

Abstract
Nerve growth factor (NGF) contributes to the development of chronic pain associated with degenerative connective tissue pathologies, such as intervertebral disc degeneration and osteoarthritis. However, surprisingly little is known about the regulation of NGF in these conditions. Toll-like receptors (TLR) are pattern recognition receptors classically associated with innate immunity but more recently were found to be activated by endogenous alarmins such as fragmented extracellular matrix proteins found in degenerating discs or cartilage. In this study we investigated if TLR activation regulates NGF and which signaling mechanisms control this response in intervertebral discs. TLR2 agonists, TLR4 agonists, or IL-1β (control) treatment increased NGF, brain-derived neurotrophic factor (BDNF), and IL-1β gene expression in human disc cells isolated from healthy, pain-free organ donors. However, only TLR2 activation or IL-1β treatment increased NGF protein secretion. TLR2 activation increased p38, ERK1/2, and p65 activity and increased p65 translocation to the cell nucleus. JNK activity was not affected by TLR2 activation. Inhibition of NF-κB, and to a lesser extent p38, but not ERK1/2 activity, blocked TLR2-driven NGF up-regulation at both the transcript and protein levels. These results provide a novel mechanism of NGF regulation in the intervertebral disc and potentially other pathogenic connective tissues. TLR2 and NF-κB signaling are known to increase cytokines and proteases, which accelerate matrix degradation. Therefore, TLR2 or NF-κB inhibition may both attenuate chronic pain and slow the degenerative progress in vivo.
Keywords: neurotrophin, NF-κB (NF-KB), nucleus pulposus, pain, toll-like receptor (TLR), intervertebral disc, low back pain, nerve growth factor
Introduction
Intervertebral disc degeneration is a major cause of chronic low back pain, for which current therapeutics are largely ineffective. Intervertebral discs have two distinct areas; the central gelatinous nucleus pulposus (NP),2 rich in proteoglycans and collagen type II, and the surrounding fibro-cartilaginous annulus fibrosus (AF), rich in collagen type I. The causes of disc degeneration are multifaceted and not well understood, yet genetics, mechanical load, or traumatic injury to the disc along with several life style choices are known to contribute to the etiologies of disc degeneration (1). During degeneration there is a large increase in catabolic proteases including matrix metalloproteinases (2, 3), cathepsins (4, 5), high temperature requirement serine protease A1 (HTRA1) (6, 7), and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs) (8, 9). Protease activity degrades the extracellular matrix (ECM), and the generated ECM fragments can potentially function as endogenous danger-associated patterns (DAMPs) also known as “alarmins” (10). Alarmins have been shown to activate toll-like receptors (TLRs) in other tissues, resulting in a robust increase of inflammatory cytokines including IL-1β and TNFα (11, 12). TLRs have recently been proposed to contribute to disc degeneration, and their activation may play an important role in the induction of inflammatory cytokines and pain mediators implicated in painful disc degeneration.
Low back pain can develop through multiple mechanisms including compression of the dorsal nerve root, dorsal root ganglia, or spinal cord and through neuronal sensitization via cytokines, chemokines and neurotrophins. Nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) are neurotrophic factors best characterized for promoting neuronal survival, maturation, and growth. However, they also induce chronic neuronal sensitization in mature peripheral afferent fibers, which results in the development of chronic pain (13). Accordingly, NGF and BDNF have both been implicated in chronic low back pain associated with degeneration. Immunohistochemical staining of NGF and BDNF is stronger in degenerating or herniated discs (14–16) and degenerating discs secrete greater amounts of NGF and BDNF compared with healthy discs (17). Healthy discs are primarily aneural, but several studies have reported that they become increasingly innervated as discs degenerate (18–20). In vitro human studies and in vivo animal studies have suggested that NGF and BDNF contribute to both innervation of degenerating discs and neuronal sensitization (17, 21, 22). These preclinical studies have made NGF and BDNF attractive therapeutic targets to treat low back pain. In fact, clinical trials using monoclonal antibodies against NGF have shown some efficacy to treat low back pain (23, 24). These prior studies demonstrate an important role for neurotrophins in the development of chronic low back pain; however, little is known about their regulation in pathologies such as painful disc degeneration.
Inflammatory mediators, such as IL-1β and TNFα, are known to increase NGF and BDNF gene expression in isolated disc cells (16, 25–27). However, it is also possible that other mechanisms increase neurotrophin expression during degeneration. For example, NGF secretion is elevated after mechanical injury to human discs or high mechanical strain to isolated disc cells (28, 29). The signaling mechanisms regulating neurotrophin transcription in pathogenic target tissues like degenerating discs are also unknown. A possible mechanism that has not been explored is that TLR signaling directly increases neurotrophin expression.
TLRs are pattern recognition receptors that were originally characterized in the innate immune system and are activated by pathogen-associated molecular patterns (PAMPs), such as bacterial cell wall components, in addition to alarmins. In humans, 10 TLRs have been described. Gene expression of TLRs 1, 2, 3, 4, 5, 6, 9, and 10 have been detected in human disc cells where expression of TLRs 1, 2, 4, and 6 are correlated with an increasing degree of degeneration (11). Alarmins include proteolysis-generated ECM fragments, such as fragmented hyaluronic acid, fibronectin, biglycan, tenacin C, versican, heparan sulfate, and aggrecan and high mobility group B1 and the heat shock proteins 60 and 70 (10, 30, 31). Many alarmins, including fibronectin and biglycan fragments and high mobility group B1, have been found in degenerating discs (32–35). TLR2 and TLR4 are thought to be the primary TLR subtypes that recognize ECM alarmins, where TLR4 functions as a homodimer and TLR2 functions as a homodimer or heterodimer with TLR1 or -6 (10). TLR activation increases the catabolic proteases matrix metalloproteinase-1 and -13 as well as IL-1β, IL-6, and IL-8 in disc cells (11, 36). Interestingly, in vivo injection of fibronectin fragments into rabbit discs induces degenerative changes (37), and exposure of NP cells to fibronectin fragments decreases proteoglycan synthesis and increases proteoglycan degradation (38). However, these studies did not investigate the mechanism responsible for these changes.
The potential role of TLR signaling in the early stages of disc degeneration led us to hypothesize that TLR activation induces neurotrophin expression either directly or as a secondary effect via cytokines. In this study we investigated TLR agonist- and cytokine-induced neurotrophin induction in human disc cells and determined that TLRs directly regulate neurotrophin expression. The signaling mechanisms regulating NGF were then explored. This study has identified novel mechanisms contributing to NGF regulation, offering alternative strategies to target NGF in disc degeneration to treat chronic pain.
Experimental Procedures
Tissue Collection and Cell Isolation
All procedures were approved by the institutional review board of McGill University (IRB# A04-M53-08B) project titled “Human Intervertebral Discs used for Culture and Extracellular Matrix.” Human lumbar spines were harvested from organ donors after donor and familial consent via a collaboration with Transplant Quebec. Donor information is presented in Table 1. NP and AF cells were isolated separately as previously described (29). Briefly, discs were excised from the lumbar spine, and NP and AF tissues were separated. Cells were cultured using “disc cell media” composed of Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum, 1× glutamax, and 25 μg/ml gentamicin (Life Technologies).
TABLE 1.
