

Abstract
Methanol expression regulator 1 (Mxr1p) is a zinc finger protein that regulates the expression of genes encoding enzymes of the methanol utilization pathway in the methylotrophic yeast Pichia pastoris by binding to Mxr1p response elements (MXREs) present in their promoters. Here we demonstrate that Mxr1p is a key regulator of acetate metabolism as well. Mxr1p is cytosolic in cells cultured in minimal medium containing a yeast nitrogen base, ammonium sulfate, and acetate (YNBA) but localizes to the nucleus of cells cultured in YNBA supplemented with glutamate or casamino acids as well as nutrient-rich medium containing yeast extract, peptone, and acetate (YPA). Deletion of Mxr1 retards the growth of P. pastoris cultured in YNBA supplemented with casamino acids as well as YPA. Mxr1p is a key regulator of ACS1 encoding acetyl-CoA synthetase in cells cultured in YPA. A truncated Mxr1p comprising 400 N-terminal amino acids activates ACS1 expression and enhances growth, indicating a crucial role for the N-terminal activation domain during acetate metabolism. The serine 215 residue, which is known to regulate the expression of Mxr1p-activated genes in a carbon source-dependent manner, has no role in the Mxr1p-mediated activation of ACS1 expression. The ACS1 promoter contains an Mxr1p response unit (MxRU) comprising two MXREs separated by a 30-bp spacer. Mutations that abrogate MxRU function in vivo abolish Mxr1p binding to MxRU in vitro. Mxr1p-dependent activation of ACS1 expression is most efficient in cells cultured in YPA. The fact that MXREs are conserved in genes outside of the methanol utilization pathway suggests that Mxr1p may be a key regulator of multiple metabolic pathways in P. pastoris.
Keywords: gene regulation, metabolism, promoter, transcription factor, yeast, acetyl-CoA synthetase, Pichia pastoris, acetate metabolism
Introduction
The ability of yeast cells to grow in the presence of diverse carbon sources offers a unique opportunity to study various metabolic pathways, which is not always feasible in higher eukaryotic systems. In addition to glucose, yeast cells can utilize acetate, ethanol, glycerol, or fatty acids as the sole source of carbon, and the study of their metabolism and regulation has been one of the fascinating areas of biochemistry. The regulation of metabolic pathways of respiratory yeasts such as Pichia pastoris has not been as well studied as that of Saccharomyces cerevisiae despite the extensive use of the former for the commercial production of recombinant proteins. P. pastoris, a methylotrophic yeast, can metabolize a number of compounds, such as glycerol, methanol, acetate, and oleic acid, in addition to glucose. However, very little information is available on the transcriptional regulation of metabolic pathways other than the methanol utilization (mut)2 pathway in this yeast species. The expression of genes of the mut pathway is regulated by at least three zinc finger proteins (1–6). Of these, methanol expression regulator 1 (Mxr1p) activates the expression of genes of the mut pathway by binding to Mxr1p response elements (MXREs) in their promoters (2, 3). Rop1p has the same DNA binding specificity as Mxr1p and functions as a repressor of genes of the mut pathway in P. pastoris cultured in nutrient-rich medium containing yeast extract, peptone, and methanol (YPM) but not minimal medium containing a yeast nitrogen base, ammonium sulfate, and methanol (YNBM) (4, 5). Trm1p is also essential for the expression of genes of the mut pathway (6). However, its mechanism of action remains unknown. The differential regulation of methanol metabolism in YNBM and YPM by Mxr1p and Rop1p led us to investigate the transcriptional regulation of other metabolic pathways in cells cultured in minimal and nutrient-rich media. In this study, we demonstrate that Mxr1p regulates acetate metabolism only in cells cultured in nutrient-rich medium containing yeast extract, peptone, and acetate (YPA) but not minimal medium containing yeast nitrogen base, ammonium sulfate and acetate (YNBA).
Experimental Procedures
Yeast and Bacterial Strains
P. pastoris (GS115, his−) was cultured in either nutrient-rich medium (1.0% yeast extract and 2.0% peptone) containing 2.0% glucose (YPD) or 2.0% acetate (YPA) or minimal yeast nitrogen base medium (0.17% yeast nitrogen base without amino acids and 0.5% ammonium sulfate) supplemented with 2.0% glucose (YNBD) or 2.0% sodium acetate (YNBA). Casamino acids (CAAs) and glutamate were added to YNBA medium to final concentrations of 1.0% and 0.5%, respectively. P. pastoris strains were grown at 30 °C in an orbital shaker at 180 rpm. For all growth and β-galactosidase assays, colonies were first cultured overnight in YNBD medium supplemented with histidine, washed with sterile water, and shifted to the respective media with an initial optical density of ∼0.1. Escherichia coli DH10β and BL21 (DE3) strains were used for plasmid isolation and recombinant protein expression, respectively. Bacterial and yeast transformations were done by electroporation (Gene Pulser, Bio-Rad) according to the instructions of the manufacturer.
Antibodies and Other Reagents
Oligonucleotides were purchased form Sigma-Aldrich (Bangalore, India). Anti-His tag, anti-c-myc tag, and anti-FLAG tag antibodies were purchased from Thermo Scientific (Bangalore, India), Merck Millipore (Bangalore, India), and Sigma-Aldrich, respectively.
Construction of the P. pastoris Δmxr1 Strain
In the Δmxr1 strain, Mxr1 encoding 320 N-terminal amino acids was replaced by a zeocin expression cassette. This deletion construct was generated by four different PCR reactions using P. pastoris genomic DNA and the pGAPZA vector (Invitrogen) as templates as well as a series of overlapping and non-overlapping primers. To begin with, the Mxr1 promoter (−997 to −1 bp) was amplified from P. pastoris genomic DNA using primer pair 1F (5′-TGTGGATCTTATCTATAGCAAGGCTATC-3′, −997 to −970 bp of the Mxr1 promoter) and 1R (5′-GCTATGGTGT GTGGGGGATCCGCAtgtgcgtgggataaagtcatcaaac-3′, 986–961 bp of the pGAPZA vector (uppercase) and −1 to −25 bp of the Mxr1 promoter (lowercase). In another PCR reaction, a 1.2-kb region of the pGAPZA vector (1419–2591 bp) comprising the TEF1 and EM7 promoters, the ShBle gene, and the CYC1 transcription termination region was amplified using the primer pair 2F (5′-GTTTGATGACTTTAT CCCACGCACA tgcggatcccccacacaccatag c-3′, −25 to −1 bp of the Mxr1 promoter (uppercase) and positions 962–986 bp of the pGAPZA vector (lowercase)) and 2R (5′-CTTCGTAAAAAGAGTAGCCATTCAAgctcacatgttggtctccagcttgc 3′, +945 to +970 bp of Mxr1 (uppercase) and 2160–2136 bp of the pGAPZA vector (uppercase)). In the third PCR reaction, 1000 bp of the Mxr1 gene between 971 and 1970 bp was amplified from P. pastoris genomic DNA using the primer pair 3F (5′-GCAAGCTGGAGACCAACATGTGAGC ttgaatggctactctttttacgaag-3′, 2136–160 bp of pGAPZA (uppercase) and 945–970 bp of Mxr1 (lowercase)) and 3R (5′-TTATCCAAAGGTTTGTTGATGTTGAAAAG-3′, 1942–1970 bp of Mxr1). All three PCR products containing overlapping regions were pooled (50 ng each) and used as templates in a final PCR reaction along with primers 1F and 3R. The 3.2-kb PCR product thus obtained was transformed into the P. pastoris GS115 strain to generate the zeocin-resistant Δmxr1 strain, in which the region encoding the 320 N-terminal amino acids of Mxr1p was replaced by a zeocin expression cassette.
