

ABSTRACT
Key steps in bacterial cell division are the synthesis and subsequent hydrolysis of septal peptidoglycan (PG), which allow efficient separation of daughter cells. Extensive studies in the Gram-negative, rod-shaped bacterium Escherichia coli have revealed that this hydrolysis is highly regulated spatially and temporally. Neisseria gonorrhoeae is an obligate Gram-negative, diplococcal pathogen and is the only causative agent of the sexually transmitted infection gonorrhea. We investigated how cell separation proceeds in this diplococcal organism. We demonstrated that deletion of the nlpD gene in strain FA1090 leads to poor growth and to an altered colony and cell morphology. An isopropyl-beta-d-galactopyranoside (IPTG)-regulated nlpD complemented construct can restore these defects only when IPTG is supplied in the growth medium. Thin-section transmission electron microscopy (TEM) revealed that the nlpD mutant strain grew in large clumps containing live and dead bacteria, which was consistent with deficient cell separation. Biochemical analyses of purified NlpD protein showed that it was able to bind purified PG. Finally, we showed that, although NlpD has no hydrolase activity itself, NlpD potentiates the hydrolytic activity of AmiC. These results indicate that N. gonorrhoeae NlpD is required for proper cell growth and division through its interactions with the amidase AmiC.
IMPORTANCE N. gonorrhoeae is the sole causative agent of the sexually transmitted infection gonorrhea. The incidence of antibiotic-resistant gonococcal infections has risen sharply in recent years, and N. gonorrhoeae has been classified as a “superbug” by the CDC. Since there is a dearth of new antibiotics to combat gonococcal infections, elucidating the essential cellular process of N. gonorrhoeae may point to new targets for antimicrobial therapies. Cell division and separation is one such essential process. We identified and characterized the gonococcal nlpD gene and showed that it is essential for cell separation. In contrast to other pathogenic bacteria, the gonococcal system is streamlined and does not appear to have any redundancies.
INTRODUCTION
Peptidoglycan (PG) is an essential component of most bacterial cell walls. PG is a polysaccharide polymer that is composed of repeating units of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) linked by a β-1-4 glycosidic bond. Peptide side chains attached to the MurNAc sugar form interpeptide cross-links and reinforce the PG structure, which is important for protecting the bacterial cell from osmotic lysis and for regulating cell size and shape. PG is a dynamic structure that undergoes constant remodeling to accommodate cell growth and division (1). Accordingly, bacteria contain a variety of peptidoglycanases that cleave the cell wall at specific locations and times.
Much of what is known about the intricacies of cell division comes from the model organism Escherichia coli, and comparatively less is known about cell division in other Gram-negative bacteria, particularly coccal organisms (2). During cell division in E. coli, the cell envelope (outer membrane, inner membrane, and peptidoglycan [PG]) undergoes a coordinated constriction at the midcell, which is orchestrated by the divisome (3). The divisome consists of cytoskeletal proteins, PG hydrolases, and associated regulatory proteins. PG hydrolases are required for septal splitting and for the final separation of daughter cells. This septal splitting and cell separation occurs through the action of three redundant PG amidases, AmiA, AmiB, and AmiC (4), which hydrolyze the amide bond between MurNAc and l-alanine. These amidases are specifically activated by the regulatory proteins EnvC and NlpD (5).
EnvC and NlpD are classified as lysostaphin-like metalloproteases (LytM proteins), which belong to the M23 peptidase family of proteins (6). The LytM domain was first identified in the autolysin lysostaphin of the Gram-positive bacterium Staphylococcus aureus (7). Although lysostaphin cleaves the pentaglycine cross-bridges found exclusively in staphylococcal PG, LytM proteins have PG hydrolase activities with a range of cleavage specificities, and some LytM proteins have no detectable PG hydrolase activity. In E. coli, these LytM-like proteins have been shown to regulate the amidases AmiA to AmiC, to contain a degenerate LytM domain possessing only two of four LytM active sites, and to have no PG hydrolytic activity themselves (5). The rod-shaped bacterium Vibrio cholerae also contains a single amidase, AmiB, which is controlled by NlpD and EnvC (8). Haemophilus influenzae contains three LytM-like factors, YebA, NlpD, and Env (9).
Significantly less is known about the process of cell division and separation in coccal bacteria (2). The diplococcal Gram-negative bacterium Neisseria gonorrhoeae divides in two planes, whereas rod-shaped bacteria divide in only one. The Min system of N. gonorrhoeae functions to regulate cell division, and minCD gonococcal mutants are heterogeneously sized with multiple, sometimes incomplete, septa (10). Gonococcal minCD mutants have reduced viability compared to E. coli minCD cells, suggesting that the Min system may be more important for N. gonorrhoeae than it is for E. coli. Finally, although E. coli has three redundant amidases, N. gonorrhoeae strain FA1090 is predicted to contain only the AmiC amidase, and a gonococcal amiC mutant shows a defect in cell separation (11).
We previously showed that the mpg gene product of N. gonorrhoeae (also called NGO1686) is a LytM protein and a bifunctional M23 metallopeptidase, possessing and exhibiting endopeptidase as well as carboxypeptidase activities on PG (12). Although an mpg mutant has no defects in cell separation, we demonstrated that Mpg alters gonococcal resistance to polymorphonuclear leukocyte (PMN)-mediated oxidative and nonoxidative killing mechanisms through its activity in type IV pilus expression (13). Mutation of the predicted M23 active sites revealed that only two of the four M23 active sites are important for Mpg-associated phenotypes and activities. Specifically, a mutation in the predicted catalytic histidine residue has no effect on phenotypes conferred by the Mpg protein (12). In this current work, we have characterized an Mpg paralog, NlpD (NG1056; E value, 2e−12). We show that a gonococcal nlpD mutant grows poorly, has an altered colony and cellular morphology, and is defective in cell separation. Purified NlpD binds to PG and does not hydrolyze PG itself, but rather it potentiates the activity of the amidase AmiC. We conclude that gonococcal NlpD has a similar interaction with AmiC as it does in E. coli; however, N. gonorrhoeae has a more streamlined system, since it contains only one amidase, AmiC.
MATERIALS AND METHODS
Bacterial strains and growth medium.
All N. gonorrhoeae strains were derivatives of FA1090. Gonococcal strains were grown at 37°C on solid GC medium base (GCB; Difco) plus Kellogg supplements I and II [22.2 mM glucose, 0.68 glutamine, 0.45 mM cocarboxylase, and1.23 mM Fe(NO3)3] (14), all from Sigma at 37°C in 5% CO2. N. gonorrhoeae strains were grown in GCB liquid (GCBL) medium (1.5% proteose peptone no. 3 [Difco], 0.4% K2HPO4, 0.1% KH2PO4, and 0.1% NaCl) with 0.042% sodium bicarbonate (all from Sigma) and Kellogg supplements I and II at 37°C with rotation. E. coli strain TOP10 (Invitrogen) was used to propagate plasmids and was grown in Luria-Bertani (LB) broth or agar at 37°C. Antibiotics (Sigma) were used at the following concentrations for N. gonorrhoeae: erythromycin (Erm), 0.75 μg/ml; kanamycin (Kan), 40 μg/ml; and tetracycline (Tet), 0.2 μg/ml. For E. coli, the following concentrations were used: Kan, 40 μg/ml; Erm, 275 μg/ml; and chloramphenicol (Cam), 25 μg/μl. Isopropyl-β-d-1-thiogalactopyranoside (IPTG) (Diagnostic Chemicals) was supplied at 1 mM in solid agar or liquid medium.
