

ABSTRACT
The squid light organ symbiont Vibrio fischeri controls bioluminescence using two acyl-homoserine lactone pheromone-signaling (PS) systems. The first of these systems to be activated during host colonization, AinS/AinR, produces and responds to N-octanoyl homoserine lactone (C8-AHL). We screened activity of a PainS-lacZ transcriptional reporter in a transposon mutant library and found three mutants with decreased reporter activity, low C8-AHL output, and other traits consistent with low ainS expression. However, the transposon insertions were unrelated to these phenotypes, and genome resequencing revealed that each mutant had a distinct point mutation in luxO. In the wild type, LuxO is phosphorylated by LuxU and then activates transcription of the small RNA (sRNA) Qrr, which represses ainS indirectly by repressing its activator LitR. The luxO mutants identified here encode LuxU-independent, constitutively active LuxO* proteins. The repeated appearance of these luxO mutants suggested that they had some fitness advantage during construction and/or storage of the transposon mutant library, and we found that luxO* mutants survived better and outcompeted the wild type in prolonged stationary-phase cultures. From such cultures we isolated additional luxO* mutants. In all, we isolated LuxO* allelic variants with the mutations P41L, A91D, F94C, P98L, P98Q, V106A, V106G, T107R, V108G, R114P, L205F, H319R, H324R, and T335I. Based on the current model of the V. fischeri PS circuit, litR knockout mutants should resemble luxO* mutants; however, luxO* mutants outcompeted litR mutants in prolonged culture and had much poorer host colonization competitiveness than is reported for litR mutants, illustrating additional complexities in this regulatory circuit.
IMPORTANCE Our results provide novel insight into the function of LuxO, which is a key component of pheromone signaling (PS) cascades in several members of the Vibrionaceae. Our results also contribute to an increasingly appreciated aspect of bacterial behavior and evolution whereby mutants that do not respond to a signal from like cells have a selective advantage. In this case, although “antisocial” mutants locked in the PS signal-off mode can outcompete parents, their survival advantage does not require wild-type cells to exploit. Finally, this work strikes a note of caution for those conducting or interpreting experiments in V. fischeri, as it illustrates how pleiotropic mutants could easily and inadvertently be enriched in this bacterium during prolonged culturing.
INTRODUCTION
Many bacteria use pheromones to regulate group behaviors (1). These pheromone-signaling (PS) systems generally require sufficiently high cell density for pheromone accumulation and are also regulated in response to the environment (2–6). Accordingly, PS outputs depend on both population density and an appropriate context. The importance of such control is easily rationalized, given the energetic cost of many PS-activated processes, such as bioluminescence (7, 8). This cost of inducing PS-controlled systems is underscored by the observation that spontaneous signal-blind “antisocial” PS-negative mutants often appear in populations, and it is thought that these mutants are enriched as “cheaters” if their lack of participation in group behavior minimizes their own costs while exploiting the presence of PS-positive relatives (9–11).
Vibrio fischeri is an excellent model for studying PS and was central to the discovery of cell-cell communication in bacteria (12). V. fischeri is a bioluminescent light organ symbiont that controls luminescence and other phenotypes using three distinct but interconnected PS systems, with the signal synthase/receptor combinations LuxI/LuxR, AinS/AinR, and LuxS/LuxPQ (13–18). Luminescence is induced largely by LuxI/LuxR, which produces and responds to N-3-oxo-hexanoyl homoserine lactone. However, luxR itself is controlled in part by the other two systems, and LuxR can be activated by the AinS-produced pheromone N-octanoyl-homoserine lactone (C8-AHL). AinS/AinR also controls motility and genes important for initiating colonization of V. fischeri's squid host, Euprymna scolopes (18, 19). The LuxS/LuxPQ system, which synthesizes and responds to autoinducer 2 (AI-2) (20, 21), uses the same core signal transduction pathway as AinS/AinR, but because LuxS/AI-2 has only modest effects in V. fischeri under the conditions tested (17), we have focused more on AinS/AinR.
The AinS/AinR PS system controls luxR and other genes through a core PS circuit (Fig. 1) that is conserved in the Vibrionaceae. Much of this PS cascade was elucidated in Vibrio harveyi, and recent studies have verified parallel functions with subtle differences in V. fischeri (17–19, 22–26). In the model that has emerged (Fig. 1A and B), at low pheromone concentrations (Fig. 1A), AinR phosphorylates LuxU, which in turn phosphorylates the σ54-dependent activator LuxO. LuxO-P activates transcription of a small RNA, Qrr, which posttranscriptionally represses the PS master regulator litR (24, 26). In contrast, when C8-AHL accumulates to higher levels (Fig. 1B), its binding to AinR is thought to decrease AinR's kinase activity, allowing AinR's phosphatase activity to dominate, resulting in more unphosphorylated LuxO, deactivation of qrr, and induction of the LitR regulon, which includes ainSR and luxR. LitR homologs are widespread master regulators of the PS-dependent phenotypes in other members of the Vibrionaceae (17, 22, 23). Spontaneous mutations in Vibrio cholerae hapR and Vibrio parahaemolyticus opaR, which encode their respective PS master regulators, have been enriched under some conditions (11, 27–29), but no parallel to these observations has been reported in V. fischeri.
FIG 1.

Model showing roles of Ain and LuxO in the pheromone regulatory circuit. The pheromone receptor AinR phosphorylates and dephosphorylates LuxU, which phosphorylates LuxO, stimulating it to activate transcription of qrr. The regulatory small RNA (sRNA) Qrr represses expression of the master regulator LitR. (A) At low C8-AHL levels, LuxO-P ultimately leads to relatively low LitR levels. (B) At high C8-AHL levels, binding of C8-AHL to AinR reduces its kinase activity, reducing the relative amounts of phosphorylated LuxU and LuxO, resulting in more LitR and induction of the LitR regulon, including luxR, bioluminescence, and ainSR. On plates, this induction also results in colony opacity through an unknown mechanism. (C) The activity of LuxO* does not require phosphorylation, although prior to this study, the effects of LuxU were untested. LuxO* essentially locks this circuit on “off,” and phenotypes resemble those at low C8-AHL levels. The colonies of luxO* mutants on plates remain translucent despite their high cell density.
The V. fischeri AinS/AinR system is activated early during colonization of its symbiotic host squid and is responsible for priming LuxI/LuxR-based symbiotic luminescence (18). Given that luminescence is only weakly induced outside the host and that AinS/AinR apparently sits atop the PS hierarchy early in infection, regulatory controls over ainSR may reveal important elements of the host environment encountered during symbiosis establishment. Only cyclic AMP receptor protein (CRP) and LitR are known to activate ainSR (17, 22, 30), and the goal of this study was to discover new regulators of ainSR. However, we unwittingly isolated spontaneous mutants in the core PS circuitry, providing insight into its function and revealing conditions under which such mutants can be enriched.
MATERIALS AND METHODS
Bacteria, growth media, and reagents.
Bacterial strains are listed and briefly described in Table 1. V. fischeri ES114 was the wild-type strain used throughout (31). Plasmids were transformed into Escherichia coli strain DH5α (32) or DH5αλpir (33) in the case of plasmids with the R6K origin of replication. E. coli was grown in LB medium (34) or brain heart infusion (BHI) medium (Bacto), and V. fischeri was grown in LB salt (LBS) medium (35), seawater-tryptone marine-osmolarity (SWTO) medium (36), or Fischeri minimal medium (FMM) (4). Solid media were prepared with 15 g liter−1 agar. For selection of E. coli, chloramphenicol (Cam) and kanamycin (Kan) were added to LB at final concentrations of 20 and 100 μg ml−1, respectively, and erythromycin (Erm) was added to BHI at a final concentration of 150 μg ml−1. For selection of V. fischeri on LBS, the concentrations of Cam, Erm, and Kan used were 2, 5, and 100 μg ml−1, respectively. For colorimetric screening of β-galactosidase activity, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was added to LBS at 100 μg ml−1. C8-AHL was obtained from Sigma-Aldrich (St. Louis, MO).
TABLE 1.
