 14–17). The last 15 C-terminal residues of TnrA interact with GS, whereas GlnK binding occurs in region 75–90 of the C terminus (9, 17). TnrA dimerization is mediated by residues 6–11 in its first N-terminal α-helix and by residues 52–67 of a hydrophobic winged helix-turn-helix motif (14). In contrast, GlnR requires the feedback-inhibited GS for dimerization and subsequent DNA binding (14, 16).
The GS of B. subtilis catalyzes the ATP-dependent amidation of glutamate to glutamine in the presence of ammonium. The biosynthesis of glutamine involves the initial phosphorylation of the γ-carboxyl group of glutamate by ATP, followed by ammonium incorporation and release of inorganic phosphate, yielding glutamine (18, 19). The enzyme forms a dodecamer, which consists of two face-to-face hexameric rings (20). The active sites are located at the interface between neighboring subunits. For each active site, the major part is made up by the C terminus of one subunit, together with a short segment from the N-terminal domain of the laterally adjacent subunit. During formation of the transition state, a loop region contributed by the N-terminal domain undergoes a major structural rearrangement (20). This catalytically induced structural change leads to significant alterations in the overall dodecamer structure of GS (20).
The activity of GS in B. subtilis is tightly regulated via feedback inhibition by glutamine and AMP (21). The recently published crystal structure also reveals the mechanism for feedback inhibition by glutamine (20). A central role in glutamine binding is played by an arginine residue from the N-terminal segment (Arg-62), which forms hydrogen bonds with glutamine. This hydrogen bonding network prevents glutamine release and locks the catalytic center in a closed state, thereby preventing substrate binding and inhibiting catalytic activity.
In addition, the biosynthetic activity of GS is tuned down by the interaction with the transcription factor TnrA, whereas GlnR does not affect the activity of GS (22). Although the GlnR-GS structure is unknown, a crystal structure between GS and a peptide corresponding to the last 36 amino acids of TnrA detected the putative TnrA binding sites in the intersubunit catalytic pores of GS mostly near the catalytic centers (14). However, the complex assembled as a tetradecamer, and the physiological relevance of this structure remains elusive.
So far, it has been assumed that only feedback-inhibited GS binds to TnrA, thereby abolishing the DNA binding activity of TnrA (9, 14). In the present study, we demonstrate that in the presence of glutamine, GS binds TnrA directly on the DNA, forming a ternary GS-TnrA-DNA complex. Through antagonistic interactions between the effector molecules, TnrA-GS complex formation is regulated by the intracellular levels of ATP, AMP, glutamine, and glutamate. Therefore, it seems that GS has a so far underestimated role as sophisticated sensor of the cellular nitrogen and energy state.
Experimental Procedures
Strains and Plasmids
B. subtilis strains and plasmids used in this study are presented in Table 1. To obtain the plasmid pDG-TnrA-ST, the tnrA gene was amplified from B. subtilis genomic DNA using primers tnrA for (AAA GTC GAC ATG ACC ACA GAA GAT CAT TCT TAT) and tnrA rev (AAA AAG CTT TCA TTA ACG GTT TTT GTA CCG AAA GTG). The PCR product was digested with SalI and HindIII and cloned into the expression vector pGP380 (23) cut with the same enzymes to obtain plasmid pGP380-TnrA. Further, the tnrA gene containing N-terminal StrepII-tag was amplified by PCR using plasmid pGP380-TnrA and primers tnrA ST for (T AAC AAG CTT AAT ACC TAG GAC TCG TTC AC) and tnrA rev. The PCR product was cloned into the HindIII site of the expression vector pDG148.
TABLE 1.
Strains and plasmids used in this study
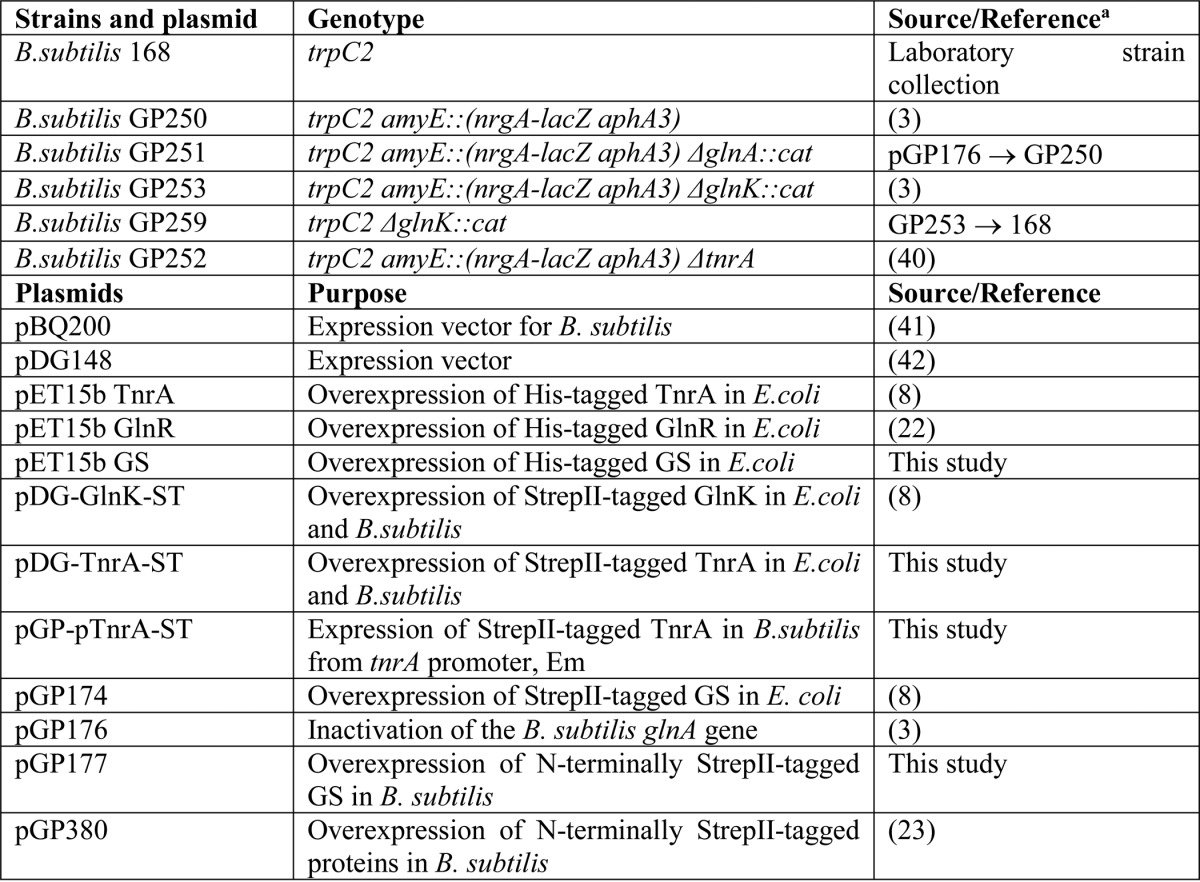
a Arrows indicate construction by transformation.
To obtain the StrepII-tagged tnrA gene under the control of its own promoter, the promoter region of tnrA was amplified using B. subtilis genomic DNA and primers ptnrA for (TC TTC GAA TTC GAT TAT CCT TCC TCC TCG) and ptnrA rev (TT TTC CCC GGG TGG ATG TCT TTT GAT AAT AG). The plasmid pGP380-TnrA was digested with EcoRI and SmaI to remove the constitutive degQ36 promoter and ligated with the PCR product containing the promoter region of tnrA.
The glnA gene was amplified from the genomic DNA B. subtilis using primers glnA for (CATCA TCATC ATCAT CACAG CAGCG GCCTG GTGCC GCGCG GCAGC CATAT GGCAA AGTAC ACTAG AGAAG) and glnA rev (GCTCA GCGGT GGCAG CAGCC AACTC AGCTT CCTTT CGGGC TTTGT TATTA ATACT GAGAC ATATA CTGTTC). The pET15b plasmid was digested with the restriction endonuclease BamHI. The PCR product and the digested plasmid were assembled using an isothermal, single-reaction method for assembling multiple overlapping DNA molecules, as described previously (24).