DNA manipulations and analysis.
Standard procedures were performed as described previously (15). Plasmid DNA was isolated from E. coli using Qiagen plasmid kits. DNA was isolated from agarose gels using QIAquick gel extraction kits (Qiagen). All DNA restriction and modification enzymes were used according to the manufacturers' instructions (Promega and New England BioLabs). E. coli strain TOP10 was transformed using heat shock according to the manufacturer. E. coli strain BL21(DE3) (Novagen) was transformed using the Gene Pulser II electroporation system (Bio-Rad Laboratories) according to the manufacturer's specifications. pilE sequences of FA1090 and derivatives were determined by amplifying pilE from the chromosome with primers PILRBS and SP3A (16) using GoTaq polymerase (Promega) and sequencing the resulting PCR product with SP3A. Sequencing products were analyzed on an ABI 3730 sequencer.
Transformation of Neisseria.
Gonococcal strains were transformed with plasmids by coculture of DNA and N. gonorrhoeae on a solid medium (spot transformation). The strain to be transformed was plated on GCB, and ∼5 to 10 μg of plasmid DNA was added to a 20-μl total reaction mixture of GCBL [containing supplements I and II and 5 mM Mg(SO4)] and was spotted on individual areas of the plate. After coincubation for 18 to 20 h at 37°C, colonies from the spotted areas were collected with a Dacron swab, resuspended in GCBL, and plated on a medium containing an antibiotic to select for transformants.
Construction of a nonvarying FA1090 (RM11.2nv) strain.
FA1090 RM11.2nv was constructed by transforming strain RM11.2 recA6 (17) with the pVD300 plasmid construct (18) containing the recA gene under the control of its endogenous promoter. Colonies were screened for those that varied in the absence of IPTG and were Tets. The pilE sequence was verified to be RM11.2. Strains were subsequently transformed with the G4 point mutant construct (19) and were screened for colonies that lost the ability to vary in the presence of IPTG (thus designated with a subscript “nv” for nonvarying). DNA sequencing verified the presence of the G4 mutation and the retention of the RM11.2 pilE sequence.
Insertional inactivation of nlpD.
To disrupt nlpD, the entire gene except for the last 3′ 29 bp was deleted, and a cat resistance gene cassette was inserted in its place. Primer pairs NlpD-3/NlpD-5 and NlpD-4/NlpD-6 (Table 1) were used to amplify the regions upstream and downstream of nlpD, respectively. The ∼460-bp product of NlpD-3/NlpD-5 and the ∼410-bp product of NlpD-4/NlpD-6 were gel-purified and ligated separately to pCR-Blunt (Invitrogen). Sequencing confirmed that no mutations were introduced. The cat gene encoding Camr from plasmid pHSS6/Cat2-9 was excised with a NotI digest, treated with T4 DNA polymerase to yield blunt ends, gel purified, and ligated to the EcoRV-digested pCR-Blunt construct containing the downstream region of nlpD. Camr colonies were selected on solid medium. A clone containing the cat gene in the same orientation as the nlpD gene was linearized with NotI and blunted with T4 polymerase. The nlpD upstream fragment was released from the pCR-Blunt construct with an EcoRI digest, blunted with T4 polymerase, and excised from an agarose gel. The fragment containing the upstream region of nlpD was subsequently ligated to the cat-containing downstream nlpD region. Proper orientation of the nlpD construct was determined by restriction enzyme digest and was confirmed by sequencing.
TABLE 1.
Primer sequences
| Primer | Sequence (5′–3′) | Reference or source |
|---|---|---|
| NlpD-13 | GGCATTAATTAACATGAAGGTTATCTTATGTTG | This work |
| NlpD-11 | CATGGAATTCTTGCAAACGGAAAGTCAGAACGCG | This work |
| NlpD-12 | GATCTTAATTAACAGGCTATATATCGGAACATGAAGG | This work |
| NlpD-10 | CATGGCTAGCATGGCTCTGTTGGGCGGTTGCGCC | This work |
| NlpD-9 | GATTCCATGGAAGCTCTGTTGGGCG | This work |
| NlpD-8 | GATTCCATGGAAATGTTGAAACAAACG | This work |
| NlpD-7 | GATTCCATGGAAATGCGGCGCAGG | This work |
| NlpD-3 | GACCGAAGCCTATCTGATGGG | This work |
| NlpD-4 | CTGAAAGACAGGACAAGCATTG | This work |
| NlpD-1 | GGGGACGGATTTTGGTGAATGCG | This work |
| NlpD-2 | AAAGACAGGACAAGCATTGTTCCG | This work |
| SP3A | CCGGAACGGACGACCCCG | 16 |
| PILRBS | GGCTTTCCCCTTTCAATTAGGAG | 16 |
Regulatable nlpD complementation construct (in Neisseria).
The product of the primer pair NlpD-3 and NlpD-4 (Table 1) was used to amplify the nlpD region, which was subsequently used as a template for the PCR using primers NlpD-13 and NlpD-11 (Table 1) with KOD polymerase (Novagen). This second PCR product contains the nlpD coding region as well as the associated ribosome binding site (RBS) and introduces a PacI site into the 5′ end of the product with primer NlpD-13. The ∼1,050-bp PCR product was cloned into vector pCR-Blunt and was sequenced for accuracy. Next, this construct was linearized with BamHI and was treated with T4 DNA polymerase to create blunt ends, and the nlpD-containing sequence was then released with a PacI digest at the 5′ end of the gene. The resulting fragment was gel purified using the QIAquick kit, was ligated overnight to PacI/PmeI-digested and ciprofloxacin (CIP)-treated vector pGCC4, and was used to transform E. coli TOP-10 cells (Invitrogen). Ermr transformants were selected and sequenced using the primer taclac 5′ to verify that the 5′ junction of the nlpD gene was intact. The resulting IPTG-inducible construct pGCC4-nlpD was spot-transformed into strain FA1090 ΔnlpD (RM11.2nv and 1-81-S2nv pilE sequences), and colonies were selected on GCB with Erm.
NlpD overexpression and purification.