Bacterial strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| E. coli strains | ||
| CC118λpir | Δ(ara-leu) araD Δlac74 galE galK phoA20 thi-1 rpsE rpsB argE(Am) recA λpir | 41 |
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 32 |
| DH5α λpir | DH5α lysogenized with λpir | 33 |
| V. fischeri strains | ||
| AKD100 | ES114 Tn7-ermR | 47 |
| AKD200 | ES114 Tn7-camR | 48 |
| CG103 | Wild-type isolate from Cleidopus gloriamaris | 84 |
| CL59 | ES114 luxO(D47E) | 19 |
| DC22 | C8-AHL bioreporter: ES114 ΔainS ΔluxR-luxI, mutant luxR (MJ1-T33A, R67M, S116A, M135I), PluxI-luxCDABEG | 22 |
| DM131 | ES114 flaJ::kanR | 48 |
| EM17 | Wild-type isolate from Euprymna morsei | 85 |
| ES114 | Wild-type isolate from E. scolopes | 31 |
| ES12 | Wild-type isolate from E. scolopes | 86 |
| ES213 | Wild-type isolate from E. scolopes | 86 |
| ES401 | Wild-type isolate from E. scolopes | 85 |
| ET101 | Wild-type isolate from Euprymna tasmanica | 87 |
| H905 | Planktonic isolate, Hawaii | 76 |
| KV1319 | ES114 with 2-bp insertion in celG | 51 |
| KV5005 | ES114 ΔrpoN | 43 |
| JB18 | ES114 litR::ermR | 22 |
| JHK057 | ES114 luxO(V106A) | This study |
| JHK061 | ES114 luxO(T335I) | This study |
| JHK062 | ES114 luxO(V106A) ΔrpoN | This study |
| JHK063 | ES114 luxO(T335I) ΔrpoN | This study |
| JHK065 | ES114 luxO(V106A) luxU::pEVS122 | This study |
| JHK066 | ES114 luxO(T335I) luxU::pEVS122 | This study |
| JHK068 | ES114 luxU::pEVS122 | This study |
| JHK069 | ES114 luxO(D47E) luxU::pEVS122 | This study |
| JHK070 | ES114 luxO(V106G) | This study |
| JHK073 | ES114 luxO(V106A) litR::ermR | This study |
| JHK074 | ES114 luxO(A91D) | This study |
| JHK075 | ES114 luxO(P41L) | This study |
| JHK076 | ES114 luxO(A91D) luxU::pEVS122 | This study |
| JHK077 | ES114 luxO(P41L) luxU::pEVS122 | This study |
| JHK087 | ES114 luxO(V106G) luxU::pEVS122 | This study |
| JHK105 | ES114 luxO(T335I) Δqrr | This study |
| NL60 | ES114 ΔainS | 4 |
| PMF8 | ES114 litR::kanR | 23 |
| PP3 | Planktonic isolate, Hawaii | 76 |
| TIM305 | ES114 Δqrr | 24 |
| TIM306 | ES114 ΔluxO | 24 |
| TME014F5 | ES114 mtsA::mini-Tn5 ermR | This study |
| TME014B6 | ES114 znuC2::mini-Tn5 ermR | This study |
| TME021D9 | ES114 yfhD::mini-Tn5 ermR | This study |
| VFS002F6 | ES114 vf0295::mini-Tn5 ermR | C. Whistler |
| VFS002F6-Tb | ES114 vf0295::mini-Tn5 luxO(A91D) ermR | C. Whistler |
| VFS012E9 | ES114 yfbQ::mini-Tn5 ermR | C. Whistler |
| VFS012E9-T | ES114 yfbQ::mini-Tn5 luxO(P41L) ermR | C. Whistler |
| VFS014F5-T | ES114 mtsA::mini-Tn5 luxO(V106A) ermR | C. Whistler |
| VFS014B6 | ES114 znuC2::mini-Tn5 ermR | C. Whistler |
| VFS014B6-T | ES114 znuC2::mini-Tn5 luxO(V106G) ermR | C. Whistler |
| VFS021D9-Tc | ES114 yfhD::mini-Tn5 luxO(T335I) ermR | C. Whistler |
| VLS2 | Wild-type isolate from E. scolopes | 84 |
| WH1 | Planktonic isolate, Massachusetts | 84 |
| WTTG0 | ES114 luxO(T107R) | This study |
| WTTG1 | ES114 luxO(F94C) | This study |
| WTTG2 | ES114 luxO(R114P) | This study |
| WTTG3 | ES114 luxO(H324R) | This study |
| WTTG4 | ES114 luxO(P98Q) | This study |
| WTTG5 | ES114 luxO(R113L) | This study |
| WTTG12 | ES114 luxO(P98L) | This study |
| WTTG17 | ES114 luxO(V108G) | This study |
| WTTG20 | ES114 luxO(V106G) | This study |
| WTTG21 | ES114 luxO(V106G) | This study |
| WTTG22 | ES114 luxO(H319R) | This study |
| WTTG24 | ES114 luxO(L205F) | This study |
| Plasmidsd | ||
| pAKD701 | Promoterless lacZ, pES213 R6Kγ oriTRP4 kanR | 37 |
| pEVS79 | pBC SK (+) oriT camR | 40 |
| pEVS104 | Conjugative helper plasmid, R6Kγ oriTRP4 kanR | 40 |
| pEVS122 | R6Kγ oriTRP4 ermR lacZα | 33 |
| pEVS170 | Mini-Tn5-ermR R6Kγ oriTRP4 kanR | 50 |
| pHK10 | PainS-lacZ reporter in pAKD701, pES213 R6Kγ oriTRP4 kanR | This study |
| pHK11 | mtsA in pVSV104, kanR | This study |
| pHK12 | PainS-gfp Pcon--mCherry in pJLS27, pES213 R6Kγ oriTRP4 kanR camR | This study |
| pHK20 | Pqrr-lacZ reporter in pAKD701, pES213 R6Kγ oriTRP4 kanR | 22 |
| pHK29 | znuC2 in pVSV104, kanR | This study |
| pHK45 | ES114 luxO in pVSV105, camR | This study |
| pHK70 | luxO(V106A) in pVSV105, camR | This study |
| pHK71 | luxO(D47E) in pVSV105, camR | This study |
| pHK73 | luxO(T335I) in pVSV105, camR | This study |
| pHK77 | luxO(V106A) and flanking sequence in pEVS79, camR | This study |
| pHK78 | luxO(T335I) and 1.5 kbp flanking sequence in pEVS79, camR | This study |
| pHK79 | 228-bp luxU fragment in pEVS122, ermR | This study |
| pHK80 | luxO(V106G) and flanking sequence in pEVS79, camR | This study |
| pHK82 | luxO(V106G) in pVSV105, camR | This study |
| pHK83 | luxO(A91D) in pVSV105, camR | This study |
| pHK84 | luxO(P41L) in pVSV105, camR | This study |
| pHK85 | luxO(P41L) and flanking sequence in pEVS79, camR | This study |
| pHK86 | luxO(A91D) and flanking sequence in pEVS79, camR | This study |
| pHK87 | luxO(T335I) and 1.1-kbp flanking sequence in pEVS79, camR | This study |
| pJLB95 | litR::ermR (opposite), ColE1 camR | This study |
| pJLS27 | Promoterless gfp, Pcon-mCherry pES213 R6Kγ oriTRP4 kanR | 38 |
| pLosTfoX | tfoX in pEVS79, camR | 42 |
| pMSM28 | ΔrpoN allele, R6Kγ oriTRP4 camR | 43 |
| pMulTfoX | tfoX in pVSV104, kanR | 42 |
| pTM267 | kan gfp PtetA-mCherry in pVSV105, camR | 24 |
| pTM268 | Pqrr-gfp PtetA-mCherry in pVSV105, camR | 24 |
| pVSV104 | Shuttle vector; pES213 R6Kγ oriTRP4 kanR lacZα | 39 |
| pVSV105 | Shuttle vector; pES213 R6Kγ oriTRP4 camR lacZα | 39 |
| Oligonucleotidese | ||
| pr_HK03 | GGGGCATGCAGAACCAAGACCTGCTCGTGCTAA | This study |
| pr_HK04 | GGCGCTAGCCATCAGTTGTTGAAGTAAATTAAAATTCTGCG | This study |
| pr_HK05 | CATGGTACCATATAGCCGTCTAGATGTAAACATTTCCAAACCG | This study |
| pr_HK06 | CATCCTAGGTTATGGCGCCTCTGTTAAATTAGTACTTTGTTTT | This study |
| pr_HK07 | CATGGTACCAAAATTAATTGTTATGTTATAACATAACAATTAAATAGCC | This study |
| pr_HK08 | CATCCTAGGCATCCAACATTCAGTACACTCCC | This study |
| pr_HK29 | GGCTCTAGACATCAGTTGTTGAAGTAAATTAAAATTCTGCG | This study |
| pr_HK73 | CATGGCATGCATATACCTATTGCAGGGAGCGTGC | This study |
| pr_HK74 | CATGGGTACCCAGCGATTTGATTAACATACTGACTCACGATAG | This study |
| pr_HK93 | CATGGGATCCAGACTATTTATGTCTCAGCCACACC | This study |
| pr_HK94 | CATGGGTACCATCGCCAAATCATGATTGG | This study |
| pr_HK95 | CATGGGATCCTCGGAAGTTGCAGAAGAAGG | This study |
| pr_HK96 | CATGGGTACCTGGTTCCACAGGCCGTATAC | This study |
| pr_HK101 | CATGGGATCCGGGACTATCGTGAGTCAGTA | This study |
| pr_HK102 | CATGGGATCCTGCCATTGTTGCAAGCTTATCT | This study |
| pr_HK110 | CATGGGATCCTGCAAATTGCGTTTTGCG | This study |
| pr_HK111 | CATGGGTACCGCTGTCAAACAAGCGGATTTAATA | This study |
Genes for drug resistance are as follows: camR, chloramphenicol resistance; ermR, erythromycin resistance; kanR, kanamycin resistance (aph).