Plasmid pGP177 was generated for overexpression of the N-terminally StrepII-tagged GS in B. subtilis. The glnA gene was amplified from plasmid pGP174 using the primer pair CD23/CD24 (AAAGG ATCCG AATGA CAAAG GAGCT GAGGA TCATG GCTAG CTGGA GCCAC CCGCAG/AAAAT GCATT CATTA ATACT GAGAC ATATA CTGTTC). The PCR product was digested with BamHI and NsiI and ligated with plasmid pBQ200 that was cut with BamHI and PstI. The sequence integrity of all genes was confirmed by DNA sequencing.
B. subtilis cells were grown in Spizizen minimal medium (25) containing glucose (0.5% (w/v)) as a carbon source. As a nitrogen source, 20 mm sodium nitrate plus 1.5 or 15 mm glutamine was used. l-Tryptophan was added to a final concentration of 50 mg/liter. For recombinant strains, kanamycin, erythromycin, or chloramphenicol was added until a final concentration of 10 mg/liter.
In Vivo Cross-linking and SPINE Assays
The in vivo cross-linking of proteins was performed as described (23). B. subtilis recombinant cells producing the StrepII-tagged TnrA, GS, or GlnK proteins (B. subtilis ΔtnrA pGP-pTnrA-ST; B. subtilis ΔglnK pDG-GlnK-ST; B. subtilis GP251 pGP177) were grown in Spizizen minimal medium supplemented with either 1.5 or 15 mm glutamine as indicated. At the late exponential phase of growth (A600 ∼0.7), paraformaldehyde was added until a final concentration of 0.6% (w/v), and incubation was continued for a further 20 min. Subsequently, cells were harvested by centrifugation and broken by FastPrep-24 (M.P. Biomedical, Irvine, CA) using 0.1-mm glass beads, and the cell-free crude extracts were prepared as described previously (17).
StrepII-tagged proteins were purified on Strep-Tactin Sepharose as recommended (IBA Life Sciences). The elution fractions were further analyzed by immunoblotting.
Protein Purification
StrepII-tagged GS was overexpressed using pGP174 plasmid (8), StrepII-tagged TnrA was overexpressed using pDG-TnrA-ST plasmid in E. coli BL21 (DE3) (Stratagene), and all proteins were purified as described previously (8). His6-tagged TnrA and GS proteins were overexpressed using pET15b vector and purified as reported previously (22).
Surface Plasmon Resonance (SPR) Detection
SPR experiments were performed using a BIAcore X biosensor system (Biacore AB, Uppsala, Sweden). The biotinylated DNA duplexes were generated as described (26) and immobilized onto the sensor surface of the streptavidin sensor chip (Biacore, GE Healthcare), with a flow rate of 10 μl/min to receive a binding signal of ∼1500 resonance units (RU). Control DNA duplexes were loaded onto flow cell 1 (FC1), and specific DNA duplexes with a TnrA-binding site were loaded onto flow cell 2 (FC2). The running buffer used for DNA immobilization contained 10 mm HEPES, pH 7.4, 100 mm NaCl, 0.2 mm EDTA, and 0.005% Nonidet P-40. Subsequently, purified TnrA was injected onto the DNA-loaded streptavidin sensor chip at a concentration of 2.5 μm (dimer) to receive a binding signal of ∼2000 RU, which corresponds to a surface concentration change of 2 ng/mm2. Subsequently, purified GS at a concentration of 0.100 μm (dodecamer) was loaded onto the TnrA-DNA chip surface with a flow rate 15 μl/min in the running buffer containing 10 mm HEPES, pH 7.4, 300 mm NaCl, 3 mm MgCl2·6H2O, 0.2 mm EDTA, and 0.005% Nonidet P-40. For regeneration of the sensor chip surface, 2 m NaCl was used.
To test TnrA interaction with different DNA fragments, the indirect capture strategy was used (27). First, a biotinylated single-stranded DNA capture linker (biotin-gcaggaggacgtagggtagg) was irreversibly bound to a streptavidin chip. Then a partially double-stranded DNA oligomer that contained the sequence of interest in the double-stranded region (Table 2) with a single-stranded overhang complementary to the capture linker was fixed onto the chip as described (27). The protein of interest was injected as described above. At the end of the experiment, the captured oligonucleotide was stripped from the linker by 1.0 m NaCl, 50 mm NaOH to regenerate the chip.
TABLE 2.
Sequences of oligonucleotides used for SPR spectroscopy
| Oligonucleotide designationa | Length | Sequence (5′–3′) |
|---|---|---|
| bases | ||
| PnrgAB_30mer_F | 30 | AAAACCATGTCAGGAAATCTTACATGAAAA |
| PnrgAB_30mer_R | 50 | TTTTCATGTAAGATTTCCTGACATGGTTTTCCTACCCTACGTCCTCCTGC |
| PglnRA_54mer_F | 54 | GATTTGATGTTAAGAATCCTTACATCGTATTGACACATAATATAACATCACCTA |
| PglnRA_54mer_R | 74 | TAGGTGATGTTATATTATGTGTCAATACGATGTAAGGATTCTTAACATCAAATCCCTACCCTACGTCCTCCTGC |
| Nonspecific DNA_30mer_F | 30 | CAGTGAGGCACCTATCTCAGCGATCTGTCT |
| Nonspecific DNA_30mer_R | 50 | AGACAGATCGCTGAGATAGGTGCCTCACTGCCTACCCTACGTCCTCCTGC |
| Nonspecific DNA_54mer_F | 54 | CAGTGAGGCACCTATCTCAGCGATCTGTCTCAGTGAGGCATCTCAGCGATCTGT |
| Nonspecific DNA_54mer_R | 74 | ACAGATCGCTGAGATGCCTCACTGAGACAGATCGCTGAGATAGGTGCCTCACTGCCTACCCTACGTCCTCCTGC |
a F, forward strand; R, reverse strand.
To immobilize purified TnrA-His6 on the Ni2+-loaded NTA sensor chip, 30 μl of a 2 μm TnrA-His6 solution (of the dimer) was injected into FC2 until a binding signal of 2000 RU was reached. To measure the binding of GS with TnrA, a 100 nm solution of GS-ST (dodecamer) was injected onto the TnrA-His6 surface at 25 °C in HBS-Mg buffer contained 10 mm HEPES, pH 7.4, 200 mm NaCl, 3 mm MgCl2·6H2O, and 0.005% Nonidet P-40, in the presence or absence of ATP, glutamine, or MSX.
To test the TnrA/GS stoichiometry, 10 μl of a 5 μm GS-His6 solution (of the dodecamer) was injected into FC2 of the Ni2+-loaded NTA sensor chip until a binding signal of 5000 RU was reached. Further, a solution of TnrA-ST with a different concentration was injected onto the GS-His6 surface at 25 °C in HBS-Mg buffer containing 10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm MgCl2·6H2O, 1 mm glutamine, and 0.005% Nonidet P-40. Prior to loading fresh TnrA-His6 or GS-His6 protein onto the NTA sensor chip, bound proteins were first removed by injecting 10 μl of 1 m imidazole, pH 7.0. The stoichiometry between GS and TnrA was calculated using Equation 1.
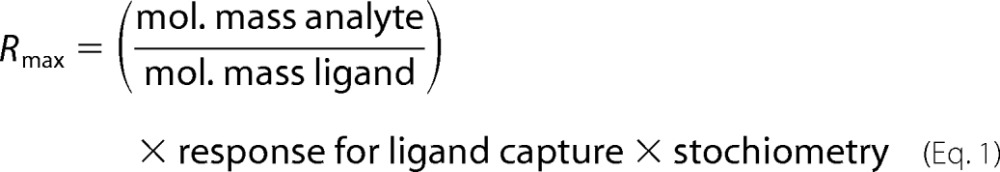 |
The resonance difference between FC2 and FC1 (ΔRU) was measured to quantify specific binding to FC2. SPR data were evaluated using BIAevaluation (Biacore AB) and GraphPad Prism (GraphPad Software, La Jolla, CA).
Isothermal Titration Calorimetry for Determination of Binding Constants
isothermal titration calorimetry (ITC) experiments were performed on a VP-ITC microcalorimeter (MicroCal, LCC) in 10 mm Hepes, pH 7.4, 5 mm MgCl2·6H2O, 50 mm KCl, 50 mm NaCl at 20 °C (17). For determination of ATP, glutamine, and AMP binding isotherms for GS, 20 or 10 μm protein solution (dodecamer) was titrated with glutamine, ATP, or AMP in concentrations ranging from 1.25 to 8 mm, as indicated. The ligand (6 μl) was injected 45 times into the 1.4285-ml cell with stirring at 155 rpm. The binding isotherms were calculated from received data and fitted to a six-site binding model with Origin (MicroCal, Northampton, MA).