Summary of the characteristics of donors used in the study
Dashes (—) indicate that both NP and AF cells were used for the indicated experiment. If only NP or AF cells were used for a particular experiment it is indicated.
| Donor | Age | Sex | Cause of death | Time course | TLR2 inhibition | Cell signaling | IL-1β inhibition | Signaling pathway inhibition |
|---|---|---|---|---|---|---|---|---|
| 1 | 19 | M | Suicide | — | — | — | ||
| 2 | 17 | M | Suicide | NP only | — | — | — | |
| 3 | 50 | F | Cerebral hemorrhage | — | — | NP only | ||
| 4 | 24 | M | Head trauma | — | — | — | ||
| 6 | 29 | M | Suicide | — | — | — | — | |
| 7 | 27 | M | Suicide | — | AF only | — | — | |
| 8 | 25 | F | MVA, head trauma | — | AF only |
In Vitro Cell Experiments
Experiments were performed with NP and AF cells separately within passages 2–4. Approximately 250,000 cells per well were seeded into 6-well plates and allowed 12–24 h to adhere. Cells were then serum-starved in serum-free disc cell media supplemented with 1× insulin-transferrin selenium (ITS, Life Technologies) for 12 h. For initial studies examining the effects of different treatments, cells were left untreated or treated with IL-1β (10 ng/ml) and TNFα (10 ng/ml, both from Peprotech, Rocky Hill, NJ), peptidoglycan (PGN; 5 μg/ml, Sigma), or lipopolysaccharide (LPS; 10 μg/ml, Sigma). RNA was extracted after 6, 12, and 24 h of treatment with TRIzol reagent (Life Technologies), and medium was collected after 12, 24, and 48 h of treatment. For cell signaling experiments, cells were treated with IL-1β or PGN. Cell lysates were collected 30, 60, 120, and 360 min after treatment using NuPage LDS loading buffer (Life Technologies).
Antibody Neutralization
Serum-free disc cell media supplemented with insulin-transferrin selenium containing polyclonal antibodies against TLR2 (5 μg/ml, Invivogen, catalogue #pab-hstlr2) or normal IgG were added to cultures 2 h before the addition of PGN. For IL-1β inhibition experiments, serum-free disc cell media supplemented with insulin-transferrin selenium containing no treatment, IL-1β, or PGN was incubated with monoclonal neutralizing antibodies against IL-1β (5 μg/ml, Invivogen, mabg-hil1b-3) or normal IgG for 1 h and then applied to cells. RNA was extracted after 6 h of treatment.
Cell Signaling Inhibition
Cells were serum-starved with disc cell media supplemented with insulin-transferrin selenium for 12 h and then pretreated with SB203580 (10 μm, p38 inhibitor; Life Technologies), PD98059 (10 μm, MEK1/2 inhibitor, Life Technologies), or BMS-345541 (5 μm, NF-κB inhibitor, Sigma) for 2 hours. Cultures were then challenged with IL-1β or PGN alone or in combination with inhibitors. RNA was extracted after 6 h of treatment, and conditioned culture media was collected after 48 h for protein analysis.
Real-time Quantitative Polymerase Chain Reaction (qRT)
qRT-PCR was performed as previously described (17). Briefly, RNA was extracted using TRIzol reagent (Life Technologies) and isolated using a chloroform extraction method as previously described (17). Approximately 500 ng of RNA was reverse-transcribed to cDNA using qSqript cDNA synthesis kits (Quanta Biosciences, Gaithersburg, MA) with an Applied Biosystems Verti thermal cycler (Life Technologies). Real-time quantitative-PCR was performed using an Applied Biosystems StepOnePlus (Life Technologies) with PerfeCTa SYBR Green FastMix (Quanta Biosciences) and previously published primers specific against 18s, IL-1β, NGF, and BDNF (39–41) (Life Technologies) or with TaqMan Gene Expression Master Mix and commercially available primers specific against 18S, TLR-1, -2 -4, and -6 (Life Technologies). Data were normalized to 18S expression or analyzed using the 2−ΔΔCt method (42).
Protein Analysis
NGF enzyme-linked immunosorbent assays (ELISAs, RayBiotech, Norcross, GA, catalogue #ELH-BNGF) and BDNF ELISAs (Millipore, Billerica, MA, catalogue #CYT306) were used to quantify protein concentrations in conditioned cell culture media according to manufacturer's instructions. For NGF Western blots, protein in the conditioned culture media was precipitated using 100% ethanol overnight at 4 °C. Samples were then spun down, and the pellet was resuspended in NuPage LDS Loading Buffer (Life Technologies). Samples were boiled and resolved on a 10–20% gradient gel (Life Technologies). For cell signaling, samples were boiled and resolved on a 10% polyacrylamide gel. Protein was then transferred to a nitrocellulose membrane (General Electric, Mississauga, ON, Canada). Membranes were blocked using 3% bovine serum albumin (BSA). A rabbit antibody against NGF (1:500, Santa Cruz, Dallas, TX; catalogue #sc-548, lot # l1113) was incubated overnight to detect NGF. For signaling proteins, phosphorylation-specific antibodies were used to detect phosphorylated p38 (Cell Signaling, Beverly, MA), ERK1/2 (Cell Signaling), p65 (recognizes phosphorylation at Ser536, Santa Cruz), and JNK (Cell Signaling). Blots were then stripped using Restore PLUS Western blot Stripping Buffer (Sigma) and reprobed with antibodies detecting total p38, ERK1/2, p65, and JNK (all from Cell Signaling). α-Tubulin was probed as a loading control. All membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies, exposed to Western Lightning Plus-ECL (PerkinElmer Life Sciences), and imaged using a LAS 4000 Image Quant system (General Electric). Densitometric analysis was carried out using the ImageQuant TL program (General Electric). Samples were normalized to the background of the blot.
Immunofluorescence
NP and AF cells in 8-well chamber slides (Nunc) were serum-starved and then treated with IL-1β, PGN, or left untreated. After 1 h cultures were fixed with ice-cold methanol for 15 min. Cultures were blocked in PBS with 1% BSA and 0.1% Triton-X100 (Sigma) for 1 h at room temperature and then incubated with an antibody specific against p65 (Santa Cruz) for 1 h at room temperature. After washing with PBS, slides were incubated with an appropriate Alexa Fluor® 488-conjugated secondary antibody for 1 h at room temperature and then counterstained with DAPI. Coverslips were mounted using Aqua Polymount (Polysciences, Inc., Warrington, PA), and images were captured with an Olympus BX51 (Olympus, Tokyo, Japan) microscope equipped with a color digital camera (Olympus DP71). Two random images of each well were taken, and p65 and DAPI images were overlaid in Photoshop.
Statistical Analysis
Data were analyzed using Graph Prism 6 (Graph Pad, La Jolla, CA). Paired t tests were used to analyze two groups, and the Kruskal-Wallis test was used to analyze data from two or more groups.
Results
Induction of Neurotrophins
Proteolysis of ECM proteins has been suggested to be an early event in disc degeneration where the resulting ECM fragments could induce TLR signaling. To determine if TLR activation induces neurotrophin expression, NP and AF cells from healthy human discs were treated with the TLR agonists PGN (TLR2 agonist) and LPS (TLR4 agonist) for 6, 12, 24, and 48 h. Cells were treated with IL-1β and TNFα to provide positive controls, and untreated cells served as the baseline. Both TLR and cytokine receptor activation are known to increase IL-1β gene expression. Therefore, IL-1β expression was measured to ensure that the cells express functional receptors for both cytokines and TLR agonists. As expected, cytokine treatment and TLR activation increased IL-1β gene expression. In some experiments baseline IL-1β expression was undetectable in untreated cells, and therefore, IL-1β expression could not be normalized to baseline levels, rendering 2−ΔΔCt values impossible to calculate for those experiments. This reduces the sample size for IL-1β gene expression to n = 3. Nevertheless, IL-1β expression was always detected in treated cells, suggesting that both NP and AF cells from non-degenerate discs express functional TLR receptors in addition to cytokine receptors (Fig. 1, A and B).
FIGURE 1.