Strains designated with the prefix “VFS” and the suffix “-T” were isolated from a mapped-transposon mutant library and represent the translucent (T) luxO*-bearing derivatives of the original transposon mutant parent. Strains designated with “WTTG” were isolated in a series of independent static culture survival experiments.
VFS021D9-T lacked the Kan resistance gene from the transposon delivery vector but displayed some Kan resistance.
All alleles cloned in this study are from V. fischeri strain ES114. Replication origins of the vectors are listed as R6Kγ, ColE1, oriV, and/or pES213. Plasmids based on pES213 are stable and do not require antibiotic selection for maintenance (39).
All oligonucleotides are shown 5′ to 3′. Restriction enzyme recognition sequences are underlined.
Molecular genetics and sequence analysis.
Oligonucleotides and plasmids are listed in Table 1, and the latter were constructed using standard techniques and materials as described previously (22). Genomic DNA (gDNA) for genome resequencing and natural transformation was purified using the Easy-DNA gDNA purification kit (Life Technologies, Grand Island, NY).
The PainS-lacZ transcriptional reporter plasmid pHK10 was generated by PCR amplifying 428 bp upstream of ainS using primers pr_HK03 and pr_HK04, digesting the resulting amplicon with SphI and NheI, and cloning this fragment between the SphI and NheI sites of pAKD701 (37). To generate the PainS-gfp transcriptional reporter, pHK12, the same promoter region used in pHK10 was amplified using primers pr_HK03 and pr_HK29, digested with SphI and XbaI, and ligated into similarly digested pJLS27 (38). To generate pHK45, pHK70, pHK71, pHK73, pHK82, pHK83, and pHK84, luxO was amplified from ES114, VFS014F5-T, CL59 (19), VFS021D9-T, VFS014B6-T, VFS002F6-T, and VFS012E9-T, respectively, using primers pr_HK73 and pr_HK74. The resulting amplicons were digested with SphI and KpnI and ligated into SphI- and KpnI-digested pVSV105 (39). To generate pHK11, mtsA was amplified from ES114 using primers pr_HK05 and pr_HK06. The resulting amplicon was digested with KpnI and AvrII and ligated into KpnI- and AvrII-digested pVSV104 (39). To generate pHK29, znuC2 was amplified from ES114 using primers pr_HK07 and pr_HK08. The resulting amplicon was digested with KpnI and AvrII and ligated into KpnI- and AvrII-digested pVSV104.
Mutant alleles were transferred from E. coli into V. fischeri on plasmids by triparental matings using the conjugative helper strain CC118λpir pEVS104 (40, 41). Recombination and marker exchange were identified by screening for antibiotic resistance, and putative mutants were tested by PCR. Transposon insertions were placed in new strain backgrounds using competence induced by overexpression of tfoX from pMulTfoX or pLosTfoX, followed by plasmid curing, as previously described (42). Strains TME014F5, TME014B6, and TME021D9 were generated using DNA from transposon mutants VFS014F5-T, VFS014B6-T, and VFS021D9-T, respectively, using tfoX-mediated transformation (42). To place spontaneous luxO mutations in fresh strain backgrounds, 1.5-kbp regions flanking the mutation sites were PCR amplified using primer sets pr_HK93 and pr_HK94 (P41L, A91D, V106A, and V106G alleles) or pr_HK95 and pr_HK96 (T335I allele), respectively. Amplicons were digested with KpnI and BamHI and ligated into similarly digested pEVS79 (40) to create pHK77 [luxO(V106A)], pHK78 [luxO(T335I)], pHK80 [luxO(V106G)], pHK85 [luxO(P41L)], and pHK86 [luxO(A91D)]. Strains JHK057, JHK061, JHK070, JHK074, and JHK075 were generated by exchanging the mutated luxO variants on pHK77, pHK78, pHK80, pHK85, and pHK86, respectively, into ES114. To generate the luxO* Δqrr double mutant, 1.1-kbp regions flanking the mutation site of luxO*(T335I) were PCR amplified using primers pr_HK110 and pr_HK111. This amplicon was digested with KpnI and BamHI and ligated into similarly digested pEVS79 to create pHK87. The insert on pHK87 is smaller than that of pHK78 and does not overlap the qrr sequence. Strain JHK105 was generated by exchanging the mutated luxO variant on pHK87 into the Δqrr strain TIM305.
To generate luxU mutants, an internal 228-bp fragment of luxU was amplified using primers pr_HK101 and pr_HK102. The resulting amplicon was digested with BamHI and ligated into BamHI-digested pEVS122 (33) to generate pHK79. The luxU::pEVS122 allele of pHK79 was introduced into JHK057, JHK061, ES114, CL59, JHK070, JHK074, and JHK075 to generate strains JHK065, JHK066, JHK068, JHK069, JHK087, JHK076, and JHK077, respectively. To generate rpoN mutants, the ΔrpoN allele on plasmid pMSM28 (43) was introduced into strains JHK057 and JHK061 to generate strains JHK062 and JHK063, respectively. To generate the luxO* litR::erm double mutant, the litR::erm allele on pJLB95 (22) was introduced into JHK057 to generate strain JHK073.
Genome resequencing.
Ten micrograms of gDNA was fragmented by sonication in iced Tris-EDTA buffer five times for 1 min with 1-s pulses at 40% duty using a W-380 ultrasonic processor (Heat Systems-Ultrasonics, Inc.) to produce ∼200-bp fragments for Illumina library construction following the Illumina Tru-seq manufacturer's protocol (San Diego, CA), except that genomic DNA adaptors were diluted 1:100 prior to ligation. Sequencing was performed using an Illumina Hi-Seq 2000 genome analyzer at the University of Missouri DNA Core Laboratory. Assembled reads were compared to the resequenced and published ES114 genomes (44, 45) as references using the Integrative Genomics Viewer (46).
Luminescence measurements.
Overnight V. fischeri cultures were diluted 1:1,000 in 25 ml SWTO medium in 125-ml flasks and incubated with shaking (200 rpm) at 24°C. At regular intervals, the optical density at 595 nm (OD595) was measured for 500-μl samples using a BioPhotometer (Brinkman Instruments, Westbury, NY). Relative luminescence was measured with a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA) immediately following shaking to aerate the sample. Specific luminescence was calculated as the luminescence per unit of optical density at 595 nm (OD595).
lacZ and gfp reporter assays.
Strains harboring the PainS-lacZ or Pqrr-lacZ reporter plasmids pHK10 and pHK20 (22), respectively, or the promoterless parent vector pAKD701, were grown overnight in LBS, subcultured 1:300 into 1.5 ml of SWTO medium in 24-well microtiter plates, and incubated with shaking (200 rpm) at 24°C. Cells were collected at an OD595 of ∼2.5 by centrifugation, the supernatant was discarded, and the cell pellet was stored overnight at −80°C. β-Galactosidase assays were then performed as previously described (7). Strains harboring the PainS-gfp or Pqrr-gfp reporter plasmids pHK12 and pTM268 (24), respectively, or the promoterless parent vectors pJLS27 and pTM267 were grown overnight in LBS, subcultured 1:1,000 into flasks containing 25 ml SWTO medium, and incubated with shaking (200 rpm) at 24°C. At regular intervals, 200-μl samples were aliquoted into clear-bottomed, black-walled, 96-well plates, where green fluorescence and OD595 were measured using a Synergy 2 plate reader (BioTek).
Motility assays.
Motility was determined by diluting overnight cultures 1:1,000 in SWTO medium and growing to an OD595 of ∼0.5. Five-microliter aliquots of culture were then spotted on the surface of 0.25% agar FMM plates containing 2.2 mM N-acetylglucosamine (19). The diameters of areas visibly covered by swimming cells were measured after 24 h of incubation at 28°C. The Erm-resistant ES114 derivative AKD100 and the ES114 flaJ::aph strain DM131 were used as positive and negative motility controls, respectively (47, 48).
C8-AHL bioassays.
C8-AHL accumulation was assessed as previously described (22). Briefly, culture supernatants were extracted with acidified ethyl acetate, extracts were dried and resuspended in SWTO medium, and C8-AHL levels were determined by comparison to standards using the bioassay strain DC22 (22, 49).
Transposition frequency.
The frequency of recovering transposon mutants following conjugative introduction of pEVS170 (50) was determined for ES114 and the luxO mutant JHK057, each mixed 1:1 with the celG mutant KV1319, which is phenotypically wild type except that it can be readily distinguished as white colonies on LBS plates containing 10 mM d-cellobiose and X-Gal (51). Performing transposon (Tn) mutagenesis on 1:1 mixtures of ES114 plus KV1319 or of JHK057 plus KV1319 enabled us to normalize values to transposition frequency in KV1319 and to test whether a luxO mutant such as JHK057 could be enriched from a mixed population during transposon mutagenesis. Conjugation spots were incubated at 28°C for 6 h before being resuspended in 500 μl Instant Ocean (Aquarium Systems, Mentor, OH), dilution plated to determine recipient strain ratios, and then stored overnight at −80°C in glycerol stocks. Stocked conjugations were thawed at room temperature and plated on LBS containing d-cellobiose, X-Gal, and Erm to select transposon-containing strains and to use blue/white screening to distinguish each mutant's parent. Erm-resistant colonies were patched on Kan plates to eliminate mutants with the Kan marker outside the transposon on pEVS170.