Characterization of the P. pastoris Δmxr1 Strain
Deletion of the region encoding the DNA binding domain was confirmed by PCR as well as Southern blotting. PCR was carried out with genomic DNA as a template and the primer pair P1 (5′-ATGAGCAATCTACCCC CAAC-3′) and P2 (5′-GCCGGCCAGTTTCTGAACTTTTCG-3′), which amplifies a region between +1 and +435 bp of Mxr1. This PCR product was then radiolabeled and used as probe A in Southern blotting. Probe B for Southern blotting was generated by PCR amplification of a region between +1201 and +1250 bp of Mxr1 using the primer pair P3 (5′-ACTTCTTCTAATGCCACAATTTCGC-3′) and P4 (5′-TAAGAAAC GGTTGGTGAATGAATC-3′). For Southern blotting, genomic DNA was digested with PstI and BamHI.
Construction of P. pastoris Expressing Chromosomally FLAG-tagged Mxr1p (Mxr1pFLAG)
A zeocin resistance expression cassette fused to the gene encoding FLAG-tagged Mxr1p was obtained by five different PCR reactions. First, a 1.2-kb zeocin expression cassette (1419–2591 bp) was amplified from the pGAPZA vector using primers pair 1F (5′-TGCGGATCCCCCACACACCATAGC-3′, 962–986 bp of the pGAPZA vector) and 1R (5′-CTATAGATAAGATCCACAA TTTTCTCAAtgctcacatgttggtctccagcttg-3′, −1007 to −979 bp of the Mxr1 promoter (uppercase) and 2161–2136 bp of the pGAPZA vector (lowercase)). In another PCR reaction, 1007 bp of Mxr1 encompassing −1007 to −1 bp were amplified from P. pastoris genomic DNA using the primer pair 2F (5′-CAAGCTGGAGACCAACATGTGAGCAttgagaaaattgtggatcttatctatag-3′, 2161-2136 bp of the pGAPZA vector (uppercase) and −1007 to −979 bp of Mxr1 (lowercase)) and 2R (5′-CGTCATGGTCTTTGTAG TCGCTCATtgtgcgtgggataaagtcatcaaac-3′, 928–953 bp encoding a 3× FLAG tag (uppercase) and −25 to −1 bp of Mxr1 (lowercase)). In the third PCR reaction, a 72-bp sequence encoding a 3× FLAG tag was amplified from the 3× FLAG vector (Sigma-Aldrich) using the primer pair 3F (5′-GTTTGATGAC TTTATCCCACGCACAatgagcgactacaaagaccatgacg-3′, −25 to −1 bp of Mxr1 (uppercase) and 928–953 bp encoding a FLAG tag (lowercase)) and 3R (5′-GAACCAAAAGTTGGGGGTAGATTGCTcttgtcatcgtcatccttgtaatc-3′, +4 to +23 bp of Mxr1 (uppercase) and 976–1000 bp encoding a FLAG tag (lowercase)). In the fourth PCR reaction, the gene encoding 797 N-terminal amino acids of Mxr1p was amplified using the primer pair 4F (5′-GATTACAAGGATGACGATGACAAGagcaatctacccccaacttttggttc 3′, 976–1000 bp encoding a FLAG tag (uppercase) and +4 to +23 bp of Mxr1 (lowercase)) and 4R (5′-tcagcatcttcaacgggcatcatttgtggg-3′, +797 to +767 bp of Mxr1). The final PCR reaction was carried out using the above four PCR products (50 ng each) together with the 1F and 4R primers to generate a 3076-bp expression cassette encoding FLAG-tagged Mxr1p, which was transformed into P. pastoris by electroporation. Zeocin-resistant colonies in which the PpFLAG-MXR1 expression cassette was integrated into the chromosomal Mxr1 locus by homologous recombination were selected by plating the transformants on YPD plates containing zeocin (100 μg/ml). The P. pastoris strain expressing chromosomally tagged Mxr1pFLAG was designated Pp-Mxr1FLAG.
Construction of P. pastoris Strains Overexpressing Mxr1p, Adr1p, Mxr1pN400, and Mxr1pS215A
For overexpression of Mxr1p and Adr1p, the pGAPBA vector containing a blasticidin resistance gene was generated by PCR amplification of the blasticidin resistance expression cassette from the pPIC6 vector (Life Technologies) using the primer pair 5′-TGCGGATCCCCCACACACCATAGCT-3′ and 5′-CTCACATGTTGGTCTCCAGCTTG-3′ and cloning it into the SmaI site of the pGAPZA vector.
The gene encoding full-length Mxr1p was obtained by PCR amplification of P. pastoris (GS115) genomic DNA using the primer pair 5′-CGGGGTACCAATGAGCAATCTACCCCCAAC-3′ and 5′-ATAAGAATGCGGCCGCAGACACCACCATCTAGTCG-3′. Similarly, the gene encoding full-length Adr1p was amplified from S. cerevisiae genomic DNA using the primer pair 5′-CGGGGTACCATGGCTAACGTAGAAAAACCAAACGATTG-3′ and 5′-ATAAGAATGCGGCCGCTACTGTTTCCCTTTAGATGATTTTCCAAAG-3′. The KpnI and NotI restriction sites are underlined. Following restriction digestion with KpnI and NotI, the Mxr1 and Adr1 genes were cloned into the KpnI- and NotI-digested pGAPBA vector to obtain the pGAPMxr1 and pGAPAdr1 vectors, respectively.
The pGAPMxr1N400 vector overexpressing 400 N-terminal amino acids of Mxr1p was generated by PCR amplification of the Mxr1 gene encoding 400 N-terminal amino acids of Mxr1p from P. pastoris genomic DNA using the primer pair 5′-CGGGGTACCAATGAGCAATCTACCCCCAAC-3′ and 5′-ATAAGAATGCGGCCGCAGCATGATAACGTGTTAGAGAAAGTCTG-3′. The KpnI and NotI restriction sites are underlined. After restriction digestion, the PCR product was cloned into the pGAPBA vector. The resultant plasmid was electroporated to the Δmxr1 strain, transformants were selected using blasticidin antibiotic, and the strain was named Pp-Mxr1pN400.
The S215A mutant of Mxr1p was generated by site-directed mutagenesis using the QuikChange method (Stratagene) using the pGAPMXR1 vector as a template and the primers 5′-CTTGGACTAAGAAGAGCTGCCTTCTCCGCCGTTAGTGG-3′ and 5′-CCACTAACGGCGGAGAAGG CAGCTCTTCTTAGTCCAAG-3′. The recombinant plasmids were electroporated into the Δmxr1 strain to generate P. pastoris strains overexpressing Mxr1p, Adr1p, Mxr1pN400, and Mxr1pS215A, which were designated PpMxr1-OE, PpAdr1-OE, Pp-Mxr1N400-OE, and Pp-Mxr1S215A-OE, respectively.
The expression levels of Mxr1p between the Pp-Mxr1FLAG and PpMxr1-OE strains were compared by Western blotting analysis using anti-myc/anti-FLAG antibodies. Mxr1 mRNA levels were examined by qPCR using the primer pair 5′-TGCTGAAACTTGGATGAAC-3′ and 5′-TCGGATATAATAGGCTCTGAAT-3′, which amplifies a 199-bp region between 792 and 991 bp of Mxr1. Because the S. cerevisiae Adr1 promoter is not active in P. pastoris (data not shown), Adr1p expression in the PpAdr1-OE strain was examined by Western blotting analysis using anti-c-myc antibodies. Adr1 mRNA was detected in the PpAdr1-OE strain by RT-PCR using the primer pair 5′-CAACCAACCTGATTTCGTCG-3′ and 5′-ATGAGGAGAAATTGGAGAGTTTGATAG-3′, which amplifies a 189-bp region between 1251 and 1440 bp of Adr1.