The nlpD gene lacking the predicted signal sequence was amplified from N. gonorrhoeae chromosomal DNA by PCR using primer NlpD-10, which introduces an NheI site, and primer NlpD-11 (Table 1) and was cloned into pCR-Blunt. Two clones with inserts of the predicted size were sequenced to verify that no mutations had been introduced. The nlpD insert was excised from pCR-Blunt with an NheI/BamHI digest and was cloned into an NheI/BamHI-digested pET28a overexpression vector (Novagen) to yield pET/His-NlpD. The 5′ junction of the insert and vector was sequenced to ensure the reading frame was maintained.
To overexpress the His-NlpD protein, the clone described above was transformed into E. coli BL21(DE3) cells (Novagen). Cells were grown at 37°C in 250 ml LB medium containing kanamycin. When the optical density at 600 nm (OD600) reached ∼0.6, protein expression was induced by the addition of 1 mM IPTG for 3 h at room temperature (RT). Cells were pelleted by centrifugation and were resuspended in 25 ml of cold His-protein purification buffer (50 mM sodium phosphate [pH 7.4], 300 mM NaCl, 10% glycerol). Cells kept on ice were disrupted by sonication using a Sonic Dismembrator (Fisher Scientific) at 25% duty cycle for 10-s intervals, with 10 s of rest in between for a total of 2 min, followed by centrifugation (10,000 × g) for 20 min to clarify the supernatant. Supernatant was passed through a 0.45-μm-pore-size filter to avoid clogging the resin. Imidazole (10 mM) was added to the cell-free supernatant and incubated with 1.5 ml Zinc Chelating Resin (G Biosciences) for 1 h with gentle shaking. Resin was collected by centrifugation, loaded on a column, and washed with protein purification buffer containing 10 mM imidazole. Protein was eluted by stepwise additions of purification buffer containing increasing amounts of imidazole (20, 40, 60, 100, and 250 mM). Fractions containing the NlpD protein, as determined by SDS-PAGE with Coomassie blue stain, were dialyzed separately against 3 changes of buffer (50 mM sodium phosphate [pH 7.4], 50 mM NaCl, 10% glycerol) containing 1 mM dithiothreitol (DTT) at 4°C and were concentrated in a Centriplus 10,000 MWCO column (Amicon). Recombinant NlpD protein was judged to be 95% pure by Coomassie gel. Fractions were frozen on liquid nitrogen.
AmiC overexpression and purification.
To purify 6×His-tagged AmiC, plasmids constructed in TAM1 E. coli were purified and transformed into E. coli BL21 Star (DE3) cells (Invitrogen). Single colonies of each strain were grown overnight in 30 ml of LB broth containing ampicillin at 37°C, and each culture was used the following day to seed one 1-liter culture/strain. One-liter cultures were grown at 37°C for ∼2.5 h until an OD600 of ∼0.6 to 0.8 was reached. The addition of 0.3 mM IPTG was used to induce protein production, and growth was continued for 5 h at 37°C. At the conclusion of growth, cells were centrifuged (8,000 × g for 10 min at 4°C) and washed once with cold phosphate-buffered saline (PBS) and were centrifuged again as portions representing 500 ml of the original culture for storage at −80°C. For protein purification, pellets were suspended in 20 ml purification buffer (20 mM sodium phosphate [pH 6], 500 mM NaCl, 10% vol/vol glycerol) and were disrupted by three passes through a cold French pressure cell (1,000 lb/in2). Unbroken cells were removed by centrifugation (20,000 × g for 30 min at 4°C), and cleared lysate was batch-bound to 500 μl of preequilibrated His-Select nickel affinity gel (Sigma) plus 20 mM imidazole for 60 min at 4°C with gentle shaking. Lysate and resin were loaded on a column, and flowthrough was collected, followed by four washes with 5 ml of purification buffer plus 20 mM imidazole. Protein was eluted in 1-ml fractions by stepwise additions of purification buffer containing increasing amounts of imidazole (40, 60, 90, and 120 mM). Samples of flowthrough, wash, and elution fractions were evaluated on a Coomassie-stained (and destained) 12% SDS-polyacrylamide gel to determine purity (>95%) and yield. Elution fractions were pooled and dialyzed into 500 ml of purification buffer (without imidazole) for 4 h and then again into 500 ml of fresh buffer overnight to remove all imidazole. Protein was stored in aliquots mixed 1:1 with 100% glycerol to produce 50% glycerol stocks for storage at −20°C.
Live/dead assay.
To examine the percentage of live versus dead bacteria in cells expressing or not expressing the nlpD gene, we employed the BacLight (Molecular Probes) viability stains SYTO9 (green) and propidium iodide (red), which stain viable and nonviable bacteria, respectively. Briefly, IPTG-inducible nlpD cells were grown on GCB plates either with or without IPTG. At ∼18 to 24 h, cells were collected by swab and were added to a GCBL medium to an OD600 of 0.5. One milliliter of the mixture was pelleted in a Microfuge (3 min, 8,000 rpm), washed in 1 ml buffer (0.1 M morpholinepropanesulfonic acid [MOPS], 1 mM MgCl2, pH 7.2), and pelleted again. Cells resuspended in 1 ml MOPS buffer were exposed to 3 μl of a 1:1 dye mixture and were mixed well and incubated at room temperature (RT) in the dark for 15 min. Five microliters of the cell-dye mixture was added to a microscope slide, mounted with a coverslip, and sealed with nail polish. Slides were visualized within 30 min of mounting using a Nikon Eclipse 90i microscope. A minimum of 4 visual fields were examined for each strain, and experiments were repeated on two separate days.
Transmission electron microscopy.
Transmission electron microscopy (TEM) of whole mounted cells was performed as described previously (17). Grids were viewed using a JEOL JEM-1220 transmission electron microscope. Thin-section TEM was performed on freshly grown (20 h) gonococci that were swabbed from plates (GCB ± IPTG), washed with PBS, and fixed with 2% paraformaldehyde for 30 min at room temperature. Cells were kept in paraformaldehyde at 4°C until use. Cell culture samples were fixed in 0.1 M sodium cacodylate buffer (pH 7.3) containing 2% paraformaldehyde and 2.5% glutaraldehyde and were postfixed with 2% osmium tetroxide in 0.1 M sodium cacodylate buffer. They were then rinsed with distilled water, en bloc stained with 3% uranyl acetate, rinsed a second time with distilled water, dehydrated in ascending grades of ethanol, transitioned with propylene oxide, embedded in the resin mixture of the EMbed 812 kit, and cured in a 60°C oven. Samples were sectioned on a Leica Ultracut UC6 ultramicrotome. Seventy-nanometer thin sections were collected on 200 mesh copper grids and were poststained with 3% uranyl acetate and Reynolds lead citrate and viewed on an FEI Tecnai Spirit G2 TEM. Digital images were captured on an FEI Eagle camera. Samples were processed for TEM by the Center for Advanced Microscopy at Northwestern University Feinberg School of Medicine.
PG purification and zymogram analysis.