Viability in static culture.
To monitor survival rates of V. fischeri strains singly, overnight V. fischeri cultures were inoculated into LBS and grown to an OD595 of ∼2.5 to simulate the inoculum density present in the construction of the transposon mutant library. These cultures were then diluted 1:1,000 in fresh LBS and grown statically in 24-well microtiter plates at 28°C. CFU were determined for the initial inoculum and after 24, 48, and 96 h. To determine mixed-culture viability, strains were grown separately as described above, and at an OD595 of ∼2.0, cultures were mixed in ratios of 1:1, 10:1, or 100:1 and cocultured until an OD595 of ∼2.5 was reached, at which point they were subcultured, grown, and sampled as described above. Strains were differentiated by colony opacity (ES114 versus the luxO* or litR::ermR strain), Erm resistance (the luxO* strain versus the litR::ermR or luxO* litR::erm strain), or Cam resistance (AKD200 versus the luxO* or luxO* Δqrr strain).
Squid colonization.
Competitive colonization of E. scolopes hatchlings was determined as previously described (51). Briefly, hatchlings were exposed to an ∼1:1 mixture of strains for 12 h, and the ratio of colonizing strains was determined 48 h after initial exposure to the inoculum. The relative competitive index (RCI) was determined by dividing the final mutant-to-wild-type ratio by the ratio of the strains in the inoculum. Unmarked luxO mutants were competed against the Camr-marked V. fischeri strain AKD200, which has wild-type colonization competitiveness (48, 52).
RESULTS
In an attempt to identify regulators of ainSR, we used blue/white screening on plates containing X-Gal to test activity of a PainS-lacZ reporter in a library of V. fischeri transposon mutants. Collectively, this library has representative mutants with insertions in approximately 2,100 distinct genes (R. Foxall and C. Whistler, personal communication). Mutants VFS021D9-T, VFS014B6-T, and VFS014F5-T, with Tn insertions in yfhD (VF_A0984), znuC2 (VF_2367), and mtsA (VF_1566), respectively, had markedly lower PainS-lacZ reporter activity. The Tn insertions from these three mutants were reintroduced into the ES114 wild-type background, and these backcrossed Tn mutants were compared to the original mutants, ES114, and the ΔainS mutant NL60, with respect to PainS-gfp reporter activity and three other phenotypes associated with loss of ainS: (i) decreased luminescence in broth, (ii) increased swimming motility, and (iii) lowered production of C8-AHL (18, 19). VFS021D9-T, VFS014B6-T, and VFS014F5-T displayed decreased PainS-gfp expression (Fig. 2A), relatively dim luminescence (Fig. 2B), hypermotility (Fig. 2C), and low C8-AHL output (Fig. 2D); however, none of these phenotypes were associated with the Tn insertions backcrossed into ES114 (Fig. 2). Moreover, wild-type copies of mtsA and znuC2 provided in trans failed to complement the mutant phenotypes of strains VFS014F5-T and VFS014B6-T (data not shown), further indicating that the Tn insertions in these mutants were not the cause of the ainS-related phenotypes.
FIG 2.

Transposon library mutants (VFS strains), but not their backcrossed-Tn counterparts (TME strains), have phenotypes consistent with deficient ainS pheromone signaling. (A) PainS-gfp transcriptional reporter activity in cultures grown in SWTO medium to an OD595 of ∼2.5. (B) Peak specific luminescence of cultures grown in SWTO medium. (C) Strain motility as measured by increase in spot diameter (on 0.25% agar plates) over 24 h. (D) C8-AHL extracted and bioassayed from cultures grown in SWTO medium. ND, none detected. In each panel, data are from single representative experiments of three independent experiments. Error bars on all panels indicate standard errors (n = 3). *, P < 0.01, and **, P < 0.001, compared to ES114.
The genome sequences of VFS014F5-T, VFS014B6-T, and VFS021D9-T confirmed their respective transposon locations and also revealed that each mutant has a unique point mutation in luxO, resulting in V106A, V106G, and T335I LuxO variants (Table 2). No other deviations from the wild-type sequence were discovered in the mutants. Given the current model of PS (Fig. 1), the phenotypes of these mutants would be consistent with constitutive LuxO activity, leading to high Qrr expression, low LitR levels, and decreased activation of ainSR. A luxO mutant with a D47E allele that effectively mimics phosphorylated LuxO shows similar phenotypes (17, 53). The mutants also displayed translucent colony morphology similar to that of litR mutants, and we appended a “T” to their strain names to indicate translucent variants. As discussed below, we eventually discovered that there were corresponding Tn library mutants that were not translucent, and adopting this strain nomenclature distinguishes between, for example, original library mutant VFS014F5 (mtsA::Tn) and what is presumably its derivative, VFS014F5-T [mtsA::Tn luxO(V106A)].
TABLE 2.
Summary of luxO* mutants described in this study
| Strain | luxO* mutation | LuxO* amino acid substitution | luxO* crossed into ES114 | luxO* function assessed in transa |
|---|---|---|---|---|
| VFS002F6-T | C272A | A91D | X | X |
| VFS012E9-T | C122T | P41L | X | X |
| VFS014F5-T | T317C | V106A | X | X |
| VFS014B6-T | T317G | V106G | X | X |
| VFS021D9-T | C1004T | T335I | X | X |
| WTTG0 | C320G | T107R | ||
| WTTG1 | T281G | F94C | ||
| WTTG2 | G341C | R114P | ||
| WTTG3 | A971G | H324R | ||
| WTTG4 | C293A | P98Q | ||
| WTTG5 | G338T | R113L | ||
| WTTG12 | C293T | P98L | ||
| WTTG17 | T323G | V108G | ||
| WTTG20 | T317G | V106G | ||
| WTTG21 | T317G | V106G | ||
| WTTG22 | A956G | H319R | ||
| WTTG24 | C613T | L205F |
luxO genes were cloned into the low-copy-number vector pVSV105, and their ability to affect ainS-controlled phenotypes (luminescence as well as PainS and Pqrr reporter activity) in the ΔluxO strain TIM306 was confirmed (24).
To test whether the luxO alleles have the predicted downstream effect on the core PS circuit, we cloned the spontaneous luxO point mutations from the Tn-mutant backgrounds, moved them into ES114 by allelic exchange, and compared Pqrr-gfp activities in the resulting strains and the original mutants. Consistent with the model in Fig. 1, the mutants encoding V106A, V106G, or T335I LuxO variants, as well as a previously described mutant encoding the LuxO(D47E) variant, yielded higher Pqrr-gfp expression than the wild type (Fig. 3). In contrast, strains where the Tn insertions had been backcrossed into ES114 with wild-type luxO displayed wild-type expression of Pqrr-gfp (Fig. 3). Thus, the luxO mutations and not the Tn insertions were causal to this mutant phenotype. Below, mutant alleles that apparently encode constitutively active LuxO are designated luxO*, consistent with the nomenclature in previous reports of similar alleles in other vibrios (54).
FIG 3.
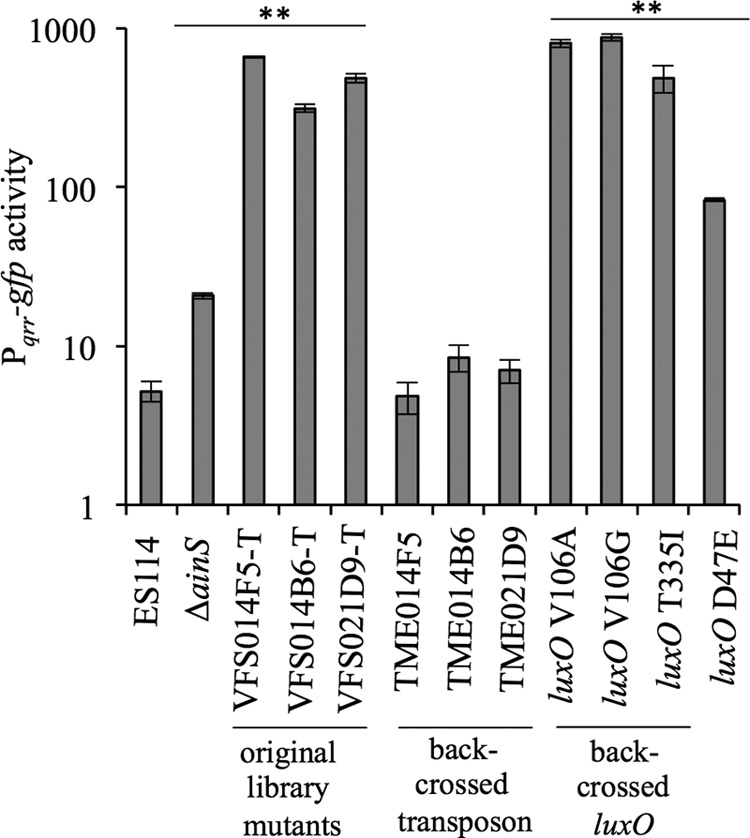
Pqrr-gfp transcriptional reporter activity is elevated in strains with luxO* mutations. GFP activity was expressed from strains harboring the Pqrr-gfp reporter on pTM268, grown in SWTO medium, and assayed at an OD595 of ∼2.5. Data are from a single representative experiment of three independent experiments. Error bars indicate standard errors (n = 3). **, P < 0.001 compared to wild-type ES114.