Construction of P. pastoris Strains Expressing His-tagged ACS1 (pACS1His)
The gene encoding ACS1 was amplified along with a 1-kb promoter from the genomic DNA of P. pastoris using the primer pair 5′-CGCGGATCCAAAACCACCAGCTAGTACAGAG-3′ and 5′-CCCAAGCTTCTAATGATGATGATGATGATGTTTGGGCATCCCTTTTAACGG-3′ and cloned into the pIB3 vector. The BamHI and HindIII restriction sites are underlined. The recombinant plasmid was transformed into different P. pastoris strains, and expression of histidine-tagged ACS1 (ACS1His) was examined by Western blotting using anti-His tag antibodies.
Expression of β-Gal from the ACS1 Promoter (pACS)
pACS-lacZ constructs consisting of the E. coli lacZ gene downstream of pACS were generated in three PCR reactions. pACS1-lacZ, consisting of 1049 bp of pACS upstream of lacZ, was generated by PCR using P. pastoris genomic DNA as a template and the primer pair 1F (5′-CGCGGATCCAAAACCACCAGCTAGTACAGAG-3′, −1049 to −1027 bp of pACS1) and 1R (5′-CGTTGTAAAACGACGGCCATaattgatcaacaactaagtcgtatc-3′,−20 to −1 bp of pACS1 (uppercase) and +1 to +25 bp of the E. coli lacZ gene (lowercase)). The BamHI site is underlined. In the second PCR reaction, the E. coli lacZ gene was amplified from pFRT/lacZeo (Life Technologies) using the primer pair 2F (5′-GATACGACTTAGTTGTTGATCAATTatggccgtcgttttacaacg-3′, −25 to −1 bp of pACS1 (uppercase) and +1 to +20 bp of lacZ (lowercase)) and 2R (5′-CCCAAGCTTCTAGTCCTGCTCCTCGGCC-3′, +3491 to +3510 bp of lacZ). The HindIII site is underlined. In the third PCR reaction, the PCR products from the first two reactions were used as templates and amplified using the 1F and 2R primers to get the pACS1-lacZ expression cassette, which was digested with BamHI and HindIII and cloned into pIB3 (Addgene) to generate pIB3-pACS1-lacZ.
To generate pIB3-pACS1-lacZ containing 809 bp of pACS upstream of lacZ, a PCR reaction was carried out using pIB3-pACS1-lacZ as a template and the primer pair 3F (5′-CGCGGATCCTCCGCCATCCGACAGCAC-3′) and 2R. To generate pIB3-pACS3-lacZ, consisting of 736 bp of pACS upstream of lacZ, a PCR reaction was carried out using the primer pair 4F (CGCGGATCCAAGTGGGATATGATTTCGTTCCTC-3′) and 2R and pIB3-pACS1-lacZ as a template. To generate pIB3-pACS4-lacZ, containing 449 bp of pACS upstream of lacZ, a PCR reaction was carried out using the primer pair 5F (5′-CGCGGATCCTACCAGCGTCTCTCTTCTGTGTG-3′) and2R and pIB3-pACS1-lacZ as a template. The BamHI sites in the primers are underlined. The PCR products were digested with BamHI and HindIII and cloned into pIB3. The resultant plasmids consisted of −809 bp (pIB3-pACS2-lacZ), −736 bp (pIB3-pACS3-lacZ), and −449 bp (pIB3-pACS4-lacZ) of pACS upstream of lacZ.
Mutations were introduced into the MXREs of pACS using the QuikChange method (Stratagene) using primer pairs carrying appropriate mutations. The primer pair 5′-GACAGCACACTACCGTCATCCCATCTTCCGTAAGACCAAAAC-3′ and 5′-GTTTTGGTCTTACGGAAGATGGGATGACGGTAGTGTGCTGTC-3′ was used to generate pIB3-pACS1-M1-lacZ. pIB3-pACS1-lacZ was used as the template for PCR. The primer pair 5′-CATCATTCTCTCTTCCCATCCTAGGTTGGTG-3′ and 5′-CACCAACCTAGGTTGGGAAGAGAGAATGATG-3′ was used to generate pIB3-pACS1-M2-lacZ. pIB3-pACS1-lacZ was used as the template for PCR. The primer pair 5′-GACAGCACACTACCGTCATCCCATCTTCCGTAAGACCAAAAC-3′ and 5′-GTTTTGGTCTTACGGAAGATGGGATGACGGTAGTGTGCTGTC-3′ was used to generate pIB3-pACS1-M3-lacZ. pIB3-pACS1-M2-lacZ was used as the template. Mutations are underlined.
A 10-bp sequence (5′-ccaaaacatc-3′) was inserted between the two MXREs of pACS1-lacZ as follows. The PCR reaction was carried out with P. pastoris genomic DNA as a template and the primers i10F1 (5′-CGCGGATCCAAAACCACCAGCTAGTACAGAG-3′ (−1049 to −1027 bp of pACS1)) and i10R1 (5′-GAAGAGAGAATGATGTTTTGGgatgttttggTCTTACGGAAGAGGGGATGAC-3′). The BamHI site is underlined. In the second PCR reaction, the pACS1 promoter along with the lacZ gene was amplified from pIB3-pACS1-lacZ using the primers i10F2 (5′-GTCATCCCCTCTTCCGTAAGAccaaaacatcCCAAAACATCATTCTCTCTTC-3′) and i10R2 (5′-CCCAAGCTTCTAGTCCTGCTCCTCGGCC 3′). The BamHI and HindIII sites are underlined. The 10-bp insertion sequence in the primers is shown in lowercase. In third and final PCR reaction, the PCR products of first and second PCR reactions were used as templates along with the i10F1 and i10R2 primer pair. The PCR product was digested with BamHI and HindIII and cloned into the pIB3 vector to obtain pIB3-pACS1(i10)-lacZ.
A 20-bp sequence (5′-ccaaaacatcccaaaacatc-3′) was inserted between the two MXREs of pACS1-lacZ as follows. The PCR reaction was carried out with P. pastoris genomic DNA as a template and the primers i10F1 (5′-CGCGGATCCAAAACCACCAGCTAGTACAGAG-3′ (−1049 to −1027 bp of pACS1)) and i20R1 (5′-GAAGAGAGAATGATGTTTTGGgatgttttgggatgttttggTCTTACGGAAGAGGGGATGAC-3′). The BamHI site is underlined. In the second PCR reaction, the pACS1 promoter along with the lacZ gene was amplified from pIB3-pACS1-lacZ using primers i20F2 (5′-GTCATCCCCTCTTCCGTAAGAccaaaacatcccaaaacatcCCAAAACATCATTCTCTCTTC-3′) and i10R2 (5′-CCCAAGCTTCTAGTCCTGCTCCTCGGCC-3′). The BamHI and HindIII sites are underlined. The 20-bp insertion sequence in the primers is shown in lowercase. In the third and final PCR reaction, the PCR products of the first and second PCR reactions were used as templates along with the i10F1 and i10R2 primer pair. The PCR product was digested with BamHI and HindIII and cloned into the pIB3 vector to obtain pIB3-pACS1(i20)-lacZ. Recombinant plasmids were transformed into GS115 and Δmxr1 strains and plated on YNBD-His− agar plates, and β-galactosidase assays were carried out with three individual colonies for each transformant essentially as described previously (7).
DNA-Protein Interactions
Recombinant Mxr1pN150 was purified from E. coli extracts, and its ability to bind to radiolabeled oligonucleotides containing pACS-MXREs was examined by EMSA essentially as described previously (2, 5).