Gene expression in NP and AF after cytokine and TLR agonist treatment. IL-1β (A and B), NGF (C and D), and BDNF (E and F) gene expression is shown in NP (A, C, and E) or AF cells (B, D, and F) after 6, 12, and 24 h of treatment with IL-1β, TNF, PGN, or LPS. Data are presented as -fold change normalized to 18S gene expression and untreated cells using the 2−ΔΔCt method. Data were analyzed using the Kruskal-Wallis test for multiple comparisons of non-parametric data. Data are presented at mean ± S.E., n = 4, except for IL-1β gene expression, where n = 3. *, p < 0.05; **, p < 0.01.
To determine if TLR2 or TLR4 activation increased neurotrophin expression, we treated cells with PGN (TLR2 agonist) or LPS (TLR4 agonist). TLR2 activation significantly increased NGF expression in NP cells compared with untreated cells after 6, 12, and 24 h of treatment. TLR4 activation also increased NGF expression, but the increase was not statistically significant (Fig. 1C). In comparison, IL-1β increased NGF expression at all time points, whereas TNFα promoted a strong early trend for increased NGF expression that was significantly increased by 24 h compared with untreated cells. IL-1β and TLR2 activation significantly increased BDNF expression after 12 and 24 h of treatment, whereas TLR4 activation and TNFα promoted less pronounced BDNF increases compared with controls (Fig. 1E). In AF cells, IL-1β significantly increased NGF after 6 h, whereas TNFα and TLR2 activation promoted an increase in NGF expression. After 12 h, IL-1β and TNFα increased NGF expression, and after 24 h IL-1β, TNFα and TLR2 activation significantly increased NGF expression (Fig. 1D). TLR2 activation never significantly increased BDNF expression in AF cells (Fig. 1F), and similarly to NP cells, TLR4 activation had a variable effect on neurotrophin induction (Fig. 1, D and F).
To further examine TLR regulation of neurotrophins, NGF and BDNF concentrations in conditioned culture media of NP and AF cells was quantified by ELISA. In NP cells, NGF was undetectable or barely detectable in untreated cells, whereas NGF was detected as early as 12 h after treatment and was significantly increased after 48 h of TLR2 activation and treatment with IL-1β. Similarly, in AF cells NGF had a low baseline expression whereas IL-1β significantly increased NGF after 48 h, and TLR2 induction of NGF showed a strong trend compared with untreated cells after 48 h (p = 0.06). TNFα also increased NGF secretion, although not to statistically significant levels. TLR4 activation increased NGF protein secretion slightly, but variably compared with untreated cells (Fig. 2). In all but a few samples BDNF was below the detection threshold (0.015 ng/ml) of the ELISA (data not shown). Therefore, we proceeded to focus on TLR2 regulation of NGF.
FIGURE 2.
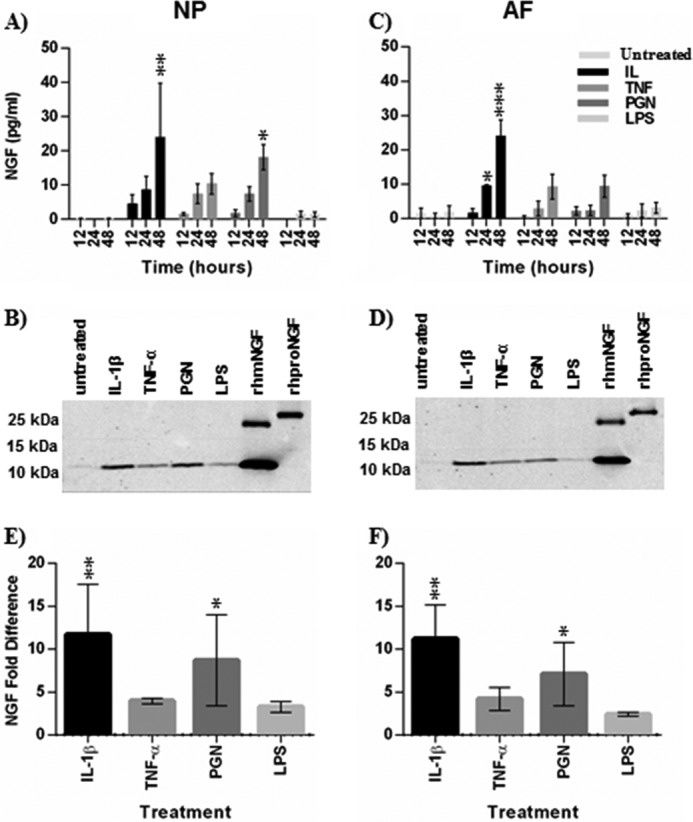
NGF protein secretion by NP and AF after cytokine and TLR agonist-treatment. Conditioned cell culture media were collected 12, 24, and 48 h after treatment with IL-1β, TNF, PGN, and LPS. NGF protein levels in the media was analyzed by ELISA (A and B) and Western blot (C–F). NP (A, C, and E) and AF (B, D, and F) media were both analyzed. Conditioned media after 48 h of treatment were used for Western blots. Recombinant human mature NGF (rhmNGF) and proNGF (rhproNGF) were used as controls for Western blots. Western blots were analyzed by densitometry and normalized to untreated cells in E (NP cells) and F (AF cells). Data were analyzed using the Kruskal-Wallis test for multiple comparisons of non-parametric data. Data are presented at the mean ± S.E. n = 4 for ELISA. n = 3 for Western blot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
NGF is post-translationally cleaved from proNGF to mature NGF (mNGF), and these two isoforms exert different biological effects (43). Therefore, we used Western blot to determine the relative predominance of proNGF or mNGF in disc cell-conditioned medium after 48 h of treatment. Only mNGF was detected in the conditioned medium (Fig. 2, C and D). Furthermore, densitometry analysis supported our findings that IL-1β and TLR2 activation more strongly increase NGF compared with other treatments (Fig. 2, E and F). Taken together, these results demonstrate that NGF transcription and translation is induced by TLR2 activation in intervertebral disc cells.
Toll-like Receptor Expression
Increased TLR1, -2, -4, and -6 gene expression has been correlated with an increasing degree of degeneration (11). TLR2 can signal as a homodimer or heterodimer with TLR1 or TLR6, whereas TLR4 signals primarily as a homodimer (10). To determine the presence of individual TLR subtypes and the level of induced gene expression, TLR1, -2, 4-, and -6 expression was analyzed in NP and AF cells after treatment with IL-1β and PGN. TLR-1, -2, -4, and -6 were all expressed in NP and AF cells, treated or untreated. The expression was variable between donors, but TLR2 or IL-1β receptor activation strongly increased TLR2 expression compared with untreated cells (Fig. 3, A and B).
FIGURE 3.

TLR gene expression and TLR2 inhibition. Gene expression of TLR-1, -2, -4, and -6 in NP (A) and AF (B) cells after 6 h of treatment with IL-1β, PGN, or left untreated is shown. Data were normalized to 18S gene expression as endogenous housekeeping gene and presented as 2−ΔΔCt, n = 4. For TLR2 inhibition (C and D), cells were pretreated for 2 h with polyclonal neutralizing antibodies against TLR2 or normal IgG and then treated with PGN. IL-1β and NGF gene expression in NP (C) and AF (D) cells was analyzed using the 2−ΔΔCt method and presented as -fold difference compared with cells that were treated with PGN and normal IgG, n = 3. To examine if IL-1β is required for TLR2 activation to induce IL-1β and NGF gene expression, NP (E and F) and AF (G and H) cells were treated with IL-1β or PGN in combination with an IL-1β neutralizing antibody or normal IgG. NGF (E and G) and IL-1β (F and H) gene expression was evaluated using the 2−ΔΔCt method and presented as -fold difference compared with cells that were treated with IL-1β or PGN and normal IgG. Differences in gene expression were assessed by ANOVA (A and B) or paired t tests (C–H). n = 4. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
TLR2 Is Required for PGN-induced NGF Expression
PGN can potentially activate other pattern recognition receptors besides TLR2, such as nucleotide-binding oligomerization domain receptors (44). Neutralizing polyclonal antibodies against TLR2 were used to confirm that PGN acts through TLR2 in disc cells. NP and AF cells treated with PGN together with TLR2 neutralizing antibodies express decreased levels of IL-1β and NGF compared with cells treated with PGN alone (Fig. 3, C and D). These results demonstrate that TLR2 activation can induce NGF expression.