PG was purified from N. gonorrhoeae with modifications as described previously (12). Zymogram analysis was performed as described previously (12, 20). E. coli BL21(DE3) cell extracts carrying pET/His-NlpD were electrophoresed on a 12% SDS gel containing 0.1% (wt/vol) N. gonorrhoeae murein sacculi using 75 V at 4°C. Following electrophoresis, gels were rinsed with water twice for 30 min at room temperature with gentle shaking to remove SDS. Gels were transferred to renaturing buffer (0.5% Triton X-100 and 25 mM Tris-HCl [pH 7.4]) twice for 30 min, and renaturing was continued overnight (∼16 h) at room temperature followed by 2.5 h at 37°C. Gels were stained with 0.1% methylene blue dissolved in 0.01% KOH for 60 min and were destained with water to visualize zones of PG clearing. Gels were subsequently stained with Coomassie brilliant blue to visualize protein bands on the gel.
PG binding assays.
PG binding assays were conducted as described previously (21). Briefly, 7.5 μg of purified protein was incubated for 2 h at 4°C with or without PG in binding buffer (30 mM Tris [pH 6.8], 50 mM NaCl, 10 mM MgCl2) in a total volume of 100 μl. Samples were centrifuged for 20 min (14,000 × g) at 4°C. The resulting pellets were washed twice in 150 μl of binding buffer, resuspended in 40 μl 2% SDS, and incubated for 1 h. Supernatants of the binding and washing steps and the resuspended pellets were analyzed by SDS-PAGE.
RBB labeling of PG.
Gonococcal PG sacculi were incubated overnight at 37°C with 20 mM Remazol brilliant blue (RBB; Sigma) in 0.25 M NaOH as described previously (5, 22). The reaction was neutralized by addition of an equal amount of 0.25 M HCl. RBB-labeled sacculi were pelleted by centrifugation (21,000 × g for 20 min) and were repeatedly washed with water and pelleted by centrifugation until the supernatant was clear. The final RBB-labeled PG pellet was resuspended in water containing 0.02% sodium azide and stored at 4°C.
Dye release assay for PG hydrolysis.
A volume of 7.5 μl of RBB-labeled (gonococcal) sacculi was incubated with 4 μM purified proteins at 37°C for various amounts of time in a total volume of 125 μl of assay buffer (50 mM sodium phosphate and 50 mM NaCl [pH 7.4]), a protocol modified from reference 5. Ten-microliter samples were removed from the reaction mixtures at various time points, and reactions were terminated by incubation for 5 min at 95°C. Samples were spun for 30 min (14,000 × g) at room temperature in a microcentrifuge. Immediately after spinning, 5 μl of the supernatant was removed, and its absorbance at 595 nm was measured with a NanoDrop spectrophotometer.
RESULTS
Genetic analysis of the nlpD ortholog of Neisseria gonorrhoeae. (i) Identification of the nlpD ortholog of N. gonorrhoeae.
We previously identified and characterized the Mpg virulence factor of N. gonorrhoeae, an M23 peptidase (LytM protein) that exhibits dual carboxypeptidase and endopeptidase activities (12) and that plays a role in PMN-mediated killing and in H2O2 sensitivity (23). An Mpg paralog, NG1056, was identified in the N. gonorrhoeae FA1090 genome (GenBank accession number AE004969) as the gonococcal ortholog of NlpD. To verify this designation, we performed a tblastn search using the amino acid sequence of the E. coli NlpD (GenBank accession number P0ADA3) to search the FA1090 genome. We confirmed that NG1056 is the only open reading frame (ORF) showing significant similarity to the E. coli NlpD protein, sharing 30% identity and 42% similarity with the E. coli NlpD over the entire length of the protein (data not shown), and we will therefore refer to NG1056 as nlpD for the remainder of this paper. The gonococcal NlpD is predicted to be a 337 amino acid, 34.5-kDa protein with a signal peptide from residue 1 to 26 and a cleavage site thereafter (http://www.cbs.dtu.dk/services/SignalP/). Moreover, the gonococcal NlpD contains a LysM domain (PG binding) from residue 106 to 148 (E value, 2.3e−13) and a LytM domain associated with PG cleavage containing a degenerate peptidase active site (E value, 1.5e−39) (Fig. 1), which is identical to the domain organization of the E. coli NlpD (data not shown).
FIG 1.

Domain structure of N. gonorrhoeae NlpD protein. The cartoon depicts the predicted domain architecture of the gonococcal NlpD protein.
(ii) Mutation of nlpD results in a growth defect and altered colony morphology and the nlpD mutation can be complemented.
We insertionally inactivated the nlpD gene by inserting a cat resistance gene cassette in place of the coding sequence. The strain FA1090nv (the subscript “nv” in the strain name indicates that a strain cannot undergo pilin antigenic variation [24]) used in this study contained either 1-81-S2 or RM11.2 pilE variant sequences. Each of the FA1090 variants were transformed with the mutant ΔnlpD::cat allele construct (here designated ΔnlpD) by spot transformation, and Camr colonies were selected on Cam plates. Camr colonies were analyzed by PCR to verify replacement of the parental allele with the mutant allele and to verify elimination of the parent allele (data not shown).
The ΔnlpD mutant colonies grew very slowly and exhibited a small and nonuniform colony morphology relative to the parent strain when viewed with a stereomicroscope (Fig. 2B and D). Serial passage of the mutant colonies was difficult since few colonies grew from these mutant colonies, indicating a loss of cell viability (data not shown). To complement the ΔnlpD mutation, we cloned the nlpD gene under lac regulatory sequences and introduced it into the NICS locus on the chromosome of N. gonorrhoeae ΔnlpD. When cultures were grown on GCB medium containing IPTG, ectopically expressed nlpD restored wild-type (FA1090) colony morphology to the ΔnlpD deletion mutant (Fig. 2E). In the absence of IPTG, the same strain containing the IPTG-inducible copy of nlpD resembled the ΔnlpD deletion mutant (Fig. 2F). Therefore, we conclude that the lack of nlpD is responsible for the changes in colony morphology and that we can complement the morphology defect by providing nlpD at an ectopic locus under the control of the lac promoter.
FIG 2.