We explored whether LuxO* variants bypass elements of the PS regulatory circuitry (Fig. 1), first by testing whether the luxO* mutants were still sensitive to C8-AHL. As shown in Fig. 1, transcription of qrr should decrease as C8-AHL levels increase. To test whether luxO* mutants also responded to C8-AHL, we assayed the activity of a Pqrr-gfp transcriptional reporter in the wild type and five luxO* mutants (some of which were isolated in experiments described below). Supplementation of cultures with C8-AHL resulted in a significant (P < 0.05) decrease in Pqrr-gfp activity in ES114 but not in any of the luxO* mutants (Fig. 4), demonstrating that they are blind to addition of the C8-AHL signal.
FIG 4.
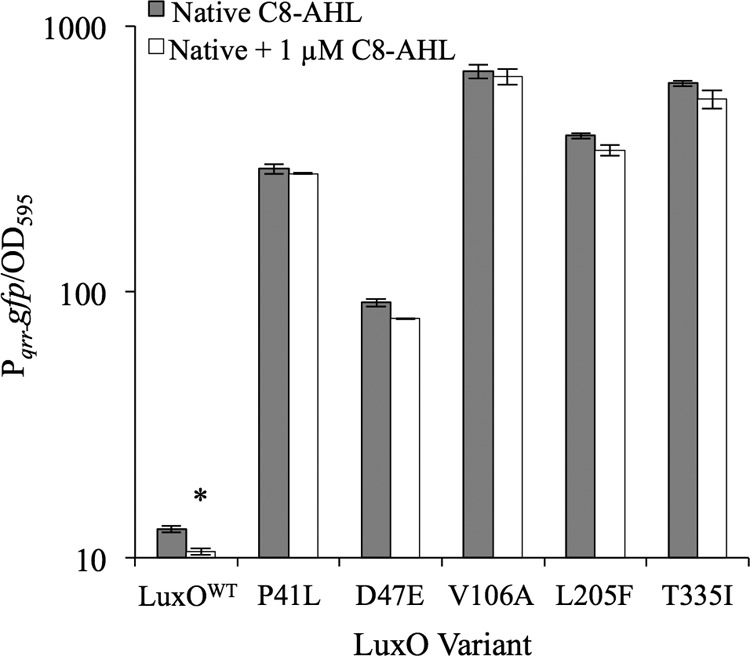
Strains bearing luxO* variants are insensitive to C8-AHL addition. GFP expression in strains ES114, JHK075 [luxO(P41L)], CL59 [luxO(D47E)], JHK057 [luxO(V106A)], WTTG24 [luxO(L205F)], and JHK061 [luxO(T335I)] harboring the Pqrr-gfp reporter plasmid pTM268, grown in SWTO medium with and without 1 μM added C8-AHL, was assayed at an OD595 of ∼2.0. Data are from a single representative experiment of three independent experiments. Error bars indicate standard errors (n = 2). *, P < 0.05 compared to the strain without added C8-AHL, as determined using Student's t test.
We further examined whether LuxO* variants are independent of the core PS circuitry (Fig. 1) by examining whether their activity required luxU or rpoN, which encode LuxU and σ54, respectively. LuxU is the only known phosphoryl donor for LuxO, which in turn activates qrr as a σ54 enhancer (Fig. 1). To test the role of luxU, we introduced a luxU::pEVS122 allele, which inactivated LuxU function, as indicated by the bright luminescence of the ES114-derived luxU::pEVS122 strain JHK068 (data not shown). Even with luxU disrupted, the three luxO* mutations described above, as well as the D47E mutation described previously, increased Pqrr-gfp expression (Fig. 5) and decreased PainS-gfp expression (see Fig. S1 in the supplemental material). In contrast, deletion of rpoN eliminated the effect of the luxO*(V106A) and luxO*(T335I) variants. For example, luxO* ΔrpoN double mutants had low Pqrr-gfp activity similar to that of the ES114 ΔrpoN mutant KV5005 (43) (see Fig. S2 in the supplemental material). Thus, as discussed below, at least for the luxO* alleles tested, their activity is independent of LuxU but remains dependent on σ54.
FIG 5.
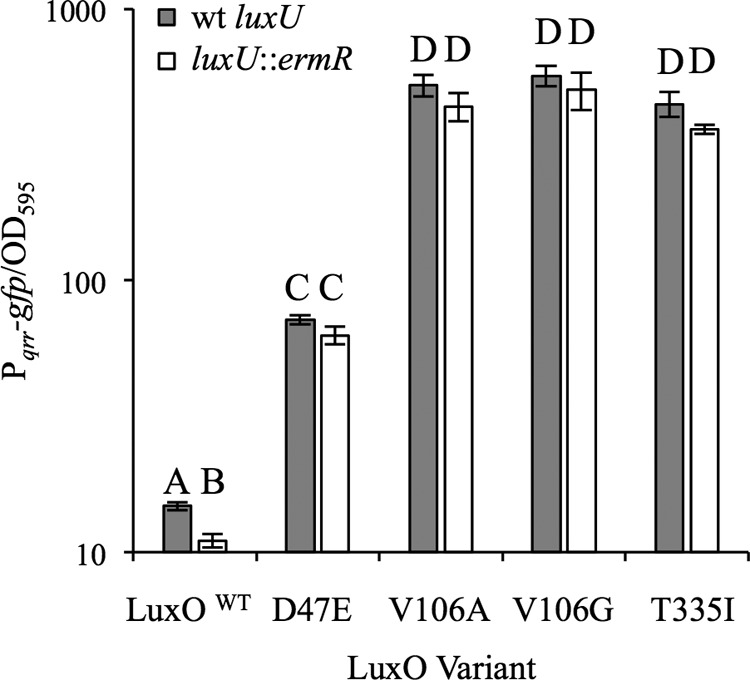
The inactivation of luxU does not eliminate the effect of different luxO* variants on Pqrr-gfp reporter activity. GFP expression in strains harboring the Pqrr-gfp transcriptional reporter pTM268 and grown in SWTO medium was assayed at an OD595 of ∼2.5. Values with the same letter are not statistically significantly different (P > 0.05), whereas different letters indicate significant differences (P < 0.01), based on a one-way analysis of variance (ANOVA) and post hoc testing using Student's t test. Data are from a single representative experiment of three independent experiments. Error bars indicate standard errors (n = 3).
After evaluating the characteristics of the newly discovered luxO* variants, we were curious about their relatively high representation in the transposon mutant library. Gain-of-function mutations resulting in constitutively active LuxO were recovered from three mutants in a library of less than 3,000, which seems remarkably frequent. Moreover, such luxO* mutations presumably ought to happen far less frequently than simple loss-of-function mutations in litR, yet based on our simplified working model (Fig. 1), both should theoretically have similar phenotypic characteristics. We therefore hypothesized that luxO* mutations provided some benefit during Tn library construction, storage, and/or propagation, thereby resulting in their enrichment. Furthermore, we speculated that this luxO* phenotype must be different than that of litR loss-of-function mutants.
To test this hypothesis, we assessed the luxO* V106A strain JHK057 for its ability to outcompete the wild type under conditions mimicking library development. For example, we tested relative viability after freezing overnight at −80°C and the frequency of transposon insertion following conjugative transfer of pEVS170 in a mixed culture. Only in the latter assay did we see a difference in the luxO* mutant (Fig. 6); however, this difference was relatively minor, and our results actually suggest that luxO* mutants are somewhat poorer recipients for conjugation-based Tn mutagenesis and if anything should be modestly enriched against as parents for Tn mutagenesis.
FIG 6.