In Vitro Cross-linking of Proteins
Proteins were dialyzed overnight in a 10 mm potassium phosphate buffer, pH 7.4, 100 mm NaCl, 3 mm MgCl2, and 20% glycerol. The reaction mixture of 1 dodecameric GS and 6 dimeric molecules of TnrA consisted of 1.5 μg/μl dialyzed GS and 0.45 μg/μl dialyzed TnrA, mixed in the presence of 1 mm Gln, 1 mm MSX, and 1 mm ATP as required. The protein mixture was treated with 0.1% freshly prepared solution of glutaraldehyde for 3 min at 37 °C. The reaction was stopped by the addition of 100 mm Tris, pH 7.4.
Size Exclusion Chromatography and Multiangle Light Scattering Analysis
Analytical size exclusion chromatography was carried out on an Äkta purifier system equipped with a Superdex 200 column PC 3.2/30 (GE Healthcare, geometric column volume of 2.4 ml) or Superose 6 Increase 3.2/300 (GE Healthcare, geometric column volume of 2.4 ml). The cross-linked sample was centrifuged for 5 min at 12,000 rpm, and 10 μl of the supernatant were injected for analysis with a flow rate 0.05 ml/min. The running buffer consisted of 10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm MgCl2, and 0.02% sodium azide. The apparent molecular weights of proteins were estimated after calibration of the column with standard proteins: thyroglobulin (670 kDa), ferritin (440 kDa), globulin (158 kDa), conalbumin (75 kDa), ovalbumin (44 kDa), carbonic anhydrase (29 kDa), RNase (13.7 kDa), and aprotinin (6.5 kDa) (Bio-Rad gel filtration standard, GE Healthcare LMW gel filtration calibration kit).
Multiangle light scattering experiments were carried out with a miniDawn Treos system (Wyatt Technology Corp.), and concentration determination was done with an Optilab rEX refractometer (Wyatt Technology). The system was connected to an HPLC system with an autosampler (Agilent 1260). Samples for analysis were centrifuged for 20 min at 12,000 rpm and subsequently filtered through a 20-nm syringe filter (GE Healthcare) before injection onto a Superose 6 10/300 column (GE Healthcare). 50 μl of the cross-linked sample were loaded with a flowrate of 0.5 ml/min. The column was equilibrated with PBS, pH 7.4, 150 mm NaCl with 0.02% sodium azide to prevent microbial growth. The resulting data were analyzed with ASTRA (Wyatt Technology). The experiments were carried out at room temperature, and the molecular weight was calculated from an average of two injections. The elution volume was plotted against the UV signal and the molecular weight derived from the light scattering data.
Results
Glutamine Synthetase Interacts in Vivo with TnrA under Both Nitrogen-rich and Nitrogen-limited Conditions
It was shown previously that a B. subtilis GS-mutant strain constitutively expresses TnrA-dependent genes under nitrogen excess conditions due to the lack of TnrA inhibition by feedback-inhibited GS (1). We raised the question of whether GS contributes to TnrA control also under nitrogen-limited conditions. To investigate this, we studied the in vivo activity of TnrA in B. subtilis reporter strains using a β-galactosidase reporter gene expressed from the TnrA-dependent nrgA promoter in a glnA− (GP251) and a glnA+ (GP250) genetic background (3). At a glutamine concentration of 1.5 mm as the sole nitrogen source, the activity of β-galactosidase was almost identical to that in nitrate-grown GP250 cells. This indicates that growth with 1.5 mm glutamine results in a similar activation of TnrA as growth with nitrate as the sole nitrogen source, considered to be nitrogen-limited (Fig. 1). With 1.5 mm glutamine, the GS-deficient mutant exhibited a 2-fold higher β-galactosidase reporter activity than wild type cells. This derepression in the GS-deficient background indicates that even under conditions of nitrogen limitation, GS partially depresses TnrA activity.
FIGURE 1.

Expression of a β-galactosidase reporter under control of the TnrA-dependent nrgA promoter reveals transcriptional activity of TnrA. B. subtilis cells were grown under nitrogen-limited conditions using either 1.5 mm glutamine (circles) or 20 mm nitrate (squares) for strain GP250 (wild type background) and using 1.5 mm glutamine (triangles) for strain GP251 (glnA− background). Error bars, S.E.
Next, the interaction of TnrA with GlnK and GS under different nitrogen conditions was evaluated using SPINE analysis (23). tnrA-, glnK-, and glnA-deficient mutants were transformed with plasmids encoding the respective StrepII-tagged proteins (TnrA-ST, GlnK-ST, and GS-ST). The recombinant proteins were then extracted with Strep-Tactin Sepharose from extracts of nitrogen-limited cells (1.5 mm glutamine) and cells shifted to nitrogen-rich conditions (15 mm glutamine). In agreement with previous studies (8, 17), GlnK was co-eluted together with TnrA-ST from extracts of nitrogen-limited cells (Fig. 2A). After shifting the cells to glutamine-rich conditions prior to paraformaldehyde cross-linking, no GlnK could be detected. However, GS was co-purified with TnrA-ST from both nitrogen-limited cells and cells supplemented with 15 mm glutamine. These observations were confirmed by the reverse SPINE experiments, using GlnK-ST- or GS-ST-producing strains. TnrA was co-purified with GlnK only from extracts of nitrogen-limited cells (Fig. 2B), whereas it was bound to GS-ST in both nitrogen excess and nitrogen-limited cells (Fig. 2B).
FIGURE 2.

SPINE analysis of TnrA interacting with GlnK and GS under limiting (1.5 mm glutamine) or sufficient (15 mm glutamine) nitrogen conditions. B. subtilis tnrA-, glnK-, and glnA-deficient mutants producing StrepII-tagged recombinant TnrA-ST, GlnK-ST, and GS-ST, respectively, were grown in Spizizen minimal medium supplemented with 1.5 mm glutamine and shifted to 15 mm glutamine as indicated. After treating the cells with 0.6% paraformaldehyde, TnrA-ST (A) and GlnK-ST and GS-ST (B) were purified from crude extracts on Strep-Tactin Sepharose; the elution fractions were analyzed by immunoblotting using anti-TnrA, anti-GlnK, and anti-GS antibodies.
Antagonistic Effects of Effector Molecules on GS-TnrA Complex Formation
The above data suggested that GS interacts with TnrA under both nitrogen-limited and nitrogen-rich conditions (Figs. 1 and 2), corroborating previous in vitro data showing that the feedback inhibitors glutamine and AMP enhance TnrA-GS interaction but are not strictly required (17). Therefore, we wanted to gain deeper insight into how GS-TnrA interaction is affected by metabolites, and we investigated GS-TnrA complex formation by SPR spectroscopy. His6-tagged TnrA was fixed onto the surface of a Ni-NTA sensor chip, and the StrepII-tagged GS was used as an analyte in the presence of various effectors (Fig. 3). In the absence of any effector molecules, a clear binding of GS to TnrA was observed. The addition of glutamine increased the amount of GS binding to TnrA by about 50%, whereas ATP almost completely abolished TnrA-GS interaction. Interestingly, in the presence of glutamine, ATP could not abrogate TnrA-GS interaction (Fig. 3A). The calculated association rate (kon) in the presence of glutamine (kon = 1.75 × 105 m−1 s−1) is very similar to the rate in the absence of any effector molecules (kon = 1.43 × 105 m−1 s−1). This reveals that the affinity of GS to TnrA is not increased by glutamine; rather, the concentration of productively binding GS molecules is increased.
FIGURE 3.

SPR analysis of GS-TnrA interplay. GS was loaded onto the TnrA chip surface either without effector molecules (continuous line) or in the presence of 1 mm ATP (dotted line), 1 mm glutamine (A) or 1 mm MSX (B) (dashed line), 1 mm ATP, and 1 mm glutamine (A)/1 mm MSX (B) (thin dashed line).