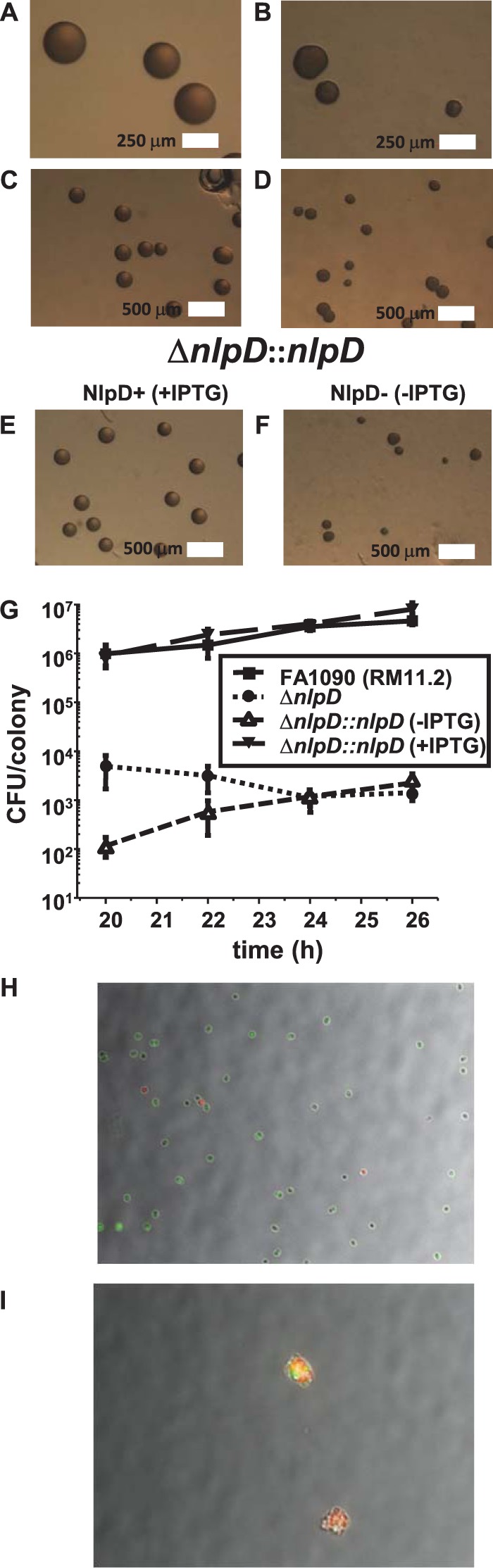
Inactivation of nlpD affects colony morphology and cell growth and viability. Representative stereomicroscope images show parental (A and C), ΔnlpD mutant (B and D), and complemented strain in the presence (E) or absence (F) of IPTG to regulate nlpD expression after 22 h of growth. Addition of IPTG recapitulates the parental colony morphology. (G) Growth curve of FA1090, ΔnlpD, and ΔnlpD::nlpD complemented strain with or without IPTG grown on a solid medium and measured as CFU per colony at the indicated times for six independent colonies of each strain. Error bars represent the standard error of the mean. Representative micrographs of NlpD+ (G) and NlpD− (H) subjected to BacLight live/dead stain. Red cells are dead, green cells are alive, and cells that appear yellow are likely two cells (one live, one dead) on top of one another in the visual field.
To quantify the growth defect of the ΔnlpD mutant and subsequent growth restoration in the complemented strain, we performed a 6-h growth curve analysis of plate-grown cells by determining the mean CFU per colony of strains FA1090nv, FA1090nvΔnlpD, and FA1090nvΔnlpD::nlpD with and without IPTG (Fig. 2G). The strains lacking NlpD showed a large reduction (2 to 4 logs) of CFU per colony relative to the parent strain. The parental strain and the complemented strain (with IPTG) showed nearly identical growth curves. The ΔnlpD strain and the complemented strain (without IPTG) showed decreased growth compared to that of the parent strain (Fig. 2G). The increase in growth over time observed in the complemented strain (without IPTG) is likely due to the lac promoter's leaky expression, which we have observed previously (12).
To probe whether the reduction of CFU per colony in the NlpD− strain was due to decreased viability, we performed live/dead assays on the complemented strain grown with or without IPTG to regulate nlpD expression. After 18 h of growth, the NlpD+ strain showed ∼17% dead cells, whereas the NlpD− strain showed between 50% and 75% dead cells. After 20 h of growth, the NlpD+ strain showed ∼8% dead cells, whereas the NlpD− strain showed >50% dead cells (all of which were found in large clumps). These data suggest that the cell masses in the NlpD− strain are largely composed of dead cells, but the presence of live cells indicate that that death is downstream of the cell septation defect.
To examine the cellular defects of the nlpD mutant, we performed transmission electron microscopy (TEM) on whole-cell mounts of FA1090 and the ΔnlpD mutant. The FA1090 parent strain exhibited the typical monococcal or diplococcal cell morphology (Fig. 3A). In contrast, the ΔnlpD mutant formed large masses of cells that appeared to be unable to separate (Fig. 3B). To more closely examine the cellular morphology of the nlpD mutant, we performed thin-section TEM on the IPTG-inducible complemented strain in the presence and absence of IPTG. The cells that expressed nlpD (with IPTG) showed mostly diplococci and monococci (Fig. 3C and E), whereas the cells that lacked nlpD (without IPTG) contained large cell masses resembling rosettes composed of ∼25 cells (Fig. 3D and F). We conclude that the loss of nlpD does not allow separation of cells after division and that this results in the decreased cell viability observed in the ΔnlpD mutant (Fig. 2G).
FIG 3.
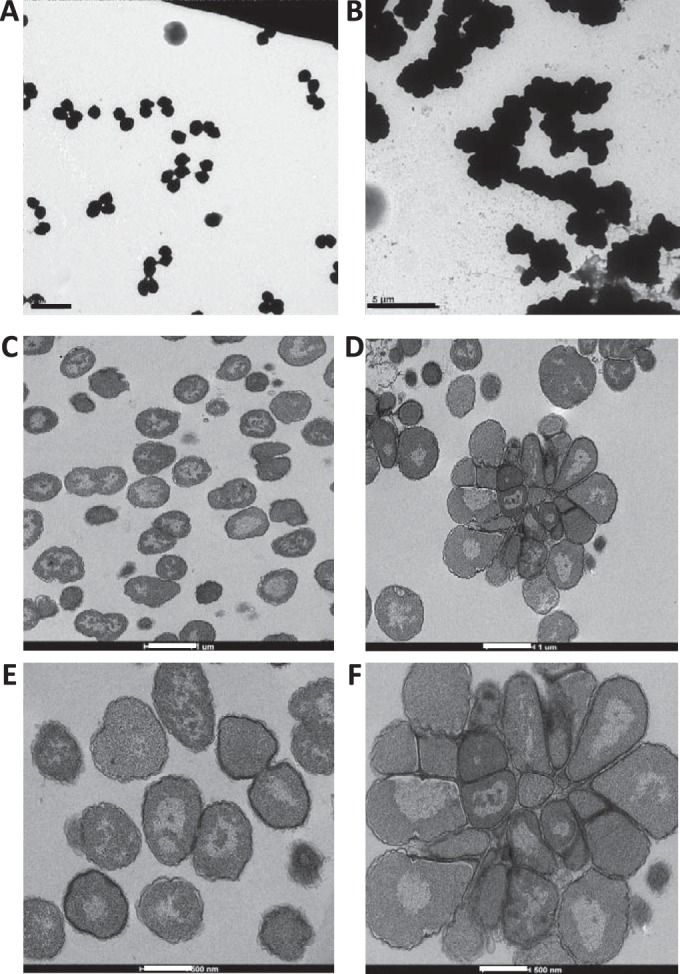
Inactivation of nlpD affects cell separation. (A and B) Representative transmission electron micrographs (TEM) of whole-cell mounts of FA1090nv and the ΔnlpD mutant. Thin-section TEM images show complemented strain ΔnlpD::nlpD with IPTG (C and E) and without IPTG (D and F). Cells not expressing nlpD grow as large aggregates (B, D, and F), whereas the parental strain and complemented strain expressing nlpD grow as either monococci or diplococci (A, C, and E). The scale bars are as follows: 2 μm (A), 5 μm (B), 1 μm (C and D), and 500 nm (E and F).