Mxr1 encoding 400 N-terminal amino acids was expressed as a GST fusion protein (Mxr1pN400) in E. coli by PCR amplification of Mxr1N400 using the primer pair 5′-CGCGGATCCATGAGCAATCTACCCCCAACT-3′ and 5′-TAAGCTAGCGGCCGCAGCATGATAACGTGTTAGAGAAAG-3′ and cloninginto the pGEX4T1 vector (GE Healthcare). The BamHI and NotI restriction sites in the primers are underlined. The recombinant plasmid was transformed into the E. coli BL21(DE3)pLysS strain, and recombinant protein expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside. Mxr1pN400 was purified by glutathione affinity chromatography according to the instructions of the manufacturer (GE Healthcare).
Subcellular Localization of FLAG-Mxr1p
Subcellular localization of FLAG-Mxr1p in P. pastoris cells cultured in YNBA and YPA was examined by immunofluorescence using a fluorescence microscope (Leica). Mouse anti-FLAG antibodies and TRITC-conjugated rabbit anti-mouse antibodies were used. Immunofluorescence was carried out essentially as described previously (5).
Northern Blotting Analysis and Quantitative Real-time PCR
RNA isolation, semiquantitative PCR, and Northern blotting analysis were carried out as described previously (5). Real-time PCR was performed using iQ SYBR Green Super Mix and a iQ5 multicolor real-time PCR thermal cycler (iCycler, Bio-Rad). The levels of mRNA expression in Δmxr1 and P. pastoris strains overexpressing Mxr1p or Mxr1pN400 relative to GS115 were normalized to tubulin mRNA. Data were analyzed by the comparative Ct method for relative quantification (ΔΔCt method), which describes the change in expression of the target genes in a test sample relative to a calibrator sample.
Statistical Analysis
Statistical tests were carried out by one-way analysis of variance followed by Tukey's multiple comparison test using GraphPad Prism 5. Data are presented as mean ± S.D. (*, p < 0.05; **, p < 0.005; ***, p < 0.0005; ns, not significant).
Results
Regulation of Acetate Metabolism by Mxr1p in P. pastoris Cultured in YPA Medium
To examine whether Mxr1p has a role in the regulation of acetate metabolism, we generated a Δmxr1 strain in which the 320 N-terminal amino acids of Mxr1p, including the zinc finger DNA binding domain, were replaced by a zeocin expression cassette (Fig. 1A). The deletion was confirmed by PCR using primers against the DNA binding domain of Mxr1 from genomic DNA (Fig. 1B) and by Southern blotting analysis using 32P-labeled probes A and B, which hybridize to regions within and outside of the region encoding the DNA binding domain, respectively (Fig. 1C). The Δmxr1 strain was unable to grow in YNBM (Fig. 1D) as expected. Deletion of Mxr1 had no effect on the growth of P. pastoris cultured in minimal medium, YNBA medium, and YNBA medium supplemented with 0.5% glutamate (YNBA + Glu) (Fig. 1, E and F). However, Δmxr1 exhibited impaired growth when cultured in YNBA medium supplemented with 1% casamino acids (YNBA +CAA) and nutrient-rich YPA medium (Fig. 1, G and H). To understand the function of Mxr1p during acetate metabolism, we examined the expression of key genes involved in acetate metabolism, such as ACS1 and ACS2, encoding acetyl-CoA synthetase (ACS), which catalyzes the first step in acetate metabolism as well as ACH encoding acetyl-CoA hydrolase catalyzing the conversion of acetyl-CoA to acetate (Fig. 1I). Northern blotting analysis revealed that Mxr1p activates the expression of ACS1, but not ACS2 and ACH, in cells cultured in YPA medium (Fig. 1J). A P. pastoris strain expressing genomically FLAG-tagged Mxr1p (Pp-Mxr1pFLAG) from its own promoter was generated, and subcellular localization of Mxr1pFLAG was studied in cells cultured in YNBA, YPA, YNBA + CAA, or YNBA +Glu media using anti-FLAG antibodies. Mxr1pFLAG was cytosolic in cells cultured in YNBA medium but localized to the nucleus of cells cultured in YPA, YNBA + CAA, and YNBA + Glu media (Fig. 1, K and L). Although Mxr1p was localized prominently in the nucleus of almost all cells cultured in YPA medium, nuclear localization of Mxr1p was highly variable among cells cultured in YNBA + CAA or YNBA + Glu media (Fig. 1L).
FIGURE 1.

Identification of Mxr1p as a key regulator of acetate metabolism and ACS1 expression. A, strategy for the generation of the Δmxr1 strain. The positions of the P1 and P2 primers used in PCR reaction are indicated. The restriction sites in Mxr1 and the size of the restriction fragments are indicated. Regions that hybridize to radiolabeled probes A and B in Southern blotting are indicated as a and b, respectively. B, generation of a 960-bp product by PCR amplification of genomic DNA isolated from GS115, but not Δmxr1, using the P1 and P2 primer pair, confirming the deletion of the region encoding the DNA binding domain. C, Southern blotting analysis of genomic DNA restricted with PstI and BamHI. Radiolabeled probes A and B hybridize to regions a (+1 to +554 bp) and b (+1200 to +1500 bp), located within and outside of the regions encoding the DNA binding domain, respectively. D–H, growth curves of the GS115 and Δmxr1 strains cultured in various media as indicated. Error bars indicate mean ± S.D. (n = 3). CAAs and Glu were added to YNBA medium to final concentrations of 1% and 0.5%, respectively. I, schematic of acetyl-CoA synthesis and degradation. J, Northern blotting analysis of ACS1, ACS2, and ACH. RNA was isolated from cells cultured in YPA medium for 12 h. K, immunofluorescence studies to examine the subcellular localization of Mxr1pFLAG in cells cultured in YNBA, YPA, YNBA + CAA, and YNBA + Glu media using mouse anti-FLAG antibodies and TRITC-conjugated goat anti-mouse antibodies. DAPI was used a nuclear marker. L, nuclear localization of Mxr1pFLAG in a large number of cells cultured in YPA, YNBA + CAA, and YNBA + Glu media.
Overexpression of Mxr1p, but Not Adr1p, Enhances ACS1 Expression and Growth of P. pastoris
To confirm the role of Mxr1p in the regulation of ACS1 expression, the Pp-Mxr1-OE strain was generated, in which Mxr1 was overexpressed as myc-tagged protein (Mxr1pMyc) from the GAPDH promoter. Mxr1pMyc overexpressed from the GAPDH promoter was present at much higher levels than Mxr1pFLAG expressed from its own promoter, as evident from Western blotting analysis (Fig. 2, A and B). The mRNA levels of Mxr1Myc were also higher than that of Mxr1FLAG, as evident from qPCR (Fig. 2C). We also generated the Pp-Adr1-OE strain to examine the ability of S. cerevisiae Adr1 encoding Adr1p (alcohol dehydrogenase II synthesis regulator) to complement Mxr1p function because Adr1p is considered to be a homologue of Mxr1p (1, 2, 8, 9). Expression of Myc-tagged Adr1p (Adr1pMyc) from the GAPDH promoter was confirmed by Western blotting using anti-c-myc antibodies (Fig. 2D) as well as semiquantitative RT-PCR (Fig. 2E). Overexpression of Mxr1p, but not Adr1p, results in a significant increase in ACS1 mRNA, as evident from Northern blotting analysis (Fig. 2F). Mxr1 overexpression results in a much higher level of ACS1His than in GS115, as evident from Western blotting (Fig. 2, G and H). An Mxr1p-mediated increase in ACS1 expression results in a significant increase in the rate of growth of P. pastoris in YPA medium (Fig. 2I). Furthermore, an Mxr1p-mediated increase in ACS1His was not observed in cells cultured in YPG medium containing glycerol as a carbon source (Fig. 2J), indicating that the presence of acetate in the nutrient-rich medium is essential for Mxr1p-mediated activation of ACS1 expression.