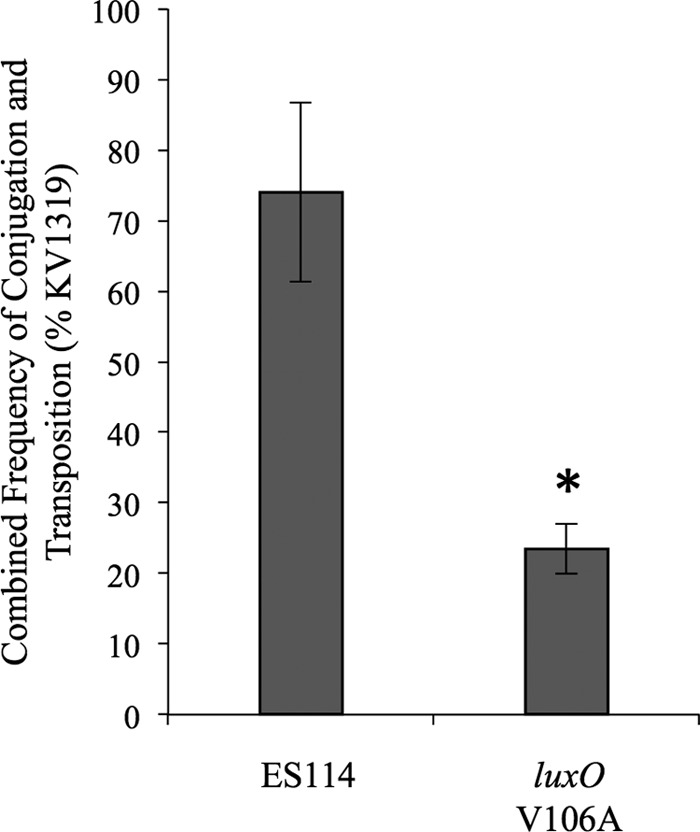
A luxO* mutation does not enhance frequency of conjugation-based transposon mutagenesis. The transposon-containing vector pEVS170 was conjugated into mixed recipient cultures of KV1319 (celG) combined ∼1:1 with either JHK057 [luxO(V106A)] or the wild type. Following conjugation, nonselective plating determined the ratio of recipient strains, and plating on Erm was used to determine the number of Tn mutants. By including 10 mM d-cellobiose and X-Gal, we differentiated recipients as either blue (JHK057 or ES114) or white (KV1319). Data are from one representative experiment of three independent experiments. Error bars indicate standard errors (n = 5). *, P < 0.01 relative to KV1319.
We discovered evidence that the spontaneous luxO* mutations occurred after Tn mutagenesis. The well occupied by VFS014B6 in the 96-well plates housing the mutant library contained a mix of translucent and opaque CFU. PCR and sequencing revealed that all colonies from this well contained the Tn insertion, but only the translucent colonies carried the luxO* mutation. Two other Tn mutants, VFS002F6 [luxO*(A91D)] and VFS012E9 [luxO*(P41L)], were similarly stocked as a mix of luxO* and wild-type luxO strains in a uniform Tn-mutant background. Each of these luxO mutants (Table 2) possesses phenotypes similar to those described above for the initial luxO* mutants, and these luxO* alleles are similarly independent of luxU (data not shown). Additionally, a mixture of opaque and translucent colonies in the well containing VFS014F5, but not the other Tn mutants, was discovered in the master Tn library, a copy of which was sent to our lab (Foxall and Whistler, personal communication). Taken together, our results suggested the luxO* mutants had arisen after transposon insertion, that freezing did not favor these mutants, and that the enrichment of luxO* mutants relative to their parent strains likely resulted during library outgrowth.
To test whether luxO* mutants could have been enriched during growth and storage of the Tn mutant library, we first grew ES114 and JHK057 [luxO(V106A)] individually during prolonged incubation in static cultures of LBS medium. The strains initially grew similarly and reached cell numbers (CFU per milliliter) at 24 h that were not significantly different from one another (P > 0.05); however, from 24 to 96 h, ES114 suffered greater decreases in CFU (see Fig. S3 in the supplemental material). Moreover, translucent colonies began to appear in ES114-inoculated wells after 48 h and eventually dominated ES114-inoculated cultures. Most of these new spontaneous translucent mutants harbored luxO* alleles (see strains with the prefix “WTTG” in Table 2); however, a small subset possessed characteristics similar to those of luxO* mutants while having wild-type luxO sequences. Interestingly, nine translucent mutants maintained wild-type Pqrr-gfp activity (and clumped in static broth culture), suggesting that there are multiple paths to translucent colony morphology and static-culture survival, including at least one independent of the central PS network.
We next competed strains to approximate the conditions under which a luxO* mutant might be enriched after arising in a wild-type culture. When JHK057 [luxO(V106A)] was grown with ES114 in static coculture at initial ES114/mutant ratios of 1:1, 10:1, or 100:1, the ratio of JHK057 to ES114 increased as much as 10-fold after 24 h (Fig. 7A to D). This change in strain ratios was most dramatic in cultures where JHK057 had initially been the most outnumbered (Fig. 7A to D). Moreover, the number of CFU per milliliter of the luxO* mutant actually increased, even during periods when the CFU/ml of ES114 dropped. In each case, the final strain ratio reflected a similar small numerical advantage for the luxO* mutant, regardless of how outnumbered it was in the initial inoculum (Fig. 7D). We observed a similar competitive advantage for JHK057 and JHK061 [luxO(T335I)] when these strains were competed against the Camr-marked ES114 derivative AKD200 (data not shown; also, see Fig. S4 in the supplemental material).
FIG 7.

Competitive advantage of luxO* mutants relative to ES114 and litR mutants during prolonged coculture. ES114 (A to D) or litR mutant JB18 (E to H) were mixed with JHK057 [luxO(V106A)] in ratios of 1:1, 10:1, or 100:1, with JHK057 always at a disadvantage. Mixes were diluted in 24-well microtiter plates and grown statically for 96 h. At intervals of 0, 24, 48, and 96 h, wells were thoroughly mixed and dilution plated to determine the ratios of viable CFU for each strain in the mixture based on translucence (JHK057 versus ES114) or Erm resistance (JHK057 versus JB18). Data are from a single representative experiment of three independent experiments. Error bars indicate standard errors (n = 3).
The advantage of luxO* mutants appeared to be dependent on prolonged stationary-phase culturing. When cultures were kept in log phase (OD595 < 0.5) for 30 generations by repeated subculturing, translucent derivatives did not appear in wild-type cultures, and in mixed competitions, the luxO* mutant was not enriched relative to the wild type (data not shown).
Given that luxO* mutants should lead to strong repression of litR (Fig. 1C), and that both litR and luxO* mutants share a translucent colony phenotype, we considered the possibility that litR mutants would have the same advantage relative to ES114 in mixed cultures; however, this was not the case (data not shown). Although translucent colonies began to dominate mixed cultures of ES114 and the litR mutant, upon closer examination, most translucent colonies lacked the erythromycin resistance of the litR::ermR mutant and were instead spontaneous mutants of ES114. These results, along with those demonstrating differences in growth and luminescence in shaking culture and Pqrr-gfp reporter activity (see Fig. S5 in the supplemental material), indicated that luxO* and litR mutants do not phenocopy.
To further explore the difference between luxO* and litR mutants, we competed JB18 (litR::ermR) and JHK057 [luxO*(V106A)] in static cultures. Here too, the luxO* mutant dominated when mixed with litR mutants (Fig. 7E to H). Similar to its competition with ES114, JHK057 outcompeted the litR mutant at all three initial strain ratios, and the experiments ended at relatively similar strain ratios, with a slight dominance by the luxO* mutant, regardless of the starting ratio (Fig. 7D and H). Similarly, the luxO* litR mutant JHK073 outcompeted the litR mutant PMF8 (data not shown) (23). Taken together, the data indicated that in prolonged static broth LBS culture, a luxO* mutant can outcompete a litR mutant, and that a competitive advantage for a luxO* mutant does not require litR. We next tested the litR mutant JB18 and the Δqrr mutant TIM305 for their abilities to give rise to spontaneous luxO* mutants when grown in static culture. We grew strains carrying the Pqrr-lacZ reporter pHK20 in static culture for up to 96 h, periodically plating on LBS containing X-Gal, and screening for blue colonies, but we were unable to isolate any mutants with increased Pqrr-lacZ activity in the JB18 or TIM305 backgrounds, indicating that litR and qrr are required for cells carrying a luxO* mutation to arise and dominate cultures. In the case of litR, this result contrasts with the observation above that a luxO* litR mutant could outcompete a litR mutant when they were coinoculated (data not shown).
Although the importance of litR was somewhat unclear, we were most interested in whether the luxO* survival advantage acts through Qrr, as qrr is the only described regulatory target of LuxO. We competed AKD200 (Camr-marked ES114) against JHK105 [luxO*(T335I) Δqrr] and found that the initial strain ratios were maintained through 96 h (Fig. 8). In this instance, it was necessary to use a marked derivative of ES114, because the JHK105 (luxO* Δqrr) lacked the distinctive translucent colony morphology of luxO* mutants. Although we used a luxO*(V106A) mutant in many of the experiments described above, we used the T335I allele here because the mutation was further from qrr, which allowed us to move the allele into a Δqrr background without also exchanging in wild-type qrr. As in other experiments, translucent mutants began to appear within 48 h, but these were solely derived from AKD200 (data not shown). The results in Fig. 8 show that the competitive advantage of the luxO* allele requires qrr.
FIG 8.