To elucidate the effect of glutamine on GS-TnrA-complex formation in more detail, we employed the GS inhibitor MSX. MSX binds to the catalytic center of GS as a glutamate analogue (28), where it is phosphorylated by ATP. The product, MSX-phosphate, irreversibly fixes the enzyme in the transition state (29). MSX in the absence of ATP stimulated GS-TnrA interaction to the same extent as glutamine (Fig. 3B), confirming that the glutamine- and MSX-bound forms of B. subtilis GS have similar conformations (30). However, when MSX was combined with ATP to generate the transition state analogue MSX-phosphate, GS binding to TnrA was completely abolished (Fig. 3B).
These results suggest two relevant conformations of GS for TnrA interaction: 1) a TnrA-binding “Q-state,” promoted by glutamine or by MSX, and 2), a non-binding “A-state,” which reflects the catalytic active form of GS in the absence of glutamine but loaded with ATP. GS seems to be arrested in the A-state by the transition state analogue MSX-phosphate.
Cooperative Versus Antagonistic Binding of Effector Molecules to GS
To characterize the binding of ATP and glutamine to GS and study their interdependence, we performed ITC (Fig. 4). In the presence of 5 mm Mg2+ ions, a strong binding of either glutamine or ATP was observed (Fig. 4, A and B). Six binding sites were predicted for both glutamine and ATP (5.6 ± 0.16 and 5.5 ± 0.1, respectively) from the titration data fitting using the “one set of sites” model, which nevertheless did not provide an exact fit to interaction signals, apparently because of the cooperativity between binding sites. Further fitting using a binding model of six sequential binding sites gave the best fit for both glutamine and ATP titrations. It resolved binding sites for glutamine with KD values of 6.9 ± 0.53, 2.7 ± 0.20, 27.7 ± 1.69, 4.4 ± 0.24, 175.7 ± 30.25, and 24.9 ± 1.3 μm for sites 1–6, respectively. The ITC signals for ATP binding could also only be fitted with a six-sequential binding site model. The corresponding KD values for sites 1–6 were calculated to be 15.2 ± 2.27, 41.3 ± 6.6, 18.2 ± 3.69, 184.4 ± 26.32, 55.2 ± 7.15, and 515.5 ± 83.33 μm. These values are consistently higher than those for glutamine, indicating that glutamine binds more tightly than ATP. As illustrated in Fig. 5, the sites are apparently sequentially occupied in an alternating order of cooperative and anti-cooperative interactions. In the case of glutamine binding, the order is cooperative followed by anti-cooperative, whereas the inverse order is observed in the case of ATP binding.
FIGURE 4.

Analysis of ligand binding properties of GS by ITC. Glutamine, ATP, or AMP in concentrations as indicated were injected (45 × 6 μl) into 20 μm (A and B) or 10 μm (C–F) GS with stirring at 155 rpm. A, titration with 2 mm glutamine. Six (5.6 ± 0.16) binding sites were predicted from the titration data. B, titration with 2 mm ATP. Six (5.5 ± 0.13) binding sites were predicted. C, titration of GS in the presence of 2 mm ATP with 2 mm glutamine. The glutamine concentration at the maximum enthalpy (highest point of the curve) was 173 ± 34.7 μm. D, titration of GS in the presence of 0.5 mm glutamine with 5 mm ATP. The ATP concentration at the maximum enthalpy (lowest point of the curve) was 421 ± 31.2 μm. E, titration of GS in the presence of 2 mm ATP with 2 mm AMP. The AMP concentration at the maximum enthalpy (highest point of the curve) was 186 ± 14.2 μm. F, titration of GS in the presence of 0.5 mm AMP with 5 mm ATP. The ATP concentration at the maximum enthalpy (lowest point of the curve) was 23 ± 4.2 μm.
FIGURE 5.

The pattern of alternating positive (synergistic) and negative (antagonistic) cooperative interactions between the sequential glutamine (dashed line) and ATP (continuous line) binding sites of GS. KD values were obtained by fitting ITC data using a binding model of six sequential binding sites.
Because glutamine prevented the negative effect of ATP on TnrA-GS interaction (Fig. 3), an ITC experiment with 2 mm glutamine was performed in the presence of 2 mm ATP (Fig. 4C), which is close to its physiological concentration (31). In the presence of ATP, binding enthalpy for glutamine was strongly decreased when compared with the titration in the absence of ATP (Fig. 4, compare C with A), and titration curves showed a complex behavior resulting from competitive binding. In a series of GS titrations with various glutamine concentrations in the presence of 2 mm ATP, a biphasic curve was observed in each case; in the first injections, the enthalpy increased, until a maximum was reached at a glutamine concentration of 173 ± 34.7 μm, which is close to the half-occupation of the fifth site (and subsequent filling of the sixth site) (Fig. 4A). Subsequently, the heat change signals decreased again, indicating gradual saturation of GS by glutamine (Fig. 4C).
The reverse experiments, where GS was titrated with ATP in the presence of glutamine, were also performed (Fig. 4D). Here again, a biphasic curve was observed. No signal was detected in the first injections. Apparently, glutamine blocked binding of low concentrations of ATP. After several injections, ATP binding resumed and reached a maximal negative enthalpy at an ATP concentration of 421 ± 31.2 μm, followed by saturation of ATP binding. In this case, the minimum point was close to the affinity of the sixth binding site for ATP: 515.5 ± 83.33 μm (see Fig. 4B). Together, these results show competition between ATP and glutamine for binding to GS.
Furthermore, the effect of AMP against ATP on GS binding was investigated. First, AMP was titrated to GS in the presence of 2 mm ATP. Again, a complex biphasic curve was observed, with a maximum point at an AMP concentration of 186 ± 14.2 μm (Fig. 4E). In a reverse experiment, when ATP was titrated to GS in the presence of 0.5 mm AMP, binding of ATP could only be observed if added in high concentrations (5 mm). Binding enthalpy was 30-fold lower than in the absence of AMP, and only a minute amount of ATP could bind to GS (Fig. 4F), reflecting only a residual interaction, possibly because of strong binding of AMP to GS (20). Therefore, it seems that elevated AMP levels prevent formation of the ATP-bound state of GS (A-state).
GS Interacts with DNA-bound TnrA
Because TnrA binds to DNA to promote the transcription of TnrA-regulated genes, we investigated how GS affects TnrA-DNA interaction by SPR spectroscopy. The TnrA-binding region of the nrgAB promoter, which is positively regulated by TnrA, and that of the glnRA promoter, which is negatively regulated by TnrA (1, 6), were used as target DNA sequences. An optimization procedure included DNA duplexes with different lengths, ranging from 17 to 163 bp, containing only the TnrA recognition sequence or additional nonspecific flanking sequences (data not shown). The best specificity and affinity were observed with a DNA duplex, which was 30 bp in length, for the nrgAB promoter (AAAACCATGTCAGGAAATCTTACATGAAAA) and a 54-bp long fragment (GATTTGATGTTAAGAATCCTTACATCGTATTGACACATAATATAACATCACCTA) for the glnRA promoter, which contains two TnrA-binding sites.
The length and environment of the TnrA-binding region of the nrgAB and glnRA promoters turned out to be very important for TnrA binding affinity. TnrA associated strongly with the nrgAB promoter fragments, whereas the affinity of TnrA to the glnRA promoter fragments was much lower, and association was followed by a rapid dissociation, independent of the glnRA fragment length (data not shown). Next, the effect of GS on TnrA-DNA binding in the presence of 1 mm glutamine was examined (Fig. 6, A and B). A mixture of TnrA and GS at a ratio of 3 (TnrA dimers) to 1 (GS dodecamer) was injected onto either nrgAB or glnRA promoter regions (Fig. 6A, continuous or dashed lines). Surprisingly, when GS was present in the analyte mixture, TnrA still stably associated with the nrgAB fragment (Fig. 6A). This result contradicts previous data indicating that the TnrA-GS complex cannot bind to DNA (9). Clearly, GS could not prevent the binding of TnrA to the nrgAB fragment. However, when a mixture of GS and TnrA was injected onto the glnRA promoter fragment, the resonance signal was 2 times lower as compared with TnrA in the absence of GS (Fig. 6A), indicating that GS partially prevented the binding of TnrA to the glnRA promoter fragment.
FIGURE 6.