IL-1β Is Not Required for TLR2-induced NGF Expression
TLR2 induction of IL-1β is well characterized in other cell types and occurs in disc cells (Fig. 1, A and B). Because IL-1β induces NGF, as does TLR2 activation, it is possible that TLR2 induces NGF through a feedback loop via IL-1β. To evaluate a potential feedback loop neutralizing antibodies against IL-1β were used in cultured NP and AF cells treated with IL-1β (control) or PGN to determine if TLR2 requires IL-1β for NGF induction. As expected, in both NP and AF cells treated with IL-1β, NGF gene expression was decreased after treatment with neutralizing IL-1β antibodies. However, neutralizing antibodies had no effect on TLR2-induced NGF (Fig. 3, E–H), indicating IL-1β is not required for TLR2-induced NGF.
PGN Induces NF-κB, p38, and ERK1/2 Signaling
Signaling mechanisms downstream of TLRs in intervertebral discs have only recently begun to be investigated. In other tissues TLR or IL-1 receptor activation can induce NF-κB, p38, ERK1/2, and JNK signaling (30). To evaluate the pathway activated in intervertebral disc cells, NP and AF cells were treated with IL-1β or the TLR2 agonist PGN, and cell lysates were analyzed after 30, 60, 120, and 360 min of treatment. Western blot analysis revealed that IL-1β and PGN strongly increased NF-κB (p65), p38, and ERK1/2 phosphorylation compared with untreated cells after 30 min. p65 and p38 phosphorylation remained elevated after 360 min, whereas ERK1/2 phosphorylation decreased after 60 min. JNK phosphorylation was undetectable or minimal after TLR2 activation, whereas IL-1β induced JNK phosphorylation after 30 min, which then declined to undetectable levels (Fig. 4, A, B, and C). These results suggest that TLR2 signaling in disc cells is predominantly through NF-κB, p38, and ERK1/2 signaling.
FIGURE 4.

Signaling pathway activity. p65, p38, ERK1/2, and JNK activity was evaluated using phosphorylated-specific antibodies and antibodies recognizing total p65, p38, ERK1/2, and JNK protein. Cell lysates of NP and AF cells were collected after 30, 60, 120, and 360 min of treatment with IL-1β, PGN, or left untreated (A). Cell lysates were probed using α-tubulin was used as a loading control. Densitometry data of phosphorylated protein is normalized to total p65, p38, ERK1/2, or JNK in NP (B) and AF (C) cells. n = 4.
To further investigate NF-κB induction, p65 translocation to the nucleus was examined. After 1 h of treatment, p65 translocation was examined by immunofluorescence. TLR2 activation and IL-1β, both, induced p65 translocation to the nucleus in NP and AF cells, whereas p65 remained dispersed throughout the cytoplasm in untreated cells (Fig. 5). These results further support that TLR2 activates NF-κB signaling in disc cells.
FIGURE 5.
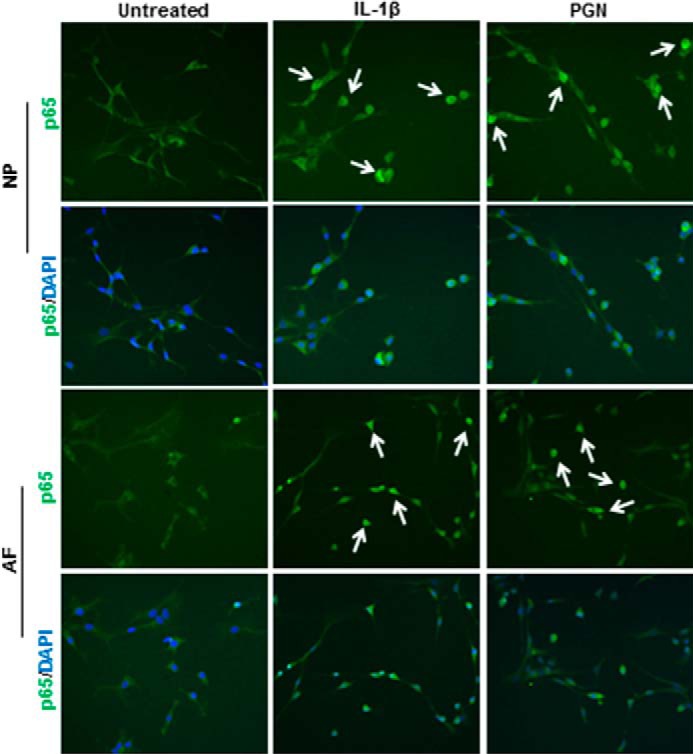
p65 Translocation to the cell nucleus. Cells were treated with IL-1β, PGN, or left untreated or left untreated for 1 h and then stained for p65 localization (green). Cultures were counterstained with the nuclear DNA stain DAPI (blue). White arrows indicate examples of p65 translocation to the nucleus. n = 3.
Signaling Mechanisms Regulating NGF Expression
Although regulatory mechanisms controlling NGF expression in the central nervous system have been investigated, little is known about its regulation in degenerating tissues such as the intervertebral disc. To evaluate if TLR2 and IL-1β regulate NGF through p38, ERK1/2, NF-κB, or another mechanism, NP and AF cells were pretreated with small molecule inhibitors that specifically block either p38, ERK1/2 (MEK), or NF-κB activity. They were then exposed to IL-1β and PGN. In NP cells treated with IL-1β, p38 inhibition significantly decreased NGF expression compared with IL-1β alone and decreased NGF expression in AF cells. Interestingly, p38 inhibition did not alter TLR2-induced NGF expression in either NP or AF cells (Fig. 6, A and B). Similarly, ERK1/2 inhibition did not effect NGF expression in NP cells (Fig. 6A) but surprisingly caused an increase in NGF expression in AF cells (Fig. 6B). To further investigate the role of MAPK signaling in NGF regulation and possible p38-ERK1/2 interactions, both pathways were inhibited together. Co-inhibition of p38 and ERK1/2 resulted in a decrease of NGF expression in IL-1β-treated but not PGN-treated NP cells (Fig. 6A). In the AF cells, co-inhibition resulted in a small increase of NGF after IL-1β treatment and a small decrease after PGN treatment. Unlike inhibition of MAPK signaling pathways, NF-κB inhibition greatly reduced NGF gene expression compared with IL-1β or PGN-treated cells without inhibitors in NP and AF cells (Fig. 6, A and B), suggesting an important role for NF-κB regulation of NGF in disc cells.
FIGURE 6.