FIGURE 2.

The effect of overexpression of Mxr1p and Adr1p on ACS1 expression and growth of cells in YPA medium. A, comparison of Mxr1p levels in GS115, Pp-Mxr1FLAG expressing Mxr1p from its own promoter and Mxr1-OE overexpressing Mxr1p from the GAPDH promoter. Mxr1p was detected using anti-FLAG/anti-c-myc antibodies. Phosphoglycerate kinase (PGK) levels served as a loading control. B, quantification of the data in A. The intensity of individual bands was quantified and expressed as arbitrary units ± S.D. relative to controls. Data are the average of three independent experiments. ***p < 0.0005. C, analysis of Mxr1 mRNA levels by qPCR. Error bars indicate mean ± S.D. ***, p < 0.0005. One-way analysis of variance, followed by Tukey's multiple comparison test was done (n = 3). D, Western blotting analysis of Adr1p in the Adr1-OE strain using anti-c-myc antibodies. PGK levels served as a loading control. E, RT-PCR analysis of Adr1 mRNA in the GS115 and Mxr1-OE strains. F, Northern blotting analysis of ACS1 expression. MS encoding methionine synthase was used as a loading control. G, analysis of ACS1His levels by Western blotting using anti-His antibodies in different P. pastoris strains. PGK served as a loading control. H, quantification of the data in G. The intensity of individual bands was quantified and expressed as arbitrary units ± S.D. relative to controls. Data are the average of three independent experiments. ***, p < 0.0005. I, growth curves of different P. pastoris strains cultured in YPA medium. Error bars indicate mean ± S.D. (n = 3). J, analysis of ACS1His levels by Western blotting using anti-His antibodies in cells cultured in YPG and YPA containing glycerol and acetate as carbon sources, respectively. PGK served as a loading control.
The N-terminal region of Mxr1p contains the zinc finger domain, the 14-3-3 protein interaction region, and a trans-activation domain (Fig. 3A) (9). To examine the role of the N-terminal trans-activation domain in the regulation of ACS1 expression and acetate metabolism, we overexpressed Mxr1pN400 in Δmxr1 and generated the Pp-Mxr1pN400-OE strain. Overexpression of Mxr1N400 was confirmed by qPCR (Fig. 3B), and protein expression was confirmed by Western blotting using anti-c-myc antibodies (Fig. 3C). Overexpression of Mxr1pN400 results in up-regulation of ACS1, resulting an increase in ACS1 mRNA (Fig. 3D) and in protein levels (Fig. 3, E and F), leading to an increase in growth rate (Fig. 3G). Therefore, the N-terminal trans-activation domain is active during acetate metabolism, and there is good correlation between the Mxr1p-mediated increase in ACS1 expression and growth rate.
FIGURE 3.
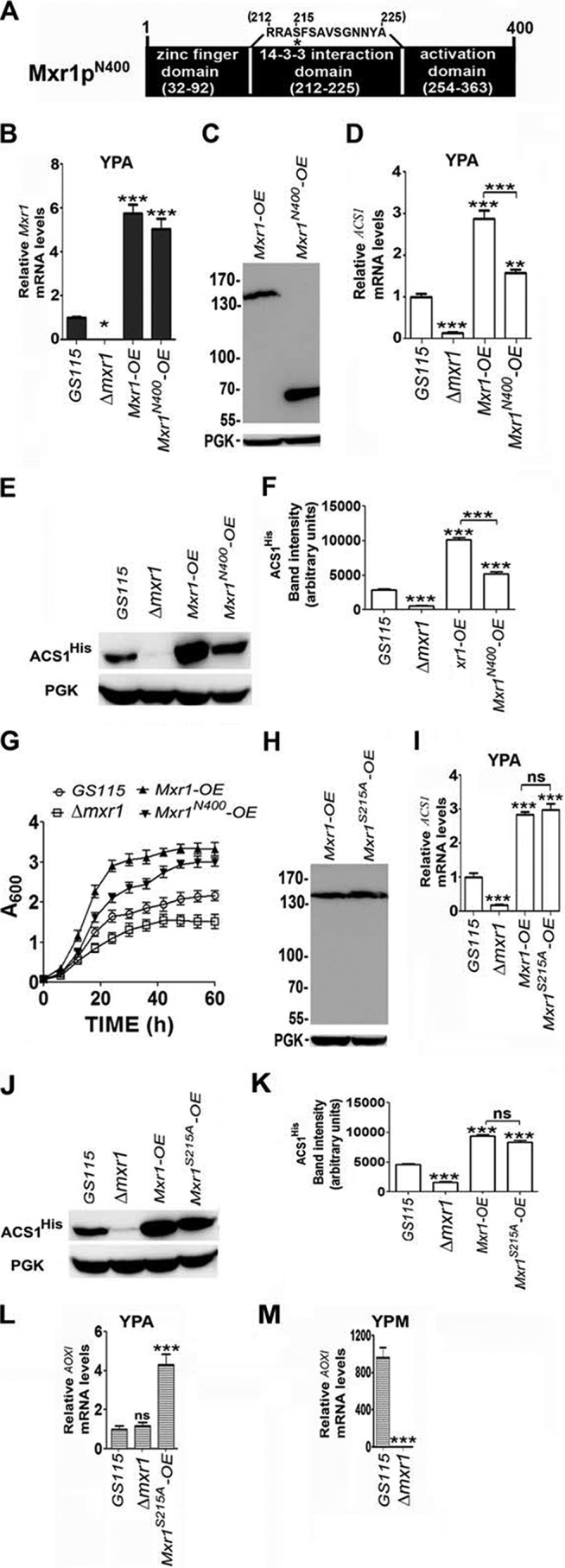
Regulation of ACS1 and AOXI expression by Mxr1pN400 and Mxr1pS215A. A, schematic of the N-terminal region of Mxr1p. Major functional domains are indicated. Serine 215, whose phosphorylation is essential for interaction with 14-3-3 protein, is indicated by an asterisk. B, analysis of Mxr1 expression by qPCR in different P. pastoris strains as indicated. C, analysis of the expression of Mxr1p and Mxr1pN400 by Western blotting using anti-c-myc antibodies in the Pp-Mxr1-OE and Pp-Mxr1N400-OE strains, respectively. PGK served as a loading control. D, analysis of ACS1 expression by qPCR in different P. pastoris strains cultured in YPA. E, Western blot analysis of ACS1His levels in different P. pastoris strains cultured in YPA. PGK was used as a loading control. F, quantification of the data in E. The intensity of individual bands was quantified and expressed as arbitrary units ± S.D. relative to controls. Data are the average of three independent experiments. G, growth curves of different P. pastoris strains cultured in YPA medium. H, analysis of the expression of Mxr1p and Mxr1pS215A by Western blotting using anti-c-myc antibodies in the Pp-Mxr1-OE and Pp-Mxr1S215A-OE strains, respectively. PGK served as a loading control. I, analysis of ACS1 expression by qPCR in different P. pastoris strains cultured in YPA. J, Western blot analysis of ACS1His levels in different P. pastoris strains cultured in YPA. PGK was used as a loading control. K, quantification of the data in J. The intensity of individual bands was quantified and expressed as arbitrary units ± S.D. relative to controls. Data are the average of three independent experiments. L, analysis of AOX1 expression by qPCR in different P. pastoris strains in YPA. M, analysis of AOX1 expression by qPCR in different P. pastoris strains cultured in YPM. Error bars indicate mean ± S.D. *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ns, not significant. One-way analysis of variance followed by Tukey's multiple comparison test was done (n = 3).