SPR analysis of TnrA binding with 30-mer nrgAB or 54-mer glnRA promoter fragment. A, TnrA was injected onto the immobilized DNA surface (thin continuous line for the nrgAB promoter, thin dashed line for the glnRA promoter) alone or in a mixture with GS in a 3:1 ratio (continuous line for the nrgAB promoter, dashed line for the glnRA promoter). B, TnrA was injected onto an immobilized nrgAB promoter fragment (continuous line) or glnRA promoter fragment (dashed line), and then GS was loaded onto the TnrA-DNA chip surface. C, TnrA was injected onto an immobilized nrgAB promoter fragment; subsequently, GS was injected onto the immobilized TnrA-DNA surface without effector molecules (continuous line), in the presence of 1 mm glutamine (dashed line) or 1 mm ATP (dotted line) and in the presence of 1 mm ATP and 1 mm glutamine (thin dashed line). D, TnrA was injected onto immobilized nrgAB promoter fragment; subsequently, GS was injected without effector molecules (continuous line), in the presence of 1 mm MSX (dashed line) or 1 mm ATP (dotted line) and in the presence of 1 mm ATP and 1 mm glutamine.
The following SPR assays were performed with two consecutive injections of proteins (Fig. 6B). First, purified TnrA was injected onto the DNA-loaded sensor chip, resulting in stable DNA-TnrA complexes. Subsequently, a second injection was performed with purified GS (Fig. 6B). Surprisingly, when TnrA was already bound to the nrgAB promoter, GS could easily form a complex with DNA-bound TnrA in the presence of 1 mm glutamine (Fig. 6B, continuous line). This complex was stable as long as 1 mm glutamine was present in the buffer and quickly dissociated when glutamine was absent (data not shown). However, when TnrA was bound to the glnRA promoter fragment, GS could not stably bind, but instead, dissociation of TnrA from the DNA fragment was observed (Fig. 6B, dashed line).
Next, the effect of metabolites on the formation of an nrgAB DNA-TnrA-GS complex was tested (Fig. 6, C and D). In the absence of any effectors, a low level of GS binding to DNA-bound TnrA was observed. The presence of 1 mm glutamine resulted in a 5-fold increase of binding (Fig. 6C, continuous and dashed lines). ATP completely abrogated the binding of GS to the TnrA-DNA complex; however, glutamine prevented the negative effect of ATP on DNA-TnrA-GS complex formation, as observed previously in the TnrA-GS interaction assay (compare Figs. 3 and 6C). MSX stimulated GS binding to TnrA in the same way as glutamine (Fig. 6D, dashed line curve). However, in the presence of MSX and ATP, GS could not bind to DNA-associated TnrA.
The Influence of Different Nucleotides on GS Interaction with DNA-bound TnrA
The same type of interaction assay was now performed to study the effect of adenyl nucleotides on the binding of GS to DNA-bound TnrA (Fig. 7). Compared with the absence of effector molecules, AMP increased the quantity of GS binding to the DNA-TnrA complex 2-fold, which is a much weaker stimulation of binding than that caused by glutamine or MSX (Fig. 7A). In the presence of AMP and MSX, the resonance signal was the same as in the presence of MSX alone (Fig. 7A). ADP prevented the binding of GS to TnrA to the same extent as ATP (Fig. 7A). However, in combination with MSX, ADP could not prevent the interaction between GS and DNA-bound TnrA, whereas ATP with MSX again led to a complete loss of binding activity. This confirms that the ATP-dependent phosphorylation of MSX is responsible for the abrogation of GS binding to TnrA (Fig. 7A). To further corroborate this hypothesis, we tested the effect of non-hydrolyzable analogues of ATP. AMP-PNP was as efficient as ATP in preventing GS binding to TnrA, indicating that hydrolysis of ATP is not necessary to shift GS into the A-state conformation. However, AMP-PNP could not counteract the binding of MSX-activated GS to TnrA, which agrees with its inability to phosphorylate GS-bound MSX (Fig. 7B), The response to ATPγS, a poorly hydrolyzable analogue of ATP, was also investigated, and an intermediary effect between ATP and AMP-PNP was expected. Indeed, in the presence of ATPγS and MSX, slow binding of GS to the TnrA-DNA surface was observed (Fig. 7B). Apparently, ATPγS slowly phosphorylates MSX, with the remaining non-phosphorylated MSX promoting complex formation between GS and TnrA. These experiments clearly establish that phosphorylation of MSX is required to shift GS into the A-state that does not bind TnrA.
FIGURE 7.

SPR analysis of GS binding to TnrA immobilized on the 30-mer nrgAB promoter fragment in the presence of different effector molecules. A, GS was loaded onto the TnrA-DNA chip surface without effector molecules or in the presence of 1 mm MSX, 1 mm ATP, 1 mm ATP plus 1 mm MSX, 1 mm ADP, 1 mm ADP plus 1 mm MSX, 1 mm AMP, and 1 mm AMP plus 1 mm MSX, as indicated. B, GS was loaded onto the TnrA-DNA chip surface without effector molecules or in the presence of 1 mm MSX, 1 mm AMP-PNP, 1 mm AMP-PNP plus 1 mm MSX, 1 mm ATPγS (dotted line), or 1 mm ATPγS plus 1 mm MSX, as indicated.
The Affinity of GS toward TnrA Depends on the Ratio between Feedback Inhibitors and Substrate Molecules
We next addressed the question of how different ratios of GS substrates and effector molecules affect GS interaction with TnrA. First, the effect of various glutamine concentrations and AMP on the ability of GS to bind DNA-bound TnrA was investigated. A titration was performed with increasing concentrations of glutamine in the presence of ATP and glutamate, and the steady-state binding of GS was plotted against the glutamine concentration (Fig. 8). In the absence of any metabolites, 22 μm glutamine resulted in half-maximal binding (EC50) of GS to the TnrA-DNA surface (Fig. 8A). In the presence of 1 mm ATP, the EC50 for glutamine increased to 90 μm (Fig. 8C). To mimic the in vivo situation, we tested the effect of glutamine on GS-TnrA interaction in the presence of both 1 mm ATP and 10 mm glutamate (Fig. 8E) (31, 32). Although glutamate alone was not inhibitory for GS-TnrA interaction (not shown), in the presence of ATP and glutamate, the EC50 for glutamine increased to 120 μm.
FIGURE 8.

Influence of glutamine or AMP on GS interaction with TnrA immobilized on the 30-mer nrgAB promoter fragment. GS in the presence or absence of various effector molecules was injected onto surface-immobilized TnrA-DNA complex. The increase in RU relative to a control injection with GS in the absence of the variable effector molecule was recorded (normalized ΔRU). Glutamine was used as a variable effector molecule in A, C, and E, and AMP was used as a variable effector molecule in B, D, and F. No additional effector molecules were present in A and B. ATP at a constant concentration of 1 mm was present in C and D, and a constant concentration of 1 mm ATP and 10 mm glutamate was present in E and F. The obtained values were fitted to the equation for one-site-specific binding with Hill slope using GraphPad Prism.
The influence of AMP on the interaction of GS with DNA-bound TnrA was also tested (Fig. 8, B, D, and F). The steady-state level of GS-TnrA interaction promoted by AMP was about 3–4 times lower than glutamine-promoted GS interaction with TnrA. Moreover, the complex was less stable than in the presence of glutamine, and the binding signal decreased following the injection phase. In the absence of other metabolites, the EC50 for AMP was 21 μm. Importantly, AMP could relieve the negative effect of ATP with an EC50 of 45 μm AMP (Fig. 8D), indicating that already, micromolar concentrations of AMP could prevent the formation of the A-state of GS. Surprisingly, in presence of 10 mm glutamate and 1 mm ATP (Fig. 8F), the protective effect of AMP on GS-TnrA interaction was more pronounced than with ATP alone (Fig. 8D), with the EC50 for AMP decreasing from 45 to 30 μm. This result is in perfect agreement with the ITC experiments above (Fig. 4F), showing that AMP quenches the binding of ATP to GS and therefore prevents formation of the A-state.