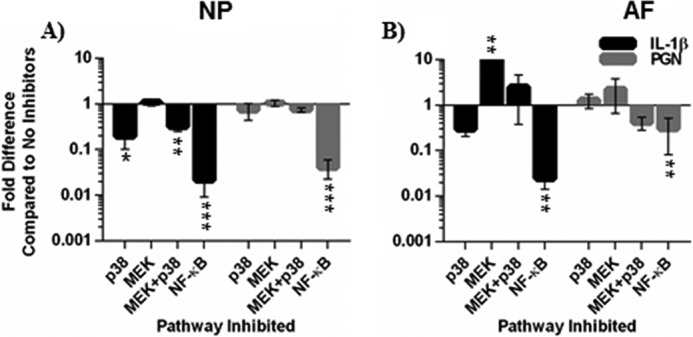
NGF gene expression after p38, ERK1/2, and NF-B inhibition. Small molecule signaling inhibitors SB203580 (10 μm, p38), PD98059 (10 μm, MEK), SB203580 and PD98059, or BMS-345541(5 μm, NF-κB) were added to cultures 2 h before treatment with IL-1β or PGN. NGF gene expression data after 6 h of treatment with IL-1β or PGN in NP (A) and AF (B) cells were normalized to GAPDH expression and analyzed using the 2−ΔΔCt. Data was presented as -fold difference compared with IL-1β or PGN treatments without signaling inhibitors. Data were analyzed using the Kruskal-Wallis test for multiple comparisons of non-parametric data. Data are presented as the mean ± S.E., n = 4. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To examine NGF protein secretion-conditioned culture, medium was probed by Western blot after 48 h. ERK1/2 inhibition had little effect or slightly increased NGF secretion in IL-1β- or PGN-treated cells. Similarly, simultaneous inhibition of p38 and ERK1/2 resulted in a small decrease in NP cells treated with IL-1β but did not affect NGF secretion induced by TLR2 activation or IL-1β in AF cells (Fig. 7). p38 inhibition resulted in small decreases of NGF secretion in NP cells (Fig. 7, A and C). Interestingly, p38 inhibition resulted in significant decreases in both IL-1β- and PGN-treated AF cells. However, similar to NGF gene expression, NF-κB inhibition reduced NGF secretion by IL-1β- or PGN-treated cells to levels similar to untreated cultures (Fig. 7, A and B). Important to note, NP and AF cells treated with cell signaling inhibitors only secreted mNGF-like cells treated with IL-1β or PGN alone. These results suggest that IL-1β and TLR2 regulate NGF through NF-κB signaling.
FIGURE 7.

NGF secretion after p38, ERK1/2, and NF-κB inhibition. Small molecule signaling inhibitors SB203580 (10 μm, p38), PD98059 (10 μm, MEK), SB203580 and PD98059, or BMS-345541(5 μm, NF-κB) were added to cultures 2 h before treatment with IL-1β or PGN. Protein was precipitated from conditioned culture media after 48 h of treatment. NGF secretion after p38 or NF-κB inhibition in NP and AF cells (A) and after ERK1/2 or ERK1/2 and p38 in NP cells (B) was assessed by Western blot. Western blots were analyzed using densitometry for both NP (C)- and AF (D)-conditioned media. Data were normalized to NGF secretion from cells treated with IL-1β or PGN but no signaling inhibitors. IKK, inhibitor of κ B kinase. *, p < 0.05.
Discussion
The results from this study demonstrate that TLR2 activation induces NGF and BDNF gene expression and NGF protein secretion in human intervertebral disc cells. To determine how NGF is regulated, p38, ERK1/2, JNK, and NF-κB signaling activity was assessed. NF-κB activity is required for NGF expression induced by TLR2 as well as IL-1β. Taken together, these results identify a new regulatory mechanism mediating pathological NGF expression in NP and AF cells (Fig. 8) that could be used to target NGF to treat low back pain associated with disc degeneration.
FIGURE 8.
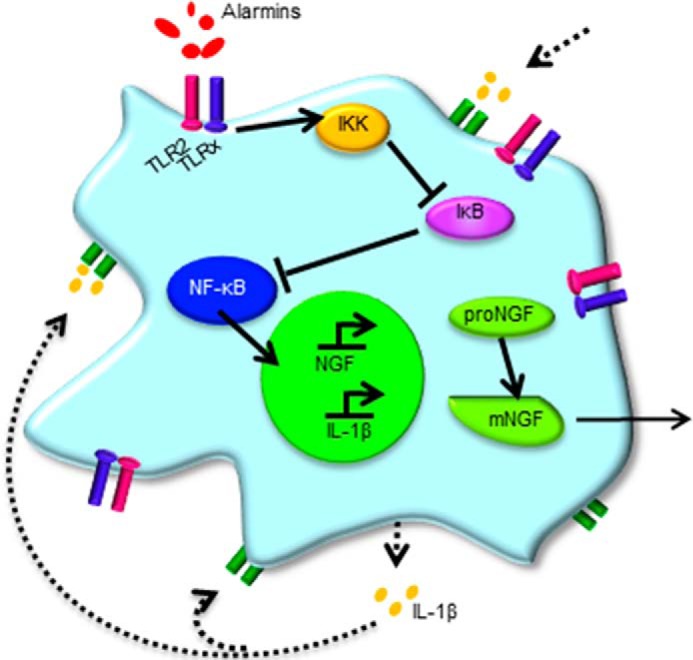
Schematic of TLR2 regulation of NGF. Alarmins activate TLR2 (blue)–TLRx (pink), where TLRx is TLR1, -2, or -6. TLR activation induces the NF-κB signaling cascade, promoting NGF synthesis. NGF is translated as proNGF and is cleaved intracellularly to mNGF and then secreted from the cell. Additionally, TLR2 activation results in increased IL-1β gene expression, which may result in increased IL-1β protein that could create a feedback loop, indicated by the dotted line, to further increase NGF. IKK, inhibitor of κ B kinase.
NGF increases during disc degeneration and has been implicated in animal models of painful disc degeneration, where it is thought to induce neuronal sensitization (15, 17, 22). NGF has also been suggested to increase innervation of degenerating discs (17, 21) and has been the target of clinical trials to treat low back pain associated with degeneration (23, 24). However, little is known about the regulation of NGF in tissues that become pathologically innervated with painful diseases like disc degeneration or osteoarthritis. Exposure of disc cells to the pro-inflammatory cytokine IL-1β increases NGF and BDNF expression. Some studies report that TNFα also increases neurotrophin expression, whereas other studies found that it does not. This discrepancy could be explained by differences in tissue source, experimental TNFα concentrations, and the time at which NGF expression was evaluated (16, 27). Our results show that TNFα induces NGF and to a lesser extent BDNF expression. However, the signaling mechanisms regulating NGF expression in pathological connective tissues like the disc were unknown until now. It was also not known that TLR2 activation increases NGF and BDNF expression in disc cells.
A current hypothesis is that ECM-produced alarmins, potentially generated by mechanical trauma, activate TLR signaling in early stages of disc degeneration (11, 12, 35). TLR activation can result in a robust increase of inflammatory cytokines and catabolic proteases, which in turn can further drive degeneration. Potential roles for TLR signaling have long been suggested in rheumatoid arthritis and osteoarthritis and most recently in disc degeneration. For example, a recent study found high mobility group B1, a TLR2 ligand, increases with the grade of degeneration in surgical specimens (35). Furthermore, TLR2 activation increases IL-1β, IL-6, IL-8, matrix metalloproteinase-1 and -13, and COX-2 gene expression and IL-6 protein in a mixed population of NP and AF cells (11, 36). These prior studies indicate that TLR2 could play an important role in the increase of inflammatory mediators and catabolic enzymes that contribute to disc degeneration.
Here, we examined the role of TLR2 and -4 in regulating the pain mediators NGF and BDNF. TLR2 activation increases NGF as early as 6 h in the NP and follows the same temporal pattern as IL-1β, demonstrating that TLR2 induces NGF directly rather than increasing NGF secondarily through cytokine activation. TLR2 also increased BDNF gene expression. To confirm PGN functions through TLR2, we inhibited TLR2 activation and found TLR2 is required for PGN-induced NGF expression. Interestingly, LPS, a TLR4 ligand, had a highly variable effect on NGF and BDNF gene expression but had little effect on NGF and BDNF protein expression. This suggests either TLR4 does not induce neurotrophins as readily as TLR2 or that there are variable levels of TLR4 on disc cell surfaces. When cells were challenged with PGN and IL-1β, only TLR2 expression increased, which agrees with other studies that found TLR2, but not TLR4, gene expression is readily inducible in disc cells (11). Taken together, these results suggest TLR2 has a more prominent role in human disc degeneration compared with TLR4. TLR2 can signal as a homo- or heterodimer, but which combination of receptors function in intervertebral discs is an area requiring further investigation. TLR2 activation leads to increased protease and cytokine levels that will contribute to ECM breakdown and degeneration. Therefore, TLR2 activation can potentially contribute to the development of both chronic low back pain via NGF synthesis and to accelerated degeneration via protease and cytokine induction.