Biochemical analysis of NlpD function. (i) Zymogram analysis of NlpD.
E. coli NlpD has been shown to function indirectly in daughter cell separation by regulating the PG hydrolysis catalyzed by the AmiC protein (5). To begin to determine the mechanism by which NlpD affects cellular processes in N. gonorrhoeae, we conducted a zymogram analysis of the NlpD protein (Fig. 4). E. coli BL21(DE3) cells containing the pET28a/His-NlpD construct were grown and were induced at an OD600 of ∼0.5 with 1 mM IPTG, and samples were taken immediately before addition of IPTG and after 1 and 2 h. We observed a zone of clearing on the zymogram (Fig. 4A) at the site where the overexpressed NlpD protein migrated on the gel (determined by subsequent Coomassie blue staining of the gel) (Fig. 4B). Since the zone of clearing may be due to either NlpD binding the PG or NlpD causing PG hydrolysis (5, 25), we performed experiments to address each of these possibilities.
FIG 4.
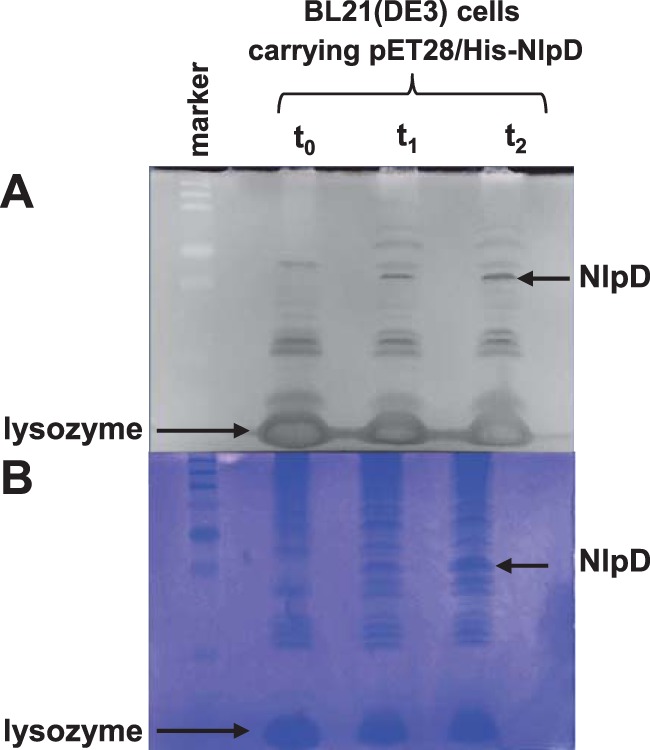
Zymogram analysis of NlpD interactions with PG. (A) E. coli cell extracts of BL21(DE3) carrying pET28/His-NlpD before (t0) and 1 and 2 h after (t1 and t2) induction with IPTG were separated on an SDS-PAGE zymogram gel containing gonococcal PG and were renatured and stained with methylene blue. Negative image of zones of clearing created by NlpD and lysozyme (seen as dark bands) are indicated with arrows. (B) Subsequent (positive) staining of the same gel with Coomassie shows migration of overexpressed NlpD and lysozyme added for cell lysis (dark bands) indicated by arrows.
(ii) NlpD binds PG in vitro.
NlpD contains a characteristic LysM (PG-binding) domain. To test whether purified NlpD is able to bind PG in vitro, we performed a series of PG pulldown experiments (Fig. 5). We incubated NlpD with and without exogenous PG for 2 h at 4°C in 100 μl of PBS buffer and collected the supernatant after centrifugation. We then performed two successive washes with PBS buffer at 4°C and analyzed each of these fractions along with the remaining pellet by SDS-PAGE. Much more NlpD protein was present in the pelleted fraction containing PG than that found in the control reactions where no PG was added (Fig. 5). Moreover, control reactions using purified E. coli RecA and bovine serum albumin (BSA) revealed no increase in these control proteins in reaction mixtures containing PG compared to reaction mixtures lacking PG (data not shown), indicating that the interaction between NlpD and PG is specific.
FIG 5.
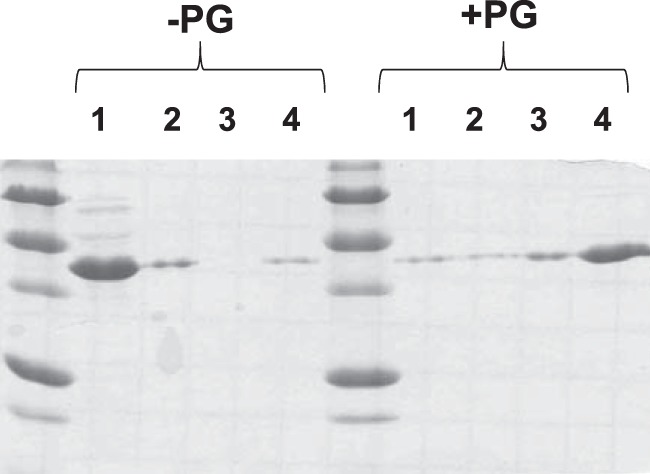
NlpD binds PG in vitro. Ten micrograms of purified NlpD protein was incubated in binding buffer with or without PG for 2 h at 4°C. Supernatant was collected by centrifugation at 14,000 × g for 20 min at 4°C (lanes 1). Two wash steps followed by centrifugation (lanes 2 and 3) and the remaining pellet (lanes 4) were analyzed by SDS-PAGE followed by staining with Coomassie blue.
(iii) NlpD requires AmiC to degrade PG in vitro.
To further characterize the function of NlpD, we tested the ability of purified NlpD protein to degrade PG using a dye release assay (22) (Fig. 6). Gonococcal PG was covalently labeled on its sugar moieties with the colorimetric dye RBB. Purified NlpD protein was incubated with RBB-labeled PG. NlpD hydrolytic activity was monitored over time by measuring the amount of color (dye) present in the supernatant after terminating the reaction and centrifugation to pellet the intact PG. No hydrolase activity was observed for NlpD, even after 20 h of incubation (Fig. 6 and data not shown).
FIG 6.