The Stoichiometry of TnrA-GS Complex Formation
The crystal structure of the TnrA C-terminal peptide in complex with GS was described as a tetradecameric structure of GS with two non-interacting 36-amino acid peptides in each intersubunit cavity of GS, where the catalytic site is located (14). To study the stoichiometry between full-length TnrA and GS, size exclusion chromatography on analytical columns was performed. Using an analytical Superose Increase 6 column, no GS-TnrA complex could be eluted, even in the presence of 10 mm glutamine, after overnight incubation of the proteins (data not shown). This indicates that the GS-TnrA complex is not stable enough to withstand gel filtration and dissociates during chromatography. For this reason, proteins were cross-linked with glutaraldehyde to stabilize the complexes. With this method, pure GS in the absence of interaction partners eluted in three peaks with apparent masses of 588 kDa (corresponding to dodecameric GS (612 kDa)), 125 kDa (corresponding to dimeric GS (102 kDa)), and 62 kDa (corresponding to monomeric GS (51 kDa)) (Fig. 9A). Pure TnrA eluted as one peak with an apparent size of 42 kDa (corresponding to dimeric TnrA (30 kDa)). To assay the GS-TnrA complex, a mixture at a ratio of 6 TnrA dimers to 1 GS dodecamer was set up and supplemented with 1 mm MSX to maximize complex formation. After cross-linking, a peak corresponding to an apparent mass of 649 kDa was obtained. This elution shift corresponds to a size difference of 61 kDa, which equals two dimeric molecules of TnrA. A comparable size shift of ∼60 kDa was also observed when the experiment was performed in the presence of glutamine (Fig. 9B). As a control, we tested the oligomeric state of the TnrA-GS complex in the presence of MSX-phosphate to inhibit TnrA-GS interaction. As expected, no shift of the dodecameric GS peak as compared with GS without effectors was observed (Fig. 9A). These data suggest that at least two dimers of TnrA associate with the dodecameric GS complex. It should be noted that after cross-linking, the elution peak tails toward lower masses, indicating that a heterogeneous population of GS with TnrA and dodecameric pure GS is eluting. Therefore, the size shift underestimates the size of fully TnrA-occupied GS.
FIGURE 9.

Size exclusion chromatography of cross-linked GS, TnrA, and GS-TnrA complexes. A, the elution profiles of GS (Ve = 1.14, 1.45, and 1.59 ml), TnrA (Ve = 1.66 ml), a mixture containing GS and TnrA (1 dodecamer:6 dimers) in the presence of 1 mm MSX (Ve = 1.12, 1.45, and 1.59 ml) or in the presence of 1 mm MSX and 1 mm ATP (Ve = 1.14, 1.45, and 1.59 ml) are shown. B, the elution profiles of GS (Ve = 1.11, 1.43, and 1.58 ml), TnrA (Ve = 1.64 ml), and a mixture of GS and TnrA (1 dodecamer:6 dimers) in the presence of 1 mm glutamine (Ve = 1.12, 1.45, and 1.59 ml). The elution peak at 0.9 ml most likely corresponds to protein aggregates. C, SEC-MALS profiles for GS and the GS-TnrA complex. Both preparations were separated by size exclusion chromatography, and the measured molar masses are shown as dotted (GS-TnrA complex) or dotted and dashed lines (GS) below the peak fractions.
To obtain more precise data about the size of the cross-linked complexes, we additionally carried out size exclusion chromatography coupled to multiangle light scattering (SEC-MALS). The calculated molar mass of the peak that corresponds to dodecameric GS is 684 ± 12 kDa (Fig. 9C, dashed line). The calculated molecular mass of GS-TnrA complex was 787 ± 3.5 kDa (Fig. 9C, straight line). This corresponds to a molecular mass difference of 100 kDa, suggesting that three TnrA dimers bind with dodecameric GS. The increase of molecular weight in relation to the theoretical size of the complex is most likely a consequence of glutaraldehyde reacting with surface-exposed lysine residues and therefore decorating the complex.
To specify this result, TnrA-GS interaction assays were performed in a quantitative manner by SPR spectroscopy (Table 3). His6-tagged GS (629,400 Da) was fixed onto the surface of a Ni-NTA sensor chip, and StrepII-tagged TnrA (29,800 Da) in various concentrations was used as an analyte in the presence of 1 mm glutamine. Saturation of GS by TnrA was only reached at a concentration exceeding 2 μm TnrA (Table 3). From the ratio between the RU of surface-bound GS and the maximal RU after TnrA injection, the stoichiometry was calculated. The results indicate that approximately three TnrA dimers bind to one dodecameric GS molecule (Table 3), which is in good agreement with the gel filtration and light scattering analysis of cross-linked proteins.
TABLE 3.
Analysis of GS/TnrA stoichiometry by SPR spectroscopy
His-tagged GS was immobilized on a sensor chip, and StrepII-tagged TnrA was used as analyte at various concentrations. The stoichiometry was calculated as described under “Experimental Procedures.”
| GS captured | TnrA | Measured ΔRU | Stoichiometry |
|---|---|---|---|
| RU | μm | ||
| 5172 | 1 | 562 | 2.3 (no saturation) |
| 5873 | 2 | 916 | 3.3 |
| 5849 | 4 | 935 | 3.4 |
| 6161 | 8 | 950 | 3.3 |
Discussion
Being a trigger enzyme, GS in B. subtilis regulates the activity of transcription factor TnrA (1, 9) as well as being a central enzyme of nitrogen assimilation. Although considerable advances have been made, many details of the dual role of GS are not yet understood. This study reveals a stunning complexity of the interaction of GS with the effector molecules ATP, AMP glutamine, and glutamate and with TnrA. In agreement with the available structural information (20), GS seems to adopt two mutually exclusive conformations. In the presence of ATP, the enzyme is catalytically competent (18, 19) but unable to interact with TnrA. In the presence of glutamine, the activity of the enzyme is inhibited (21), but affinity toward TnrA is maximal. In the absence of any effector molecules, GS can interact with TnrA, although quantitatively less binding was observed as compared with GS in the glutamine state. According to the kinetic constants, this difference is due to a lower concentration of productively binding GS molecules. These data agree with a model according to which GS in the absence of effectors resides in an equilibrium between the A- and Q-state. The addition of ATP or glutamine shifts this equilibrium either to the A- or Q-state in a concentration-dependent manner. When both effectors are present simultaneously, the equilibrium position depends on the relative concentrations of these metabolites (discussed in more detail below).
In the Q-state, dodecameric GS can bind up to three TnrA dimers according to our SEC and MALS analysis. This result contradicts a recently published work (14) that suggested a major rearrangement of the oligomeric structure of GS upon binding of TnrA into a tetradecameric complex. SEC and MALS analysis in solution clearly preclude a tetradecameric structure. SPR analysis of GS-TnrA interaction provides an additional strong argument in favor of the formation of GS-TnrA complexes in the dodecameric state; dodecameric His-tagged GS was immobilized on the Ni-NTA surface of the sensor chip, thereby fixing the structure in an irreversible manner, precluding the interconversion of a GS dodecamer into a tetradecamer. Nevertheless, TnrA bound perfectly to the dodecameric immobilized GS. The tetradecameric structure of GS co-crystallized with the C-terminal peptide of TnrA might be caused by the particular crystallographic conditions. Nonetheless, this structure shows that the interface between opposing subunits of the double hexameric ring constitutes the binding site for the TnrA C terminus. This corresponds to six possible binding sites surrounding the dodecamer. However, we detected only up to three TnrA dimers binding to dodecameric GS, implying that every second site should be occupied by TnrA according to symmetry considerations. The model of three TnrA dimers binding to dodecameric GS corresponds to a previously published nondenaturing PAGE analysis of GS-TnrA complexes, where three additional protein bands of higher mass were clearly resolved after adding TnrA to GS (9).
The crystal structure of B. subtilis GS revealed that the active sites are located at the interface between two laterally adjacent subunits. Because the enzyme is made up of two face-to-face hexameric rings, each half-enzyme contains six active sites (20, 33, 34). Interestingly, ITC data of GS titration with glutamine or ATP predicted six sites in each case instead of the expected 12 sites. In accordance with this, a model of six binding sites gave the best fit of the binding data (Fig. 4). The ITC data evaluation revealed a remarkable pattern of alternating positive (synergistic) and negative (antagonistic) cooperative interactions between the metabolite binding sites of GS (Fig. 5). This indicates a high degree of cooperativity between the subunits. This pronounced intramolecular communication between binding sites could be intimately linked to the sensory properties of this trigger enzyme.
The concept of the two mutually exclusive A- and Q-states of GS, as derived from binding of TnrA, is fully supported by the ITC metabolite binding experiments. It also agrees with the crystal structure of GS (20), which showed that the same binding site cannot be occupied by both ATP and glutamine simultaneously. Both effectors were able to displace one another from the active site, with glutamine binding being more efficient than ATP.