TLR2 and IL-1 receptor (IL-1β receptor) both contain an intracellular Toll-interleukin receptor (TIR) intracellular domain. As expected, the downstream signal mechanisms are similar and include the NF-κB pathway and p38, ERK1/2, and JNK MAP kinase signaling, which can cause a variety of transcription factors to translocate to the nucleus (10, 30). Activation of these pathways in discs results in an increase in inflammatory mediators and proteases (45). Although signaling mechanisms of TLR2 have been extensively investigated in other cell types, they are only now being elucidated in intervertebral disc. Quero et al. (36) found that fragmented hyaluronic acid activation of TLR2 increases JNK, p38, and ERK1/2 phosphorylation but not p65 phosphorylation or translocation, and Pam3CSK4 (TLR2/TLR1 agonist) also did not stimulate p65 (11). These studies suggest TLR2 activation functions through MAP kinase signaling and not NF-κB signaling in disc cells. However, the current study found TLR2 activation in NP and AF cells increased p38, ERK1/2, and NF-κB activity and induced p65 translocation, whereas JNK phosphorylation did not increase compared with controls. Therefore, although the current study confirms previous findings that TLR2 activates p38 and ERK1/2 signaling, it conflicts with previous data suggesting that NF-κB is not activated in disc cells. This contradiction may be due to several factors: 1) the current study uses NP and AF cells from non-degenerating discs, whereas the previous studies used surgical samples from patients with symptomatic disc degeneration, disc herniation, or trauma; 2) previous studies did not separate NP and AF cells; 3) different ligands were used. Due to the tissue sources, there was likely different TLR expression between tissues at baseline. This, along with different ligands, could influence what signaling pathways are activated by TLR2 signaling. Importantly, TLR2 activation provides another mechanism for activating p38, ERK1/2, and NF-κB signaling during disc degeneration. Furthermore, phosphorylation of NF-κB is sustained (>30 min), which agrees with other studies that have found prolonged NF-κB activation in NP and AF cells (46, 47). For other pathologies, both p38 (rheumatoid arthritis) and NF-κB (atopic dermatitis) inhibitors have gone to clinical trials (45). If successful, these inhibitors may also positively affect disc degeneration.
Until now, signaling mechanisms regulating NGF in pathogenic connective tissues had not been investigated. However, NGF synthesis has been investigated in other tissues including the central nervous system. In astrocytes, PKC activation was associated with increased NGF, whereas increased cAMP levels did not affect NGF expression (48). In astrocytoma cells, c-fos activation increases NGF and inhibiting c-fos-reduced β-adrenergic receptor activation of NGF (49). In glioma cells PKA, PKC, or increased Ca2+ mediates AP-1 (c-fos/c-jun) binding a consensus sequence in the NGF gene (50). Similarly in fibroblasts, AP-1 binds a consensus sequence in the first intron of NGF, and mutating this binding site reduces NGF promoter activity (51). NGF mRNA synthesis has also been shown to be mediated by cAMP in Schwan cells (52). Aside from AP-1-mediated NGF transcription, β-adrenergic receptor stimulation also activates PKA and induces NGF via C/EBPδ and CREB (cAMP-response element-binding protein) in glioma cells and in the cerebral cortex. Importantly, a C/EBPδ binding motif is also present in the NGF promoter region (53). Taken together, these studies demonstrate central nervous system or fibroblast NGF synthesis is likely mediated by a number of different pathways that converge on AP-1, C/EBPδ, or CREB.
Here we investigated NGF synthesis in peripheral target tissues when induced by inflammatory mediators. We found that both TLR2 and IL-1β increase p38 and ERK1/2 activity, which can cause AP-1 translocation to the nucleus, and others have found evidence of CREB and C/EBPδ activity in intervertebral disc cells (55, 56). However, despite these trans-activating factors, we found that NF-κB signaling is required for TLR2 and IL-1β to increase NGF expression. Furthermore, NF-κB inhibition had the largest effect on NGF synthesis of all pathways inhibited. TLR2-induced NF-κB-dependent NGF synthesis in both NP and AF cells differs from other cell types, suggesting that NGF is regulated in a context-dependent or cell type-dependent manner. This has important implications when considering NGF as a therapeutic target. Diseases of the central nervous system such as Alzheimer disease are associated NGF dysregulation, whereas increased NGF can cause chronic pain in peripheral tissues. Thus, being able to target NGF in a context-dependent manner is advantageous.
NGF is translated into the ∼26-kDa proNGF, which is then cleaved to the ∼13-kDa mature form of NGF. ProNGF and mNGF exert different biological effects, whereas ProNGF has apoptotic functions, and mNGF promotes neurite growth and survival. mNGF is classically thought to sensitize neurons via the TrkA receptor, and proNGF may also affect inflammatory pain via the p75NTR-sortillin receptor complex in addition to having apoptotic effects (55). In the central nervous system evidence suggests that proNGF is secreted and processed extracellularly by plasmin to mNGF (54). In contrast, others have found proNGF is cleaved to mNGF by Furin before secretion in the central nervous system (56). The mechanisms and location of NGF maturation have not yet been investigated in pathological connective tissues such as degenerating discs. In this study only mNGF was detected in conditioned culture media, and preliminary data have also detected mNGF in cell lysates of disc cells, suggesting that proNGF can be cleaved intracellularly. However, additional investigation is required to develop a complete understanding of NGF maturation in discs.
The results from this study show for the first time that TLR2 activation directly increases NGF gene expression and protein levels in human cells. Furthermore, NF-κB signaling is a novel regulatory mechanism of NGF. This novel mechanism of NGF regulation is outlined in Fig. 7. NGF undoubtedly plays an important role in the development of low back pain and many other painful degenerative connective tissue disorders. As clinical trials with monoclonal antibodies against NGF have shown, targeting NGF could be an effective therapeutic strategy. By better understanding how NGF is regulated, new therapeutics could be developed to target NGF production and to treat low back pain. An additional benefit to targeting TLR2 is that its activation also increases cytokines and catabolic proteases, which further contribute to disc degeneration. By using TLR2 as a therapeutic target, not only could NGF levels be potentially reduced, levels of inflammatory cytokines and catabolic proteases could also be similarly reduced, thus slowing the progression of disc degeneration in addition stopping the associated low back pain.
Author Contributions
E. K., J. B. C., and D. H. R. performed experiments and collected data. E. K., L. S. S., D. H. R., and L. H. designed the study and wrote the manuscript. M. H. W. and J. A. O. assisted in sample collection and results interpretation. All authors reviewed, edited, and approved the final manuscript.
Acknowledgments
We thank members of the Ribeiro-da-Silva laboratory for advice concerning NGF Western blots and Dr. Peter Roughley for critical review and discussion of the manuscript.
This work was funded by Canadian Institutes of Health Research (CIHR) Operating Grant CIHR MOP-119564 (to L. H., L. S. S., and J. A. O.) and a studentship from the McGill Faculty of Medicine and a Fonds de Recherche du Québec–Santé doctoral fellowship (to E. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- NP
- nucleus pulposus
- AF
- annulus fibrosus
- ECM
- extracellular matrix
- TLR
- toll-like receptor
- PGN
- peptidoglycan
- proNGF
- pro-form nerve growth factor
- mNGF
- mature NGF
- CREB
- cAMP-response element-binding protein.