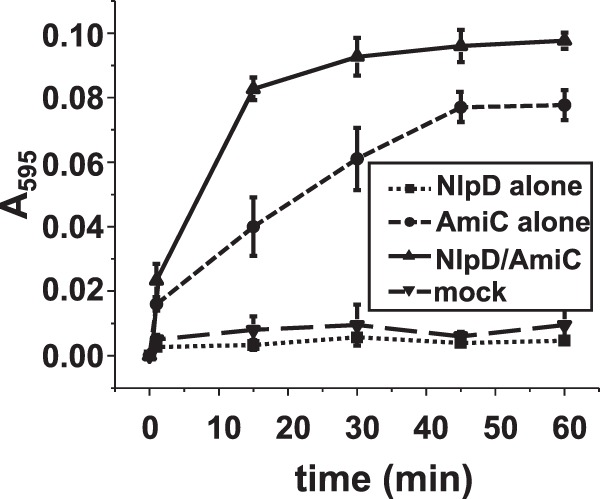
NlpD itself does not cleave PG but rather potentiates PG cleavage by AmiC. Data shown are from an RBB dye release assay measuring PG hydrolysis by NlpD, AmiC, NlpD and AmiC, and mock treatment. Reaction mixtures containing a 4 µM concentration of each indicated protein (or no protein) were incubated at 37°C for the indicated amounts of time. Samples were removed, heat inactivated at 95°C for 5 min, and centrifuged at 14,000 × g for 30 min. The absorbance of the supernatants was measured at 595 nm. The error bars represent the standard error of the mean from three experiments.
In E. coli, the NlpD protein acts in conjunction with the AmiC protein to effect PG hydrolysis. Our currently observed ΔnlpD mutant phenocopies a previously described amiC mutant in N. gonorrhoeae (11). Therefore, we sought to determine whether the NlpD and AmiC proteins were functional partners in N. gonorrhoeae as they are in E. coli. To do this, we performed an RBB-release assay incubating labeled PG with the two proteins individually and together. With the addition of AmiC and NlpD to the reaction, we observed the largest amount of PG hydrolysis; however, AmiC alone exhibited modest PG hydrolysis (Fig. 6). Therefore, we conclude that the function of NlpD in the gonococcal cell is to potentiate the hydrolytic activity of AmiC.
DISCUSSION
Cell division and separation are fundamental processes; however, the present knowledge of bacterial separation stems largely from studies on rod-shaped bacteria. Several recent articles have focused on the relatively complex process of cell separation and its spatiotemporal regulation (5, 26, 27) by LytM factors. We sought to investigate one of these genes, nlpD, from N. gonorrhoeae. The gonococcal nlpD gene was cloned, insertionally inactivated, and found to profoundly affect viability, growth rate, and colony and cell morphology, all of which likely stem from a defect in cell separation. Further experiments revealed that, although NlpD binds PG, it has no hydrolytic activity by itself, but it can stimulate AmiC's PG hydrolytic activity. This work clearly shows that NlpD acts to potentiate the activity of the amidase AmiC in cell separation in N. gonorrhoeae.
The purified NlpD protein has no hydrolytic activity by itself on PG. However, even in the absence of NlpD, AmiC is able to hydrolyze quite a bit of PG in vitro (Fig. 6). Interestingly, although AmiC is present in the ΔnlpD mutant neisserial cells, the cells have a large defect in cell separation, which phenocopies the defect in cell separation identified previously in an amiC mutant (11). This demonstrates that AmiC exhibits activity in vitro that it lacks in the neisserial cell. The E. coli AmiC was also able to hydrolyze PG in the absence of its activator NlpD (5). One possible explanation for this is that the autoinhibitory helix at the C terminus of AmiC, which NlpD displaces in E. coli (21), does not have the same degree of autoinhibition in vitro as it does inside the bacterial cell.
The rod-shaped bacteria E. coli, V. cholerae, H. influenzae, and Yersinia pestis all exhibit various degrees of functional redundancy of septal PG cleavage enzymes and their activators, and differing phenotypes result from inactivation of nlpD in these four bacteria. E. coli contains three amidases, AmiA, AmiB, and AmiC, which are regulated by NlpD (AmiC) and EnvC (AmiA and AmiB) (27). The situation is slightly more streamlined in V. cholerae, which has a single amidase, AmiB, which is regulated by NlpD and EnvC (8). Because of the redundancy of these regulatory genes in E. coli and V. cholerae, deletion of nlpD has only minor, if any, effects on cellular morphology (8, 27, 28). A recent report on LytM proteins in H. influenzae showed that an nlpD mutant not only exhibited defects in cell separation and growth rate but also showed increased outer membrane vesicle release, indicating a role for NlpD in membrane stability (9). In contrast, we did not observe an increase in vesicle release in the gonococcal ΔnlpD strain (data not shown). Finally, although an nlpD mutant of Y. pestis exhibited normal growth, it showed defects in cell separation and was attenuated in virulence in mice (29). In N. gonorrhoeae, inactivation of nlpD alone is sufficient to cause gross changes in cell separation, and the nlpD mutant phenocopies the amiC mutant (11). Thus, N. gonorrhoeae has the most streamlined cell separation system that has been described to date, consisting only of AmiC, NlpD, and LtgC (11, 30). Further work will determine whether other coccal organisms also exhibit this pared-down cell separation machinery and will also identify the genes required for cell separation.
Although N. gonorrhoeae has an EnvC homolog (NGO571), deletion of the gene had no effect on cellular or colonial morphology (data not shown). However, several gonococcal genes have been identified whose products affect the degree of cell separation as well as other assorted phenotypes. The tpc, rdgC, and minD mutants in N. gonorrhoeae all result in defects in cell separation (10, 31, 32) The minD mutant shows an aberrant cell morphology, with cells of many sizes growing in clumps, and is deficient in growth and viability (10). Although growth of the tpc mutant is not affected, it shows a specific tetracoccal form (two diplococci fused together), where the outer membrane appears to surround the entire tetracoccal structure. This mutant also shows defects in transformation competence and epithelial cell invasion, which are virulence-associated traits (31). rdgC mutants (32) are negatively affected in growth or cell viability and have subtle defects in cell separation. The rdgC mutant also shows decreased pilin antigenic variation, a Neisseria-specific process whereby the amino acid sequence of the main pilus component, pilin, is altered.
Elucidating the details of cell division in the pathogenic Neisseria spp. will provide essential information about this process in a coccal bacterium. Studies of cell division in additional coccal organisms will further elaborate the differences and similarities between coccal and rod-shaped bacteria and will enrich our understanding about this fundamental cellular process.
ACKNOWLEDGMENTS
We thank Lennell Reynolds in the Center for Advanced Microscopy at Northwestern University Feinberg School of Medicine for assistance with TEM. We thank Alice Chateau and John Brooks for assistance with cell imaging using the Nikon Eclipse 90i microscope.
The NIH provided funding to H. Steven Seifert under grant numbers R01-AI044239 and R37-AI033493, to Joseph P. Dillard under grant numbers R01-AI097157 and R21-AI099539, and to Jonathan D. Lenz under grant number T32-AI055379.