Competition between ATP and glutamine for GS is likely to be relevant for the in vivo situation by adjusting the equilibrium between the A- and Q-state of GS. In the competitive titration experiments with glutamine in the presence of ATP, biphasic curves were obtained with a maximum point at an ATP/Gln ratio of about 10:1 (Fig. 4C). At this point, maximal binding of glutamine to GS at a single injection occurred. In the following injections, gradual saturation of the signal occurred. This glutamine/ATP ratio seems to be well tuned to the physiologically relevant concentrations of these metabolites. The intracellular concentration of glutamine may vary, depending on the nitrogen source, from 0.3 to 3 mm, whereas the cellular ATP level is considered to be maintained at concentrations in the low millimolar range (31). Remarkably, a recent study demonstrated unexpected heterogeneity of ATP concentrations in an E. coli cell population, ranging from 0.5 to 6.0 mm (35). It seems likely that at elevated concentrations of glutamine (corresponding to nitrogen excess conditions), GS is strongly impaired in ATP binding and is predominantly present in the Q-state. Under these conditions, interaction of GS with TnrA is maximal. In accord, ATP was not able to quench the interaction between GS and TnrA in the presence of 1 mm glutamine (e.g. see Figs. 3A and 6C). Conversely, at low cellular glutamine levels (nitrogen-limited conditions), the A- and Q-states of GS are balanced. This explains why in cells grown under nitrogen-limited conditions, there is still some residual GS-TnrA interaction (Fig. 2A). This residual interaction also explains the 2 times higher TnrA activity in a GS mutant strain in nitrogen-limited cells (Fig. 1). Furthermore, the in vitro data indicate that the balance between the A- and Q-state is slightly affected by the glutamate concentration (Fig. 8E), due to competition with glutamine for the same binding site. Considering all of these parameters, it appears that a small shift at the lower end of the intracellular glutamine concentrations (between 0.2 and 0.5 mm) could drastically change the ratio between the A- and Q-state of GS and thus regulate the activity of the enzyme and its interaction with transcription factor TnrA in response to the nitrogen availability.
In addition to the ratio of glutamine, glutamate, and ATP, the interaction between TnrA and GS is highly sensitive to AMP (Fig. 8, D and F). AMP itself did not stimulate the GS-TnrA complex formation on the DNA as strongly as glutamine (Fig. 7 A); however, it successfully prevented the negative effect of 1 mm ATP (Fig. 8D). AMP stimulated binding of GS to the TnrA-DNA complex more strongly when both glutamate and ATP were present (Fig. 8F). Together, the balance between the A- and Q-state depends on the ratio between ATP, AMP, glutamine, and glutamate. The amount of glutamate, which is an abundant compound in the cell, is maintained at a constantly high level (12, 31). The ATP concentration varies in the millimolar range (31), and AMP and glutamine levels are prone to strong nutrition-dependent fluctuations. Therefore, the balance between the A- and Q-state of GS signals the relative concentration of these decisive effector molecules in a very sensitive manner.
The present SPR data on GS-TnrA interaction in the presence of DNA requires reconsideration of the mechanism of how GS regulates the activity of the TnrA transcription factor. Previously, complex formation with GS was considered as a mechanism to prevent TnrA binding to DNA (9). This conclusion was based on EMSAs, which only resolve highly stable complexes that are maintained during electrophoresis (36). Here we used SPR, which monitors the formation and dissociation of the complex in real time (36). The SPR study clearly revealed that GS-complexed TnrA can still bind to the nrgAB promoter, even in the presence of excess glutamine. In contrast, GS diminished TnrA-DNA interaction on the glnRA promoter, probably due to a lower affinity of TnrA to this fragment. When TnrA was preloaded onto the nrgAB promoter fragment, GS in the Q-state could easily bind to the TnrA-DNA complex. Because GS is a huge dodecameric molecule, which is 600 kDa in size, the binding of this large oligomeric machine to TnrA on the DNA should impair transcription. The formation of the TnrA-GS complex on the DNA probably results in the inactivation of the transcriptional activity of TnrA, because the large GS complex will sterically shield it from RNA polymerase. Our refined model of TnrA control by GS thus assumes that under nitrogen excess conditions, where GS is mainly in the Q-state, it forms a complex with TnrA on positively regulated promoters and sterically blocks transcriptional activation. However, the negatively regulated promoter glnRA is liberated from TnrA and can now efficiently bind the GlnR-GS complex.
Taken together, our results suggest that B. subtilis GS can integrate the energy status (AMP/ATP ratio) and the nitrogen availability (using glutamine as a nitrogen status reporter) of the cell, thereby modifying the transcriptional outcome through interaction with the central regulator of nitrogen metabolism, TnrA. These sensory properties of GS have striking analogies to the signaling properties of the trimeric PII signal transduction proteins (37–39). In contrast, the B. subtilis PII protein GlnK appears to have acquired more specialized functions. Unlike other bacterial PII proteins, B. subtilis GlnK only interacts with ATP and lacks a clear 2-oxoglutarate response (8, 17). The physiological role of the GlnK-TnrA complex formation might be as a mechanism to protect TnrA from proteolytic degradation (17) and/or preserve GS biosynthetic activity from inhibitory interactions with TnrA (22). However, it plays only a minor role in the control of TnrA activity. Conversely, in B. subtilis, GS might have taken over the role of PII proteins as central integrator of the nitrogen and energy status of the cells. Determination of whether this is a specialized adaptation of B. subtilis or a general trait of Firmicutes bacteria requires further investigation.
Author Contributions
K. H. performed the SPR assays and size exclusion analysis. A. K. performed ITC experiments, in vivo experiments, and SPINE analysis. F. G. performed multiangle light scattering analysis. K. F. conceived and coordinated the study and interpreted the results. All authors analyzed the results, wrote the manuscript, and approved the final version of the manuscript.
Acknowledgments
Prof. Jörg Stülke and Dr. Fabian M. Commichau (Göttingen University) are gratefully acknowledged for providing B. subtilis strains and plasmids.
This work was supported by Deutsche Forschungsgemeinschaft Grant Fo195/9-2; German Academic Exchange Service (DAAD) Russian-German Program “Michail Lomonosov” Grants A/12/75478, A/12/25777, and 91583699; DAAD research fellowship for Ph.D. students and young scientists 50015537; and Russian Foundation for Basic Research (RFBR) Grant 15-04-02583a. The authors declare that they have no conflicts of interest with the contents of this article.
- GS
- glutamine synthetase
- SPR
- surface plasmon resonance
- ITC
- isothermal titration calorimetry
- SPINE
- Strep-protein interaction experiment
- MSX
- methionine sulfoximine
- AMP-PNP
- adenosine 5′-(β,γ-imido)triphosphate
- ATPγS
- adenosine 5′-(γ-thio)triphosphate tetralithium salt
- RU
- resonance unit(s)
- FC1 and FC2
- flow cell 1 and 2, respectively
- Ni-NTA
- nickel-nitrilotriacetic acid
- SEC
- size exclusion chromatography
- MALS
- multiangle light scattering.