References
- 1. Adams M. A., and Roughley P. J. (2006) What is intervertebral disc degeneration, and what causes it? Spine 31, 2151–2161 [DOI] [PubMed] [Google Scholar]
- 2. Weiler C., Nerlich A. G., Zipperer J., Bachmeier B. E., and Boos N. (2002) 2002 SSE award competition in basic science: expression of major matrix metalloproteinases is associated with intervertebral disc degradation and resorption. Eur. Spine J. 11, 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Le Maitre C. L., Freemont A. J., and Hoyland J. A. (2004) Localization of degradative enzymes and their inhibitors in the degenerate human intervertebral disc. J. Pathol. 204, 47–54 [DOI] [PubMed] [Google Scholar]
- 4. Konttinen Y. T., Kääpä E., Hukkanen M., Gu X. H., Takagi M., Santavirta S., Alaranta H., Li T. F., and Suda A. (1999) Cathepsin G in degenerating and healthy discal tissue. Clin. Exp. Rheumatol. 17, 197–204 [PubMed] [Google Scholar]
- 5. Ariga K., Yonenobu K., Nakase T., Kaneko M., Okuda S., Uchiyama Y., and Yoshikawa H. (2001) Localization of cathepsins D, K, and L in degenerated human intervertebral discs. Spine 26, 2666–2672 [DOI] [PubMed] [Google Scholar]
- 6. Akhatib B., Onnerfjord P., Gawri R., Ouellet J., Jarzem P., Heinegård D., Mort J., Roughley P., and Haglund L. (2013) Chondroadherin fragmentation mediated by the protease HTRA1 distinguishes human intervertebral disc degeneration from normal aging. J. Biol. Chem. 288, 19280–19287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tiaden A. N., Klawitter M., Lux V., Mirsaidi A., Bahrenberg G., Glanz S., Quero L., Liebscher T., Wuertz K., Ehrmann M., and Richards P. J. (2012) Detrimental role for human high temperature requirement serine protease A1 (HTRA1) in the pathogenesis of intervertebral disc (IVD) degeneration. J. Biol. Chem. 287, 21335–21345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel K. P., Sandy J. D., Akeda K., Miyamoto K., Chujo T., An H. S., and Masuda K. (2007) Aggrecanases and aggrecanase-generated fragments in the human intervertebral disc at early and advanced stages of disc degeneration. Spine 32, 2596–2603 [DOI] [PubMed] [Google Scholar]
- 9. Pockert A. J., Richardson S. M., Le Maitre C. L., Lyon M., Deakin J. A., Buttle D. J., Freemont A. J., and Hoyland J. A. (2009) Modified expression of the ADAMTS enzymes and tissue inhibitor of metalloproteinases 3 during human intervertebral disc degeneration. Arthritis Rheum. 60, 482–491 [DOI] [PubMed] [Google Scholar]
- 10. Schaefer L. (2014) Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 289, 35237–35245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klawitter M., Hakozaki M., Kobayashi H., Krupkova O., Quero L., Ospelt C., Gay S., Hausmann O., Liebscher T., Meier U., Sekiguchi M., Konno S., Boos N., Ferguson S. J., and Wuertz K. (2014) Expression and regulation of toll-like receptors (TLRs) in human intervertebral disc cells. Eur. Spine J. 23, 1878–1891 [DOI] [PubMed] [Google Scholar]
- 12. Krock E., Rosenzweig D. H., and Haglund L. (2015) The inflammatory milieu of the degenerate disc: is mesenchymal stem cell-based therapy for intervertebral disc repair a feasible approach? Curr. Stem Cell Res. Ther. 10, 317–328 [DOI] [PubMed] [Google Scholar]
- 13. Pezet S., and McMahon S. B. (2006) Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci. 29, 507–538 [DOI] [PubMed] [Google Scholar]
- 14. Gruber H. E., Ingram J. A., Hoelscher G., Zinchenko N., Norton H. J., and Hanley E. N. Jr. (2008) Brain-derived neurotrophic factor and its receptor in the human and the sand rat intervertebral disc. Arthritis Res. Ther. 10, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freemont A. J., Watkins A., Le Maitre C., Baird P., Jeziorska M., Knight M. T., Ross E. R., O'Brien J. P., and Hoyland J. A. (2002) Nerve growth factor expression and innervation of the painful intervertebral disc. J. Pathol. 197, 286–292 [DOI] [PubMed] [Google Scholar]
- 16. Purmessur D., Freemont A. J., and Hoyland J. A. (2008) Expression and regulation of neurotrophins in the nondegenerate and degenerate human intervertebral disc. Arthritis Res. Ther. 10, R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krock E., Rosenzweig D. H., Chabot-Doré A.-J., Jarzem P., Weber M. H., Ouellet J. A., Stone L. S., and Haglund L. (2014) Painful, degenerating intervertebral discs up-regulate neurite sprouting and CGRP through nociceptive factors. J. Cell Mol. Med. 18, 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wuertz K., and Haglund L. (2013) Inflammatory mediators in intervertebral disk degeneration and discogenic pain. Global Spine J. 3, 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Freemont A. J., Peacock T. E., Goupille P., and Hoyland J. A., O'Brien J., and Jayson M. (1997) Nerve ingrowth into diseased intervertebral disc in chronic back pain. Lancet 350, 178–181 [DOI] [PubMed] [Google Scholar]
- 20. Miyagi M., Millecamps M., Danco A. T., Ohtori S., Takahashi K., and Stone L. S. (2014) ISSLS Prize winner: Increased innervation and sensory nervous system plasticity in a mouse model of low back pain due to intervertebral disc degeneration. Spine 39, 1345–1354 [DOI] [PubMed] [Google Scholar]
- 21. Richardson S. M., Purmessur D., Baird P., Probyn B., Freemont A. J., and Hoyland J. A. (2012) Degenerate human nucleus pulposus cells promote neurite outgrowth in neural cells. PLoS ONE 7, e47735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aoki Y., Ohtori S., Ino H., Douya H., Ozawa T., Saito T., Moriya H., and Takahashi K. (2004) Disc inflammation potentially promotes axonal regeneration of dorsal root ganglion neurons innervating lumbar intervertebral disc in rats. Spine 29, 2621–2626 [DOI] [PubMed] [Google Scholar]
- 23. Kivitz A. J., Gimbel J. S., Bramson C., Nemeth M. A., Keller D. S., Brown M. T., West C. R., and Verburg K. M. (2013) Efficacy and safety of tanezumab versus naproxen in the treatment of chronic low back pain. Pain 154, 1009–1021 [DOI] [PubMed] [Google Scholar]
- 24. Leite V. F., Buehler A. M., El Abd O., Benyamin R. M., Pimentel D. C., Chen J., Hsing W. T., Mazloomdoost D., and Amadera J. E. (2014) Anti-nerve growth factor in the treatment of low back pain and radiculopathy: a systematic review and a meta-analysis. Pain Physician 17, E45–E60 [PubMed] [Google Scholar]
- 25. Gruber H. E., Hoelscher G. L., Bethea S., and Hanley E. N. (2012) Interleukin 1β upregulates brain-derived neurotrophic factor, neurotrophin 3 and neuropilin 2 gene expression, and NGF production in annulus cells. Biotech. Histochem. 87, 506–511 [DOI] [PubMed] [Google Scholar]
- 26. Hayashi S., Taira A., Inoue G., Koshi T., Ito T., Yamashita M., Yamauchi K., Suzuki M., Takahashi K., and Ohtori S. (2008) TNF-α in nucleus pulposus induces sensory nerve growth: a study of the mechanism of discogenic low back pain using TNF-α-deficient mice. Spine 33, 1542–1546 [DOI] [PubMed] [Google Scholar]
- 27. Abe Y., Akeda K., An H. S., Aoki Y., Pichika R., Muehleman C., Kimura T., and Masuda K. (2007) Proinflammatory cytokines stimulate the expression of nerve growth factor by human intervertebral disc cells. Spine 32, 635–642 [DOI] [PubMed] [Google Scholar]