When P. pastoris is cultured in media containing ethanol as the sole source of carbon, genes of the mut pathway remain repressed because of the interaction of Mxr1p with the 14-3-3 protein, in which serine 215 of Mxr1p has a key role (9). In cells expressing the S215A mutant, Mxr1p-14-3-3 protein interaction is abrogated, resulting in derepression of genes of the mut pathway when cultured in a medium containing ethanol (9). We generated Mxr1pS215A and expressed it in Δmxr1 (Fig. 3H). ACS1 mRNA and ACSHis protein levels are comparable between Pp-Mxr1-OE and Pp-Mxr1S215A-OE (Fig. 3, I–K). Overexpression of Mxr1pS215A results in slight derepression, resulting in an ∼4-fold increase in AOXI mRNA in cells cultured in YPA (Fig. 3L). This increase is not significant because AOXI expression in GS115 cultured in YPM is several hundred-fold higher than that observed in Pp-Mxr1S215A-OE cultured in YPA (Fig. 3M).
Identification and Characterization of the MxRU in pACS
To identify cis-acting elements in pACS involved in Mxr1p-mediated regulation of ACS1 expression, −1049 bp, −809 bp, −736 bp, and −449 bp of pACS were cloned upstream of E. coli lacZ encoding β-galactosidase to generate the pACS1-lacZ, pACS2-lacZ, pACS3-lacZ, and pACS4-lacZ plasmids, respectively (Fig. 4A). These constructs were introduced into GS115 as well as Δmxr1, and β-galactosidase activity was measured in cells cultured in YPA and YNBA. Mxr1p-dependent activation of pACS was observed in the cases of pACS1-lacZ and pACS2-lacZ, but not pACS3-lacZ and pACS4-lacZ, in cells cultured in YPA but not YNBA (Fig. 4B), indicating that the region between −809 and −736 bp is essential for Mxr1p function. Analysis of the pACS sequence revealed the presence of two putative MXREs (MXRE1 and MXRE2) at −780 and −742 bp, which were designated MXRE1 and MXRE2, respectively (Fig. 4C). Point mutations that abolish Mxr1p binding to pAOXI-MXREs (2) were introduced into either (M1 and M2) or both (M3) MXREs of pACS1 (Fig. 4D), and reporter gene expression was examined. The results indicate that mutations in either or both MXREs abrogate Mxr1p-dependent activation of pACS1 (Fig. 4E). Furthermore, insertion of a 10- or 20-bp sequence between the two MXREs abrogates Mxr1p-dependent activation of pACS1 (Fig. 4, F and G). Mxr1p-dependent activation of ACS1 expression is more efficient in cells cultured in YPA than in those cultured in YNBA + CAA or YNBA + Glu (Fig. 4H). Therefore, in addition to acetate and amino acids, other components present in YPA also contribute to the transcriptional activation of ACS1 by Mxr1p.
FIGURE 4.
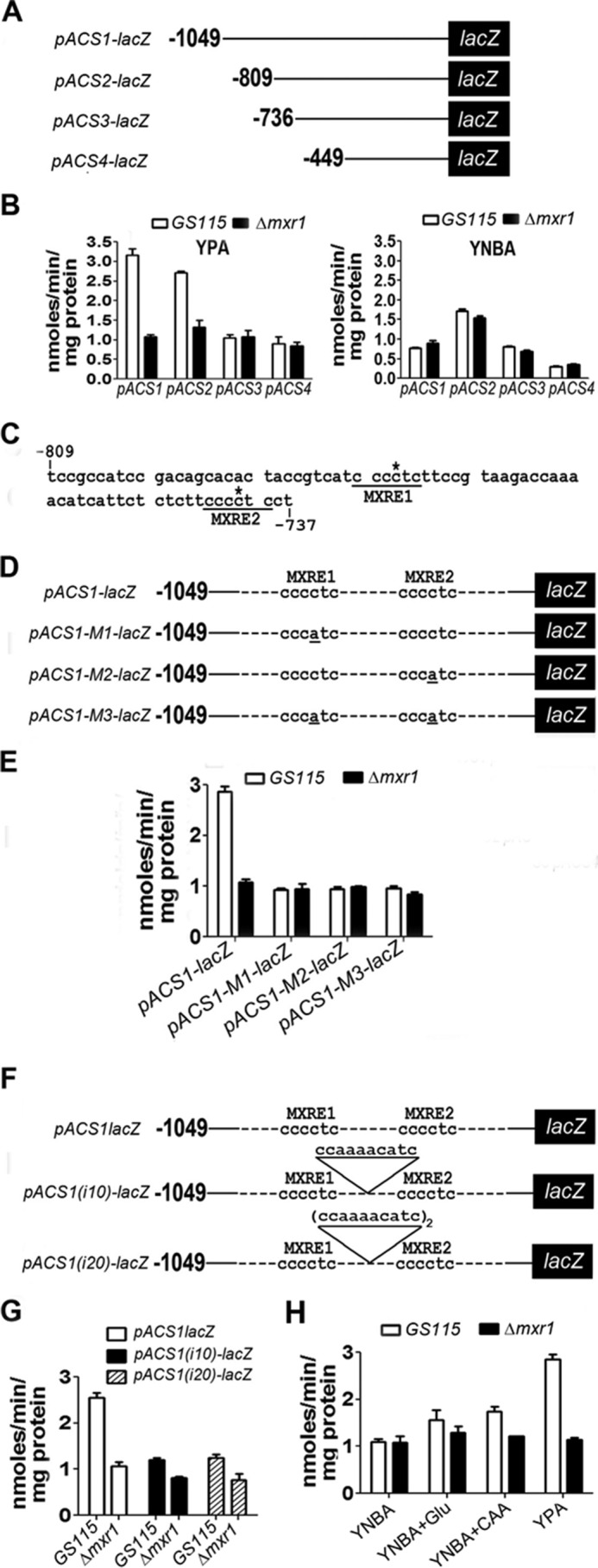
Study of lacZ expression from pACS and identification of pACS-MXREs. A, schematic of pACS-lacZ constructs. B, estimation of β-galactosidase activity in lysates of cells transformed with pACS-lacZ constructs. Cells were cultured in YPA or YNBA. C, nucleotide sequence of pACS between −809 and −737 bp. MXRE1 and MXRE2 are underlined. The cytosine residue within the MXRE crucial for Mxr1p binding is indicated by an asterisk. D, schematic of pACS1-lacZ constructs containing wild-type and mutant MXREs. Point mutations within MXREs are underlined. E, estimation of β-galactosidase activity in lysates of cells transformed with pACS-lacZ constructs containing wild-type and mutant MXREs. F, schematic of pACS1-lacZ constructs carrying 10- or 20-bp insertions between the two MXREs. G, effect of insertion of 10- and 20-bp insertions between the two MXREs on β-galactosidase activity in lysates of cells transformed with pACS-lacZ constructs. β-Galactosidase activity measurements represent the mean ± S.D. of data from three independent experiments. H, lacZ expression from pACS1 in cells cultured in various media as indicated.
To demonstrate Mxr1p binding to pACS-MXREs, oligonucleotides carrying mutations in either or both the MXREs were synthesized (Fig. 5A) and radiolabeled, and their ability to bind to recombinant Mxr1pN150 and Mxr1pN400 encoding 150 and 400 N-terminal amino acids, respectively (Fig. 5, B and C), was examined in an EMSA. Two DNA-protein complexes (I and II) were generated when these proteins were incubated with the pACS1-WT probe (Fig. 5D). Only complex I was formed with pACS1-M1 and M2 probes. Protein-DNA complex formation was abrogated when incubated with pACS1-M3 (Fig. 5D). Addition of anti-His or anti-GST antibodies resulted in either a supershift or abrogation of DNA-protein complexes, confirming the presence of recombinant Mxr1pN150 or Mxr1pN400 in these complexes, respectively (Fig. 5, E and F).