References
- 1. Wray L. V. Jr., Ferson A. E., Rohrer K., and Fisher S. H. (1996) TnrA, a transcription factor required for global nitrogen regulation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 93, 8841–8845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fisher S. H. (1999) Regulation of nitrogen metabolism in Bacillus subtilis: vive la difference! Mol. Microbiol. 32, 223–232 [DOI] [PubMed] [Google Scholar]
- 3. Detsch C., and Stülke J. (2003) Ammonium utilization in Bacillus subtilis: transport and regulatory functions of NrgA and NrgB. Microbiology 149, 3289–3297 [DOI] [PubMed] [Google Scholar]
- 4. Nakano M. M., Hoffmann T., Zhu Y., and Jahn D. (1998) Nitrogen and oxygen regulation of Bacillus subtilis nasDEF encoding NADH-dependent nitrite reductase by TnrA and ResDE. J. Bacteriol. 180, 5344–5350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wray L. V. Jr., Zalieckas J. M., and Fisher S. H. (2000) Purification and in vitro activities of the Bacillus subtilis TnrA transcription factor. J. Mol. Biol. 300, 29–40 [DOI] [PubMed] [Google Scholar]
- 6. Yoshida K., Yamaguchi H., Kinehara M., Ohki Y. H., Nakaura Y., and Fujita Y. (2003) Identification of additional TnrA-regulated genes of Bacillus subtilis associated with a TnrA box. Mol. Microbiol. 49, 157–165 [DOI] [PubMed] [Google Scholar]
- 7. Mirouze N., Bidnenko E., Noirot P., and Auger S. (2015) Genome-wide mapping of TnrA-binding sites provides new insights into the TnrA regulon in Bacillus subtilis. Microbiologyopen 4, 423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heinrich A., Woyda K., Brauburger K., Meiss G., Detsch C., Stülke J., and Forchhammer K. (2006) Interaction of the membrane-bound GlnK-AmtB complex with the master regulator of nitrogen metabolism TnrA in Bacillus subtilis. J. Biol. Chem. 281, 34909–34917 [DOI] [PubMed] [Google Scholar]
- 9. Wray L. V. Jr., Zalieckas J. M., and Fisher S. H. (2001) Bacillus subtilis glutamine synthetase controls gene expression through a protein-protein interaction with transcription factor TnrA. Cell 107, 427–435 [DOI] [PubMed] [Google Scholar]
- 10. Fisher S. H., and Wray L. V. Jr. (2008) Bacillus subtilis glutamine synthetase regulates its own synthesis by acting as a chaperone to stabilize GlnR-DNA complexes. Proc. Natl. Acad. Sci. U.S.A. 105, 1014–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Commichau F. M., Gunka K., Landmann J. J., and Stülke J. (2008) Glutamate metabolism in Bacillus subtilis: gene expression and enzyme activities evolved to avoid futile cycles and to allow rapid responses to perturbations of the system. J. Bacteriol. 190, 3557–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gunka K., and Commichau F. M. (2012) Control of glutamate homeostasis in Bacillus subtilis: a complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol. Microbiol. 85, 213–224 [DOI] [PubMed] [Google Scholar]
- 13. Brown S. W., and Sonenshein A. L. (1996) Autogenous regulation of the Bacillus subtilis glnRA operon. J. Bacteriol. 178, 2450–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schumacher M. A., Chinnam N. B., Cuthbert B., Tonthat N. K., and Whitfill T. (2015) Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Genes Dev. 29, 451–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wray L. V. Jr., and Fisher S. H. (2007) Functional analysis of the carboxy-terminal region of Bacillus subtilis TnrA, a MerR family protein. J. Bacteriol. 189, 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wray L. V. Jr., and Fisher S. H. (2008) Bacillus subtilis GlnR contains an autoinhibitory C-terminal domain required for the interaction with glutamine synthetase. Mol. Microbiol. 68, 277–285 [DOI] [PubMed] [Google Scholar]
- 17. Kayumov A., Heinrich A., Fedorova K., Ilinskaya O., and Forchhammer K. (2011) Interaction of the general transcription factor TnrA with the PII-like protein GlnK and glutamine synthetase in Bacillus subtilis. FEBS J. 278, 1779–1789 [DOI] [PubMed] [Google Scholar]
- 18. Meek T. D., and Villafranca J. J. (1980) Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry 19, 5513–5519 [DOI] [PubMed] [Google Scholar]
- 19. Wedler F. C., and Horn B. R. (1976) Catalytic mechanisms of glutamine synthetase enzymes: studies with analogs of possible intermediates and transition states. J. Biol. Chem. 251, 7530–7538 [PubMed] [Google Scholar]
- 20. Murray D. S., Chinnam N., Tonthat N. K., Whitfill T., Wray L. V. Jr., Fisher S. H., and Schumacher M. A. (2013) Structures of the Bacillus subtilis glutamine synthetase dodecamer reveal large intersubunit catalytic conformational changes linked to a unique feedback inhibition mechanism. J. Biol. Chem. 288, 35801–35811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deuel T. F., and Prusiner S. (1974) Regulation of glutamine synthetase from Bacillus subtilis by divalent cations, feedback inhibitors, and l-glutamine. J. Biol. Chem. 249, 257–264 [PubMed] [Google Scholar]
- 22. Fedorova K., Kayumov A., Woyda K., Ilinskaja O., and Forchhammer K. (2013) Transcription factor TnrA inhibits the biosynthetic activity of glutamine synthetase in Bacillus subtilis. FEBS Lett. 587, 1293–1298 [DOI] [PubMed] [Google Scholar]
- 23. Herzberg C., Weidinger L. A., Dörrbecker B., Hübner S., Stülke J., and Commichau F. M. (2007) SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics 7, 4032–4035 [DOI] [PubMed] [Google Scholar]
- 24. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 25. Saxild H. H., and Nygaard P. (1987) Genetic and physiological characterization of Bacillus subtilis mutants resistant to purine analogs. J. Bacteriol. 169, 2977–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hart D. J., Speight R. E., Cooper M. A., Sutherland J. D., and Blackburn J. M. (1999) The salt dependence of DNA recognition by NF-kappaB p50: a detailed kinetic analysis of the effects on affinity and specificity. Nucleic Acids Res. 27, 1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stevenson C. E., Assaad A., Chandra G., Le T. B., Greive S. J., Bibb M. J., and Lawson D. M. (2013) Investigation of DNA sequence recognition by a streptomycete MarR family transcriptional regulator through surface plasmon resonance and x-ray crystallography. Nucleic Acids Res. 41, 7009–7022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liaw S. H., and Eisenberg D. (1994) Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry 33, 675–681 [DOI] [PubMed] [Google Scholar]
- 29. Krajewski W. W., Jones T. A., and Mowbray S. L. (2005) Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. U.S.A. 102, 10499–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wray L. V. Jr., and Fisher S. H. (2010) Functional roles of the conserved Glu304 loop of Bacillus subtilis glutamine synthetase. J. Bacteriol. 192, 5018–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., and Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ikeda T. P., Shauger A. E., and Kustu S. (1996) Salmonella typhimurium apparently perceives external nitrogen limitation as internal glutamine limitation. J. Mol. Biol. 259, 589–607 [DOI] [PubMed] [Google Scholar]
- 33. Almassy R. J., Janson C. A., Hamlin R., Xuong N. H., and Eisenberg D. (1986) Novel subunit-subunit interactions in the structure of glutamine synthetase. Nature 323, 304–309 [DOI] [PubMed] [Google Scholar]
- 34. Eisenberg D., Gill H. S., Pfluegl G. M., and Rotstein S. H. (2000) Structure-function relationships of glutamine synthetases. Biochim. Biophys. Acta 1477, 122–145 [DOI] [PubMed] [Google Scholar]
- 35. Yaginuma H., Kawai S., Tabata K. V., Tomiyama K., Kakizuka A., Komatsuzaki T., Noji H., and Imamura H. (2014) Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 4, 6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matos R. G., Barbas A., and Arraiano C. M. (2010) Comparison of EMSA and SPR for the characterization of RNA-RNase II complexes. Protein J. 29, 394–397 [DOI] [PubMed] [Google Scholar]
- 37. Forchhammer K. (2008) P(II) signal transducers: novel functional and structural insights. Trends Microbiol. 16, 65–72 [DOI] [PubMed] [Google Scholar]
- 38. Huergo L. F., Chandra G., and Merrick M. (2013) PII signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiol. Rev. 37, 251–283 [DOI] [PubMed] [Google Scholar]
- 39. Forchhammer K., and Luddecke J. (2015) Sensory properties of the PII signalling protein family. FEBS J. 10.1111/febs.13584 [DOI] [PubMed] [Google Scholar]
- 40. Blencke H. M., Reif I., Commichau F. M., Detsch C., Wacker I., Ludwig H., and Stülke J. (2006) Regulation of citB expression in Bacillus subtilis: integration of multiple metabolic signals in the citrate pool and by the general nitrogen regulatory system. Arch Microbiol. 185, 136–146 [DOI] [PubMed] [Google Scholar]
- 41. Martin-Verstraete I., Débarbouillé M., Klier A., and Rapoport G. (1994) Interactions of wild-type and truncated LevR of Bacillus subtilis with the upstream activating sequence of the levanase operon. J. Mol. Biol. 241, 178–192 [DOI] [PubMed] [Google Scholar]
- 42. Stragier P., Bonamy C., and Karmazyn-Campelli C. (1988) Processing of a sporulation σ factor in Bacillus-subtilis: how morphological structure could control gene expression. Cell 52, 697–704 [DOI] [PubMed] [Google Scholar]