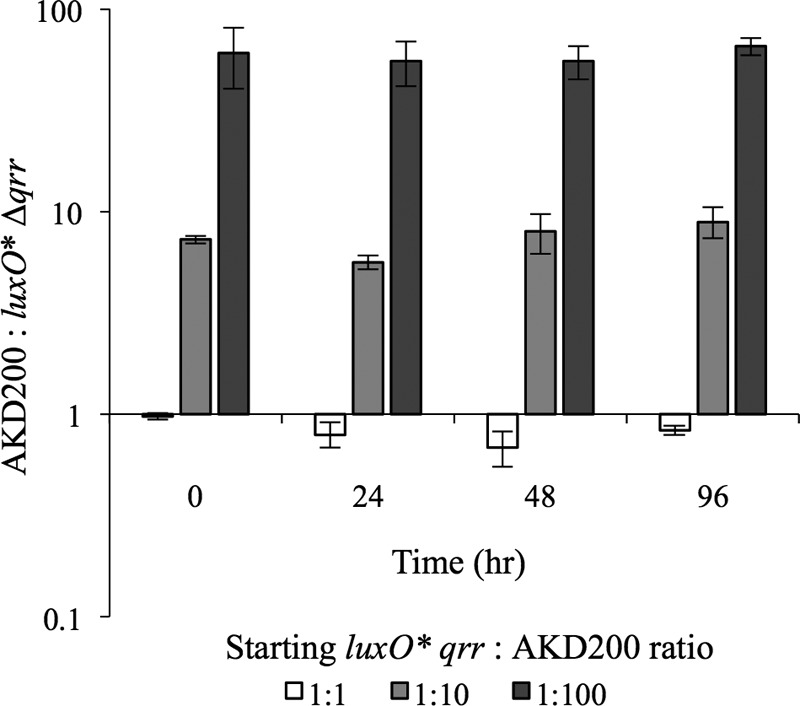
Qrr is required for a luxO*-mediated survival advantage. Cam-marked ES114 (AKD200) and JHK105 [luxO(T335I) Δqrr] were grown overnight and subcultured into fresh LBS medium until reaching an OD595 of ∼2.0, at which time they were mixed in ratios of 1:1, 1:10, or 1:100, with JHK061 always at a disadvantage. Mixes were outgrown to an OD595 of ∼2.5 and then diluted 1:1,000 in fresh LBS medium in 24-well microtiter plates grown statically for 96 h. At intervals of 0, 24, 48, and 96 h, wells were thoroughly mixed and dilution plated to determine the ratios of viable cells of each mixture by Cam resistance. Data are from a single representative experiment of two independent experiments. Error bars indicate standard errors (n = 3).
In contrast to its competitive advantage in broth culture (see Fig. S4 in the supplemental material), mutant JHK061 [luxO*(T335I)] was significantly (P < 0.001) outcompeted by the Camr-marked ES114 derivative AKD200 during host infection and colonization (Fig. 9). The competitive defect of the luxO* mutant in symbiosis does not stem from differential survival of the inoculum in artificial seawater (data not shown). The results shown in Fig. 9 are another example where a luxO* mutant phenotype is dissimilar from that of litR mutants, which actually outcompete the wild type during colonization of E. scolopes (23). Our results are consistent with the attenuated colonization previously reported for the luxO(D47E) mutant CL59 (19), but the effects appear to be more severe, potentially owing to the higher relative activity of the luxO* T335I allele in JHK061 than of the luxO*(D47E) allele in CL59 (Fig. 3, 4, and 5).
FIG 9.
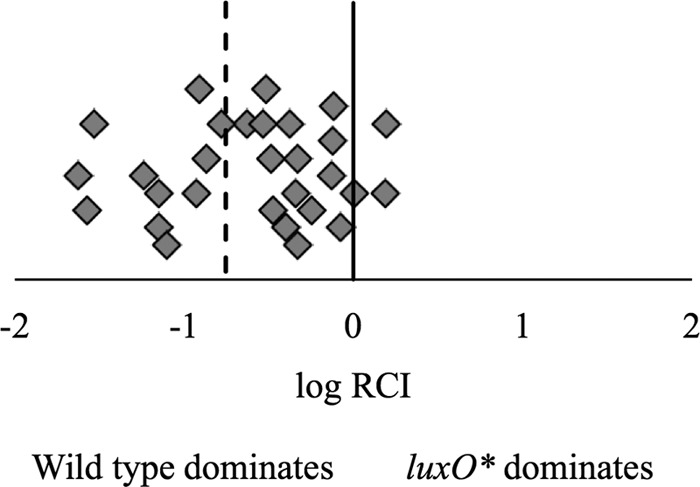
The luxO* strain JHK061 shows a defect when competed 1:1 against the wild-type ES114 derivative AKD200 during colonization of E. scolopes. Newly hatched squid (n = 29) were exposed to a mixed inoculum of JHK061 [luxO*(T335I)] and AKD200 (∼2,000 CFU) in filter-sterilized Instant Ocean for 12 h with subsequent water change at 24 h. After 48 h, squid were homogenized, dilution plated on LBS agar, and patched to determine Cam resistance. The log relative competitive index (RCI) was calculated as the log of the ratio of JHK061 to AKD200 in the host homogenate to the ratio of the two strains in the inoculum. The solid line represents an equal ability to colonize the host; the dashed line represents the mean log RCI of −0.75. Data from a single representative experiment of three independent experiments is shown. Statistical significance was determined using a one-sample Student's t test where the null hypothesis is a mean log RCI of 0. Each diamond represents the log RCI in one hatchling.
Given the identification of luxO* alleles other than the previously reported D47E allele, we were curious if certain sites were more prone to mutation than others and/or if additional allelic variants were possible. Using the strategy of static-culture luxO* enrichment, we grew 26 independent cultures for 48 h, dilution plated, and picked translucent colonies for luxO sequencing. In all, 10 more luxO* alleles were identified (F94C, P98L, P98Q, T107R, V108G, R113L, R114P, L205F, H319R, and H324R alleles) and luxO*(V106G) was recovered two more times in our screen (Fig. 10). Taken together, these data show that alleles were distributed across LuxO, with the exception of the DNA-binding domain.
FIG 10.
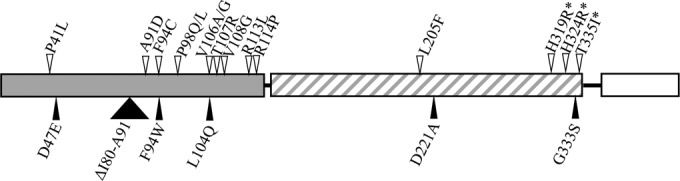
Distribution of luxO* variants obtained in this study (open triangles) and those from previous studies in other vibrios aligned to the V. fischeri sequence (filled triangles) (53, 54, 57, 58, 60). Rectangles represent LuxO domains, including the regulatory (gray), AAA+/RpoN interaction (hatched), and DNA-binding (white) domains. The asterisk indicates a potential site of interaction with LuxU (78).
Finally, given that some components of the PS circuitry, including ainSR and luxIR, have diverged between V. fischeri lineages (55), we wondered whether spontaneous luxO* mutants, or other mutants that upregulate qrr, would be similarly enriched during prolonged growth in static culture of other wild-type strains besides ES114. We tested wild-type V. fischeri isolates CG103, ES12, ES114, ES213, ES401, EM17, ET101, H905, PP3, VLS2, and WH1 (Table 1), by growing strains carrying the Pqrr-lacZ reporter pHK20 in static culture for up to 96 h, as described above. We further tested blue colonies by displacing pHK20 with the Pqrr-gfp reporter pTM268 and screening for high fluorescence to ensure that mutants had genomic changes that increased qrr reporter expression. Strains ES12, ES114, ES213, ES401, EM17, and ET101 gave rise to mutants with increased qrr expression.
DISCUSSION
In this study, we sought to identify regulators of the ainSR PS system using a library of Tn insertion mutants; however, the effects we observed on ainSR expression were unrelated to the Tn insertions and were instead due to spontaneous mutations in the core PS-signaling circuit (Fig. 1C). The most notable mutants had constitutively active variants of the regulator LuxO and are referred to as luxO* mutants (54), which outcompeted the wild type during prolonged static coculture in LBS medium (e.g., see Fig. 7).
Our data suggest that these luxO* mutations arose after Tn mutagenesis and that mutants were probably enriched in subsequent steps. Although the 96-h duration of our competition experiments is longer than that of any incubations used during library construction and propagation, >10-fold enrichment of luxO* mutants was observed in as little as 24 h (Fig. 7), and three successive rounds of 12-h static culture incubations followed by subculturing was sufficient to generate translucent mutants (data not shown). Moreover, overnight incubations in LBS were used in stocking the original Tn mutants prior to sequencing, in generating the defined master library, and finally in propagating the library prior to introduction of the PainS-lacZ reporter. Knowing now that luxO* (or phenotypically similar) mutants are prevalent in most cultures of ES114 after 48 h of incubation, and that luxO* mutants are significantly enriched by 24 h, it seems plausible that during construction and handling of the Tn mutant library, three in approximately 2,000 wells became significantly contaminated by a luxO* derivative. In a similar vein, Petrun and Lostroh reported a growth advantage during stationary phase for 7-day-old cultures of V. fischeri over 1-day-old cultures (56), and it would be interesting to assess such cultures for the presence of luxO* variants. Overall, our findings serve as a cautionary tale, encouraging the prompt use of cultures and the verification of phenotypes using complementation and/or backcrossing. In this circumstance, examining the opacity or translucence of newly made strains also provides a convenient screen against these unexpected mutations.