FIGURE 5.
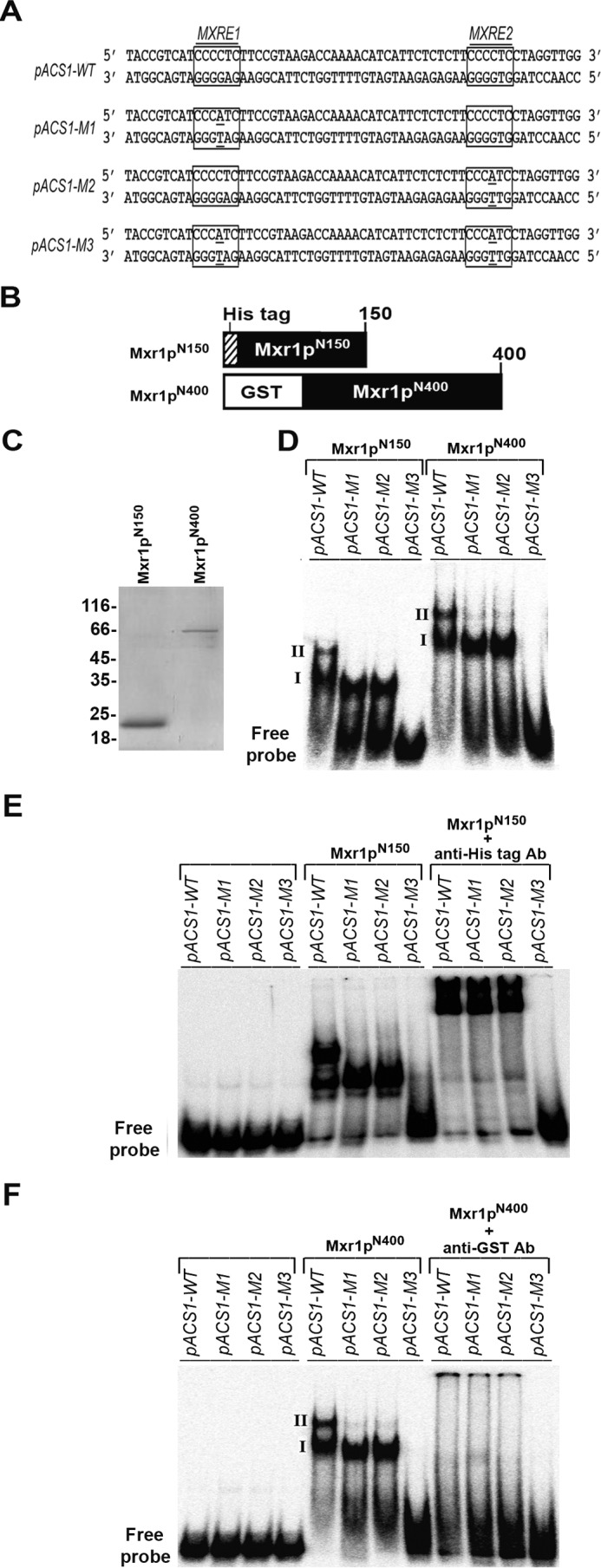
Analysis of binding of recombinant Mxr1pN150 and Mxr1pN400 to pACS-MXREs by EMSA. A, nucleotide sequence of oligonucleotides containing wild-type and mutant MXREs. MXREs are boxed. B, schematic of histidine-tagged Mxr1pN150 and GST-tagged Mxr1pN400. C, SDS-PAGE profile of Mxr1pN150 and Mxr1pN400 purified from E. coli lysates by nickel-nitrilotriacetic acid and glutathione affinity chromatography, respectively. D, analysis of binding of Mxr1pN150 and Mxr1pN400 to 32P-labeled oligonucleotides containing wild-type and mutant MXREs by EMSA. Complexes I and II are generated when Mxr1pN150 and Mxr1pN400 bind to both MXREs. Only complex I is obtained when Mxr1pN150 and Mxr1pN400 bind to only one of the MXREs. E, supershift of complexes I and II formed by His-tagged Mxr1pN150 by the addition of anti-His antibodies. F, abrogation of the formation of complexes I and II by GST-tagged Mxr1pN400 by addition of anti-GST antibodies.
Discussion
In this study, we demonstrate that Mxr1p, a transcriptional activator of genes of the mut pathway, is a key regulator of the acetate utilization pathway in P. pastoris. Mxr1p activates the expression of ACS1 by binding to two MXREs present in pACS1. Mutations in either of the pACS1-MXREs and a 10- or 20-bp increase in the distance between the two MXREs abrogate trans-activation by Mxr1p. Therefore, occupation of both MXREs by Mxr1p and close proximity (30 bp) between two MXREs is essential for trans-activation of pACS1 by Mxr1p. Therefore, two MXREs separated by a 30-bp spacer function as an MxRU of pACS1. Although recombinant Mxr1pN150 and Mxr1pN400 bind to both pACS1-M1 and M2 probes in vitro, formation of both complex I or II is essential for trans-activation by full-length Mxr1p in vivo. It will be interesting to examine the binding of full-length Mxr1p to pACS1-MXREs in vitro. So far, we have not been successful in generating recombinant full-length Mxr1p, and DNA binding studies with P. pastoris cell extracts have not been conclusive. Taken together, the results of this study indicate that Mxr1p binding to two pACS1-MXREs separated by a 30-bp spacer is essential for trans-activation from pACS1, as shown schematically in Fig. 6.
FIGURE 6.
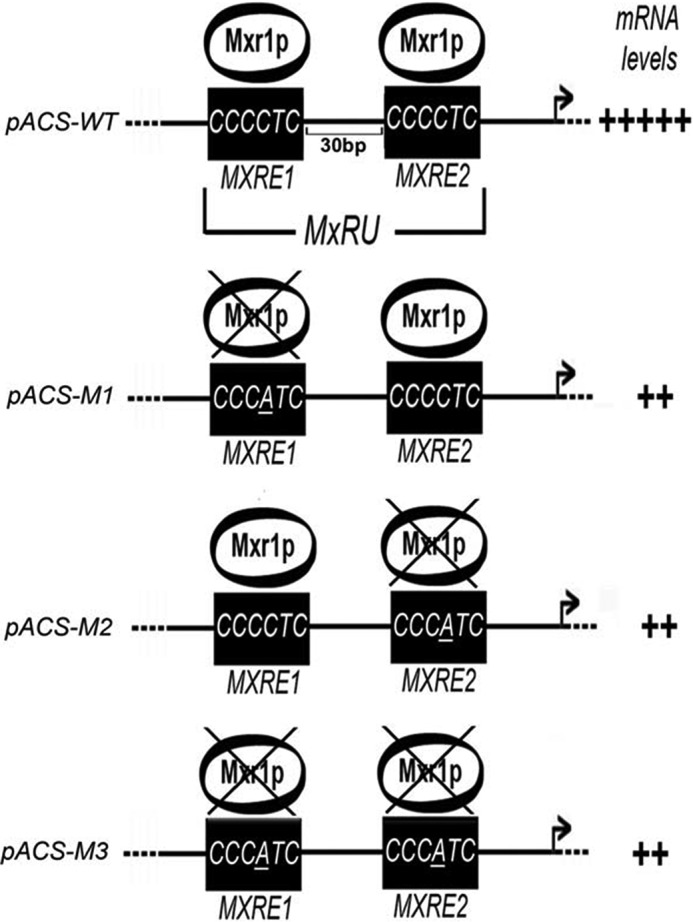
Schematic of Mxr1p-mediated trans-activation of pACS1 in P. pastoris cells cultured in YPA. A point mutation in MXRE1 or MXRE2 abrogates Mxr1p binding to pACS1 in vitro and trans-activation of pACS1-lacZ in vivo. An increase in the distance between the two MXREs also abolishes trans-activation in vivo. Therefore, the two MXREs separated by a 30-bp region functions as an MxRU.