- 28. Alkhatib B., Rosenzweig D. H., Krock E., Roughley P. J., Beckman L., Steffen T., Weber M. H., Ouellet J. A., and Haglund L. (2014) Acute mechanical injury of the human intervertebral disc: link to degeneration and pain. Eur. Cell Mater. 28, 98–110 [DOI] [PubMed] [Google Scholar]
- 29. Gawri R., Rosenzweig D. H., Krock E., Ouellet J. A., Stone L. S., Quinn T. M., and Haglund L. (2014) High mechanical strain of primary intervertebral disc cells promotes secretion of inflammatory factors associated with disc degeneration and pain. Arthritis Res. Ther. 16, R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawai T., and Akira S. (2006) TLR signaling. Cell Death Differ. 13, 816–825 [DOI] [PubMed] [Google Scholar]
- 31. Lees S., Golub S. B., Last K., Zeng W., Jackson D. C., Sutton P., and Fosang A. J. (2015) Bioactivity in an Aggrecan 32-mer fragment is mediated via toll-like receptor 2. Arthritis Rheumatol. 67, 1240–1249 [DOI] [PubMed] [Google Scholar]
- 32. Oegema T. R. Jr., Johnson S. L., Aguiar D. J., and Ogilvie J. W. (2000) Fibronectin and its fragments increase with degeneration in the human intervertebral disc. Spine 25, 2742–2747 [DOI] [PubMed] [Google Scholar]
- 33. Ruel N., Markova D. Z., Adams S. L., Scanzello C., Cs-Szabo G., Gerard D., Shi P., Anderson D. G., Zack M., An H. S., Chen D., and Zhang Y. (2014) Fibronectin fragments and the cleaving enzyme ADAM-8 in the degenerative human intervertebral disc. Spine 39, 1274–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melrose J., Smith S. M., Fuller E. S., Young A. A., Roughley P. J., Dart A., and Little C. B. (2007) Biglycan and fibromodulin fragmentation correlates with temporal and spatial annular remodelling in experimentally injured ovine intervertebral discs. Eur. Spine J. 16, 2193–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gruber H. E., Hoelscher G. L., Bethea S., Ingram J., Cox M., and Hanley E. N. Jr. (2015) High-mobility group box-1 gene, a potent proinflammatory mediators, is upregulated in more degenerated human discs in vivo and its receptor upregulated by TNF-α exposure in vitro. Exp. Mol. Pathol. 98, 427–430 [DOI] [PubMed] [Google Scholar]
- 36. Quero L., Klawitter M., Schmaus A., Rothley M., Sleeman J., Tiaden A. N., Klasen J., Boos N., Hottiger M. O., Wuertz K., and Richards P. J. (2013) Hyaluronic acid fragments enhance the inflammatory and catabolic response in human intervertebral disc cells through modulation of toll-like receptor 2 signalling pathways. Arthritis Res. Ther. 15, R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greg Anderson D., Li X., Tannoury T., Beck G., and Balian G. (2003) A fibronectin fragment stimulates intervertebral disc degeneration in vivo. Spine 28, 2338–2345 [DOI] [PubMed] [Google Scholar]
- 38. Aota Y., An H. S., Homandberg G., Thonar E. J., Andersson G. B., Pichika R., and Masuda K. (2005) Differential effects of fibronectin fragment on proteoglycan metabolism by intervertebral disc cells: a comparison with articular chondrocytes. Spine 30, 722–728 [DOI] [PubMed] [Google Scholar]
- 39. Burbach G. J., Kim K. H., Zivony A. S., Kim A., Aranda J., Wright S., Naik S. M., Caughman S. W., Ansel J. C., and Armstrong C. A. (2001) The neurosensory tachykinins substance P and neurokinin A directly induce keratinocyte nerve growth factor. J. Invest. Dermatol. 117, 1075–1082 [DOI] [PubMed] [Google Scholar]
- 40. Zhao P., Qiao J., Huang S., Zhang Y., Liu S., Yan L. Y., Hsueh A. J., and Duan E. K. (2011) Gonadotrophin-induced paracrine regulation of human oocyte maturation by BDNF and GDNF secreted by granulosa cells. Hum. Reprod. 26, 695–702 [DOI] [PubMed] [Google Scholar]
- 41. Dussault A. A., and Pouliot M. (2006) Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol. Proced. Online 8, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 43. Bradshaw R. A., Pundavela J., Biarc J., Chalkley R. J., Burlingame A. L., and Hondermarck H. (2015) NGF and ProNGF: regulation of neuronal and neoplastic responses through receptor signaling. Adv. Biol. Regul. 58, 16–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Caruso R., Warner N., Inohara N., and Núñez G. (2014) NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wuertz K., Vo N., Kletsas D., and Boos N. (2012) Inflammatory and catabolic signalling in intervertebral discs: the roles of NF-κB and MAP kinases. Eur. Cell Mater. 23, 103–119; discussion 119–120 [DOI] [PubMed] [Google Scholar]
- 46. Li J., Yuan W., Jiang S., Ye W., Yang H., Shapiro I. M., and Risbud M. V. (2015) Prolyl-4-hydroxylase domain protein 2 controls NF-κB/p65 transactivation and enhances the catabolic effects of inflammatory cytokines on cells of the nucleus pulposus. J. Biol. Chem. 290, 7195–7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang X. H., Hong X., Zhu L., Wang Y. T., Bao J. P., Liu L., Wang F., and Wu X. T. (2015) Tumor necrosis factor α promotes the proliferation of human nucleus pulposus cells via nuclear factor-κB, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. Exp. Biol. Med. (Maywood) 240, 411–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miklic S., Juric D. M., and Carman-Krzan M. (2004) Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int. J. Dev. Neurosci. 22, 119–130 [DOI] [PubMed] [Google Scholar]
- 49. Mocchetti I., De Bernardi M. A., Szekely A. M., Alho H., Brooker G., and Costa E. (1989) Regulation of nerve growth factor biosynthesis by β-adrenergic receptor activation in astrocytoma cells: a potential role of c-Fos protein. Proc. Natl. Acad. Sci. U.S.A. 86, 3891–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Colangelo A. M., Pani L., and Mocchetti I. (1996) Correlation between increased AP-1NGF binding activity and induction of nerve growth factor transcription by multiple signal transduction pathways in C6–2B glioma cells. Brain Res. Mol. Brain Res. 35, 1–10 [DOI] [PubMed] [Google Scholar]
- 51. Hengerer B., Lindholm D., Heumann R., Rüther U., Wagner E. F., and Thoenen H. (1990) Lesion-induced increase in nerve growth factor mRNA is mediated by c-fos. Proc. Natl. Acad. Sci. U.S.A. 87, 3899–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matsuoka I., Meyer M., and Thoenen H. (1991) Cell-type-specific regulation of nerve growth factor (NGF) synthesis in non-neuronal cells: comparison of Schwann cells with other cell types. J. Neurosci. 11, 3165–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Colangelo A. M., Johnson P. F., and Mocchetti I. (1998) β-adrenergic receptor-induced activation of nerve growth factor gene transcription in rat cerebral cortex involves CCAAT/enhancer-binding protein delta. Proc. Natl. Acad. Sci. U.S.A. 95, 10920–10925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bruno M. A., and Cuello A. C. (2006) Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. U.S.A. 103, 6735–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lewin G. R., and Nykjaer A. (2014) Pro-neurotrophins, sortilin, and nociception. Eur. J. Neurosci. 39, 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lim K. C., Tyler C. M., Lim S. T., Giuliano R., and Federoff H. J. (2007) Proteolytic processing of proNGF is necessary for mature NGF regulated secretion from neurons. Biochem. Biophys. Res. Commun. 361, 599–604 [DOI] [PubMed] [Google Scholar]