REFERENCES
- 1.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 2.Pinho MG, Kjos M, Veening JW. 2013. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 3.de Boer PA. 2010. Advances in understanding E. coli cell fission. Curr Opin Microbiol 13:730–737. doi: 10.1016/j.mib.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Holtje JV. 2001. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol 41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- 5.Uehara T, Parzych KR, Dinh T, Bernhardt TG. 2010. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J 29:1412–1422. doi: 10.1038/emboj.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rawlings ND, Tolle DP, Barrett AJ. 2004. MEROPS: the peptidase database. Nucleic Acids Res 32:D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odintsov SG, Sabala I, Marcyjaniak M, Bochtler M. 2004. Latent LytM at 1.3A resolution. J Mol Biol 335:775–785. doi: 10.1016/j.jmb.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Moll A, Dorr T, Alvarez L, Chao MC, Davis BM, Cava F, Waldor MK. 2014. Cell separation in Vibrio cholerae is mediated by a single amidase whose action is modulated by two nonredundant activators. J Bacteriol 196:3937–3948. doi: 10.1128/JB.02094-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ercoli G, Tani C, Pezzicoli A, Vacca I, Martinelli M, Pecetta S, Petracca R, Rappuoli R, Pizza M, Norais N, Soriani M, Arico B. 2015. LytM proteins play a crucial role in cell separation, outer membrane composition, and pathogenesis in nontypeable Haemophilus influenzae. mBio 6:e02575-14. doi: 10.1128/mBio.02575-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szeto J, Ramirez-Arcos S, Raymond C, Hicks LD, Kay CM, Dillon JA. 2001. Gonococcal MinD affects cell division in Neisseria gonorrhoeae and Escherichia coli and exhibits a novel self-interaction. J Bacteriol 183:6253–6264. doi: 10.1128/JB.183.21.6253-6264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia DL, Dillard JP. 2006. AmiC functions as an N-acetylmuramyl-l-alanine amidase necessary for cell separation and can promote autolysis in Neisseria gonorrhoeae. J Bacteriol 188:7211–7221. doi: 10.1128/JB.00724-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stohl EA, Chan YA, Hackett KT, Kohler PL, Dillard JP, Seifert HS. 2012. Neisseria gonorrhoeae virulence factor NG1686 is a bifunctional M23B family metallopeptidase that influences resistance to hydrogen peroxide and colony morphology. J Biol Chem 287:11222–11233. doi: 10.1074/jbc.M111.338830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stohl EA, Dale EM, Criss AK, Seifert HS. 2013. Neisseria gonorrhoeae metalloprotease NGO1686 is required for full piliation, and piliation is required for resistance to H2O2- and neutrophil-mediated killing. mBio 4:e00399-13. doi: 10.1128/mBio.e00399-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kellogg DS Jr, Peacock WL Jr, Deacon WE, Brown L, Pirkle DI. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J Bacteriol 85:1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 16.Seifert HS, Wright CJ, Jerse AE, Cohen MS, Cannon JG. 1994. Multiple gonococcal pilin antigenic variants are produced during experimental human infections. J Clin Invest 93:2744–2749. doi: 10.1172/JCI117290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long CD, Tobiason DM, Lazio MP, Kline KA, Seifert HS. 2003. Low-level pilin expression allows for substantial DNA transformation competence in Neisseria gonorrhoeae. Infect Immun 71:6279–6291. doi: 10.1128/IAI.71.11.6279-6291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koomey JM, Falkow S. 1987. Cloning of the recA gene of Neisseria gonorrhoeae and construction of gonococcal recA mutants. J Bacteriol 169:790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cahoon LA, Seifert HS. 2009. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science 325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zahrl D, Wagner M, Bischof K, Bayer M, Zavecz B, Beranek A, Ruckenstuhl C, Zarfel GE, Koraimann G. 2005. Peptidoglycan degradation by specialized lytic transglycosylases associated with type III and type IV secretion systems. Microbiology 151:3455–3467. doi: 10.1099/mic.0.28141-0. [DOI] [PubMed] [Google Scholar]
- 21.Rocaboy M, Herman R, Sauvage E, Remaut H, Moonens K, Terrak M, Charlier P, Kerff F. 2013. The crystal structure of the cell division amidase AmiC reveals the fold of the AMIN domain, a new peptidoglycan binding domain. Mol Microbiol 90:267–277. [DOI] [PubMed] [Google Scholar]
- 22.Zhou R, Chen S, Recsei P. 1988. A dye release assay for determination of lysostaphin activity. Anal Biochem 171:141–144. doi: 10.1016/0003-2697(88)90134-0. [DOI] [PubMed] [Google Scholar]
- 23.Stohl EA, Criss AK, Seifert HS. 2005. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol 58:520–532. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sechman EV, Rohrer MS, Seifert HS. 2005. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol 57:468–483. doi: 10.1111/j.1365-2958.2005.04657.x. [DOI] [PubMed] [Google Scholar]
- 25.Kohler PL, Hamilton HL, Cloud-Hansen K, Dillard JP. 2007. AtlA functions as a peptidoglycan lytic transglycosylase in the Neisseria gonorrhoeae type IV secretion system. J Bacteriol 189:5421–5428. doi: 10.1128/JB.00531-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uehara T, Bernhardt TG. 2011. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr Opin Microbiol 14:698–703. doi: 10.1016/j.mib.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uehara T, Dinh T, Bernhardt TG. 2009. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J Bacteriol 191:5094–5107. doi: 10.1128/JB.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lange R, Hengge-Aronis R. 1994. The nlpD gene is located in an operon with rpoS on the Escherichia coli chromosome and encodes a novel lipoprotein with a potential function in cell wall formation. Mol Microbiol 13:733–743. doi: 10.1111/j.1365-2958.1994.tb00466.x. [DOI] [PubMed] [Google Scholar]
- 29.Tidhar A, Flashner Y, Cohen S, Levi Y, Zauberman A, Gur D, Aftalion M, Elhanany E, Zvi A, Shafferman A, Mamroud E. 2009. The NlpD lipoprotein is a novel Yersinia pestis virulence factor essential for the development of plague. PLoS One 4:e7023. doi: 10.1371/journal.pone.0007023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cloud KA, Dillard JP. 2004. Mutation of a single lytic transglycosylase causes aberrant septation and inhibits cell separation of Neisseria gonorrhoeae. J Bacteriol 186:7811–7814. doi: 10.1128/JB.186.22.7811-7814.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fussenegger M, Kahrs AF, Facius D, Meyer TF. 1996. Tetrapac (tpc), a novel genotype of Neisseria gonorrhoeae affecting epithelial cell invasion, natural transformation competence and cell separation. Mol Microbiol 19:1357–1372. doi: 10.1111/j.1365-2958.1996.tb02479.x. [DOI] [PubMed] [Google Scholar]
- 32.Mehr IJ, Long CD, Serkin CD, Seifert HS. 2000. A homologue of the recombination-dependent growth gene, rdgC, is involved in gonococcal pilin antigenic variation. Genetics 154:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]