The inability of Adr1p to restore ACS1 expression and growth in Δmxr1 indicates that Adr1p is not a functional homologue of Mxr1p. This is consistent with our earlier observation where we demonstrated that the DNA binding specificity of Mxr1p is different from that of Adr1p (2). Mxr1p overexpression studies clearly demonstrate that the growth rate of P. pastoris in YPA medium is directly proportional to the level of expression of Mxr1p and ACS1. Furthermore, the trans-activation domain in the N-terminal region has an important role in the regulation of ACS1 expression and growth, as evident from the ability of Mxr1pN400 to restore ACS1 expression and growth of Δmxr1. The differences in ACS1 expression levels and growth rates of Pp-Mxr1-OE and Pp-Mxr1N400-OE indicate that another trans-activation domain, present between amino acids 401 and 1155, also has a key role in the activation of ACS1 expression. Studies with Pp-Mxr1S215A-OE indicate that the serine 215 residue of Mxr1p has no role in the regulation of ACS1 expression.
Mxr1p localizes to the nuclei of cells cultured in several non-fermentable carbon sources. However, its target genes in the mut pathway are activated only in selected non-fermentable carbon sources such as methanol. Repression of Mxr1p target genes in other non-fermentable carbon sources such as ethanol is facilitated by its interaction with 14-3-3 protein (9). Phosphorylation of serine 215 of Mxr1p is crucial for this interaction, and S215A mutation abrogates the Mxr1p/14-3-3 protein interaction (9). Having identified ACS1 as a novel target gene of Mxr1p, it was of interest to examine the role of the S215A mutation on ACS1 expression. The results indicate that the S215A mutation does not affect Mxr1p-mediated trans-activation of ACS1 in cells cultured in YPA.
The differential localization of Mxr1p in cells cultured in YNBA, YNBA + Glu, YNBA + CAA, and YPA led us to investigate the role of amino acids in the regulation of ACS1 expression by Mxr1p. Although Mxr1p localizes to the nucleus of cells cultured in YNBA + Glu, YNBA + CAA, and YPA, Mxr1p-dependent growth was observed only in cells cultured in YNBA + CAA and YPA. Furthermore, Mxr1p-dependent lacZ expression from the ACS1 promoter is more efficient in cells cultured in YPA than in those cultured in YNBA + CAA. Therefore, in addition to amino acids, other components in YPA medium may contribute to the regulation of ACS1 expression by Mxr1p. The fact that acetate, together with glutamate or casamino acids, triggers the nuclear translocation of Mxr1p opens up new avenues of study of the mechanism of nuclear translocation of Mxr1p.
The differential regulation of acetate metabolism by Mxr1p in minimal and nutrient-rich media is similar to that of the Rop1p-mediated regulation of methanol metabolism. Rop1p translocates to the nucleus in cells cultured in YPM and represses transcription of genes of the mut pathway by competing with Mxr1p for binding to MXREs. However, Rop1p is cytosolic in cells cultured in YNBM and has no role in the regulation of methanol metabolism. The physiological significance of differential regulation of methanol metabolism by Rop1p can be explained as follows. Unlike S. cerevisiae, P. pastoris can utilize amino acids as a source of carbon. In cells cultured in YPM, utilization of amino acids is preferred to that of methanol, and, therefore, to minimize methanol utilization, Rop1p translocates to the nucleus and represses the expression of genes of the mut pathway. In YNBM, methanol is the sole source of carbon, and, therefore, Rop1p remains in the cytosol, facilitating the efficient activation of genes of the mut pathway by Mxr1p. Therefore, Mxr1p and Rop1p function as nutrient sensors and differentially regulate acetate and methanol metabolism, respectively, in cells cultured under minimal and nutrient-rich conditions.
Author Contributions
U. S. and P. N. R. designed the study, and U. S. performed all experiments. P. N. R. wrote the paper. U. S. and P. N. R. analyzed the results and approved the final version of the manuscript.
Acknowledgment
We thank G. Padmanaban for support and advice.
This study was supported by Grant BT/PR3889/BRB/10/996/2011 from the Department of Biotechnology, New Delhi, India (to P. N. R.). The authors declare that they have no conflicts of interest with the contents of this article.
- mut
- methanol utilization pathway
- MXRE
- Mxr1p response element
- YPM
- yeast extract, peptone, and methanol
- YNBM
- yeast nitrogen base, ammonium sulfate, and methanol
- YNBA
- yeast nitrogen base, ammonium sulfate, and acetate
- CAA
- casamino acid
- qPCR
- quantitative PCR
- TRITC
- tetramethylrhodamine isothiocyanate
- Glu
- glutamate
- ACS
- acetyl-CoA synthetase
- MxRU
- Mxr1p response unit
- PGK
- phosphoglycerate kinase
- YPA
- yeast extract, peptone, and acetate.
References
- 1. Lin-Cereghino G. P., Godfrey L., de la Cruz B. J., Johnson S., Khuongsathiene S., Tolstorukov I., Yan M., Lin-Cereghino J., Veenhuis M., Subramani S., and Cregg J. M. (2006) Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris. Mol. Cell. Biol. 26, 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kranthi B. V., Kumar R., Kumar N. V., Rao D. N., and Rangarajan P. N. (2009) Identification of key DNA elements involved in promoter recognition by Mxr1p, a master regulator of methanol utilization pathway in Pichia pastoris. Biochim. Biophys. Acta 1789, 460–468 [DOI] [PubMed] [Google Scholar]
- 3. Kranthi B. V., Kumar H. R., and Rangarajan P. N. (2010) Identification of Mxr1p-binding sites in the promoters of genes encoding dihydroxyacetone synthase and peroxin 8 of the methylotrophic yeast Pichia pastoris. Yeast 27, 705–711 [DOI] [PubMed] [Google Scholar]
- 4. Kumar N. V., and Rangarajan P. N. (2011) Catabolite repression of phosphoenolpyruvate carboxykinase by a zinc finger protein under biotin and pyruvate carboxylase-deficient conditions in Pichia pastoris. Microbiology 157, 3361–3369 [DOI] [PubMed] [Google Scholar]
- 5. Kumar N. V., and Rangarajan P. N. (2012) The zinc finger proteins Mxr1p and repressor of phosphoenolpyruvate carboxykinase (ROP) have the same DNA binding specificity but regulate methanol metabolism antagonistically in Pichia pastoris. J. Biol. Chem. 287, 34465–34473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sahu U., Krishna Rao K., and Rangarajan P. N. (2014) Trm1p, a Zn(II)2Cys6-type transcription factor, is essential for the transcriptional activation of genes of methanol utilization pathway, in Pichia pastoris. Biochem. Biophys. Res. Commun. 451, 158–164 [DOI] [PubMed] [Google Scholar]
- 7. Rose M., and Botstein D. (1983) Construction and use of gene fusions lacz which are expressed in yeast. Methods Enzymol. 10, 167–180 [DOI] [PubMed] [Google Scholar]
- 8. Beier D. R., Sledziewski A., and Young E. T. (1985) Deletion analysis identifies a region, upstream of the ADH2 gene of Saccharomyces cerevisiae, which is required for ADR1-mediated derepression. Mol. Cell. Biol. 5, 1743–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parua P. K., Ryan P. M., Trang K., and Young E. T. (2012) Pichia pastoris 14-3-3 regulates transcriptional activity of the methanol inducible transcription factor Mxr1 by direct interaction. Mol. Microbiol. 85, 282–298 [DOI] [PMC free article] [PubMed] [Google Scholar]