The prevalence of luxO* in the Vibrionaceae.
LuxO is part of a PS network conserved throughout the Vibrionaceae that has been well studied over the last 3 decades. Other luxO* mutants have been reported in the Vibrionaceae, isolated both from natural populations and during laboratory experiments (54, 57–61). Recently, phenotypic discrepancies between two ΔluxU mutants of V. cholerae were discovered to be due to one strain harboring a previously undetected luxO* mutation (58, 59), which is reminiscent of our own discovery of spontaneous luxO* causing phenotypes we initially attributed to defined transposon insertions. In another parallel to our study, luxO* mutants were recovered in strains subjected to transposon mutagenesis (54, 58, 62, 63); however, we found that luxO* mutants of V. fischeri were not enriched by this procedure specifically (Fig. 6), and luxO* mutants were readily recovered from strains that had not been genetically manipulated or subjected to Tn mutagenesis. If our observations in V. fischeri hold true for other Vibrio species, it may simply be that rounds of growth into stationary phase during genetic manipulations can give rise to luxO* mutants. In this regard, if we consider the reports by Keynan and Hastings (64) and Silverman et al. (65) of “luminescence variation” in V. harveyi resulting in genetically stable dim and dark mutants in old, statically grown cultures, it is tempting to speculate that at least some of these may have been luxO* mutants. One such dark mutant was used in the first description of the luxO locus (66), raising the possibility that a spontaneous luxO* mutant contributed to the discovery of luxO almost 30 years ago (65).
Are LuxO* mutants cheaters?
One area of recent interest in the study of bacterial PS and group behaviors is the appearance of cheaters in populations. These cheaters are PS-negative mutants that benefit from the PS-induced behaviors of nearby cells while minimizing their own costs and thus are enriched in the population (9–11). Given that luxO* mutants make little C8-AHL and remain locked in a noninduced state (e.g., dim) (Fig. 1C and 2) but can also take over mixed cultures with the wild type (Fig. 7), such mutants might fit the description of PS cheaters. Although such cheating mutants have been described in a variety of bacterial systems, to our knowledge, this would be the first description of cheaters arising spontaneously in V. fischeri and may shed light on a more general method of survival among vibrios and the importance of tight regulation of the pheromone systems of V. fischeri in its natural habitats.
On the other hand, luxO* mutants did show some survival benefits even in the absence of the wild type (see Fig. S3 in the supplemental material), and they may simply be better adapted to broth culture environments without needing to exploit a nearby wild-type population. In this sense, it seems possible that these mutants, and perhaps others, are “antisocial” without being cheaters. In situations where social behaviors have costs without benefits, being antisocial could simply be viewed as “prudent” or “efficient.” The terminal or master regulators of LuxO-containing PS circuits (e.g., LitR in V. fischeri) control a diverse array of phenotypes, including opacity (67), swarming (68), protease production (29), biofilm formation (27), symbiosis (23), and luminescence (69). These behaviors may present fitness costs without compensatory benefits under certain circumstances, and luxO* mutants would have the effect of damping expression of these costly systems.
If luxO* mutations are advantageous due to their repression of PS master regulators like litR, this raises the question of why presumably rarer gain-of-function mutations in luxO should dominate instead of loss-of-function mutations in litR. In fact, such loss-of-function mutations to these regulators have repeatedly been observed in other members of the Vibrionaceae. For example, in V. cholerae, mutants disrupted in the litR homolog hapR account for cheaters arising in populations of that bacterium and have been frequently isolated (11, 27, 29). One reason luxO* mutants may be enriched rather than litR mutants may be that, in some instances, fitness is maximized by decreasing the master regulator without completely eliminating it. At least in V. fischeri, in terms of static culture survival, luxO* mutants outcompete either their wild-type parents or litR mutants (Fig. 7). Thus, while luxO* mutations may be more rare, in this circumstance they confer greater fitness. Others have suggested that PS systems may have evolved in ways that make the appearance of cheaters difficult (70, 71), and perhaps the V. fischeri system has similarly evolved such that the simple loss of litR is not advantageous.
Insights into the LuxO-Qrr regulatory module.
Based on a simple model of the LuxO-containing PS regulatory circuit (Fig. 1A and B), luxO* mutants and litR loss-of-function mutants should have a similar phenotype, with the caveat noted above that LuxO* should diminish but may not eliminate LitR. In addition to luxO* mutants outcompeting litR mutants (Fig. 7), prolonged growth in stationary phase selected for luxO* mutants but not litR mutants, and a luxO* litR double mutant outcompeted a litR mutant (data not shown). Furthermore, luxO* and litR mutants do not phenocopy with respect to their ability to compete with the wild type in host colonization (Fig. 9), growth and luminescence in broth culture (see Fig. S5A and B in the supplemental material), and Pqrr-gfp reporter activity (see Fig. S5C in the supplemental material). On the other hand, consistent with our model of the PS circuitry (Fig. 1), the survival advantage of luxO* mutations is Qrr dependent (Fig. 8). Given these observations, we speculate that the Qrr regulon extends beyond litR. There is precedent for such regulation, as Qrr has multiple targets in other Vibrio species (72, 73), although there are also usually multiple semiredundant copies of qrr, whereas V. fischeri has only one qrr gene (24). Our data seem consistent with a model where Qrr regulates an unknown target that affects survival and competitiveness during prolonged culturing.
Our results also point to mutants that could help reveal alternative targets for Qrr and perhaps novel inputs to qrr control. As noted above, prolonged culturing of ES114 yields not only luxO* mutants but also other translucent-colony mutants with wild-type luxO. These mutants fell into two classes. One set was relatively abundant, quickly settled in clumps out of static broth culture, and had wild-type Pqrr-gfp activity. Analyses of these mutants might provide insight into the key targets for stationary-phase survival downstream of Qrr. The second set was less common and had high Pqrr-gfp activity, similar to luxO* mutants. These mutants might reveal other regulatory inputs into qrr control. While we have shown that our luxO* mutants act independently of their only known phospho-donor, LuxU (Fig. 5), this does not preclude the existence of some hitherto-unknown input into the system acting at or downstream of LuxU. Alternative inputs into LuxU have been shown in V. harveyi and V. cholerae through HqsK and VpsS, respectively, and perhaps there exist similar pheromone-independent inputs phosphorylating LuxO (74, 75).
Symbiotic phenotypes of luxO* and litR mutants.
Our data are also consistent with previous reports that suggest luxO* and litR mutants have different symbiotic phenotypes. Fidopiastis et al. showed a litR mutant outcompeted its wild-type parent (23); however, Lupp and Ruby reported a colonization defect at 12 h postinoculation for a luxO*(D47E) mutant (19). Understanding the role of each gene was further complicated by findings that minor competition defects for luxO and qrr mutants are not rescued by the addition of a litR mutation (24). This observation suggested a requirement for luxO and qrr for full colonization of the host. Our results are consistent with those of Lupp and Ruby, and it also seems possible that the more active the luxO* allele is (e.g., see Fig. 3), the greater the competitive defect is.
Taken together, the results suggest that increased qrr expression may be detrimental to V. fischeri's ability to colonize E. scolopes. In that regard, litR mutants did not display increased qrr reporter expression (see Fig. S5C in the supplemental material), despite LitR feedback regulating the ainSR pathway (17, 22, 30). Given the symbiotic disadvantage of luxO* mutations, it seems unlikely that they would be enriched in natural symbiotic populations of V. fischeri, although they might be found in free-living populations or in mixed-species communities in the guts of fishes. At least in the shallow sandy reefs of Hawaii, where symbiosis apparently enhances V. fischeri populations (76, 77), symbiotic fitness presumably filters out luxO* mutants.
New insights into LuxO structure and function.
The many different luxO* alleles that we found (Fig. 10) indicate that there are multiple paths to LuxO* activity. Classical D47E mutations alter the amino acid (D47) that is usually phosphorylated to activate the protein, and presumably the longer side chain at this site in some way mimics the conformational changes imposed by phosphorylation at this residue. This study contributes to a body of work describing mutations showing that less intuitive mechanisms of generating LuxO* variants are possible (Fig. 10). These mechanisms could include deactivation of negative autoregulation by the N-terminal receiver domain, modulation of the predicted site of interaction with the phosphor-donor LuxU (78), promotion of new interactions with other phosphate donors, or other mechanisms. Previous studies of luxO and other σ54-dependent regulators have described similar effects of mutations in approximately the same positions within the protein as we isolated (53, 79). LuxO belongs to a subclass of σ54-dependent regulators wherein the N-terminal receiver domain represses the catalytic activity of the central AAA+ domain, and deletion of the receiver domain results in constitutive activity of the protein (53, 79). Furthermore, the majority of the variant mutations we isolated were clustered within a putative helix that, when LuxO dimerizes, serves to stabilize the “off” state conformation in other systems (79–81). Mutagenesis within these helices, like phosphorylation of the conserved aspartate residue, may destabilize this interaction, promoting a more active state (79–82). It is noteworthy, and perhaps surprising, that each of the alleles we tested appeared to be active independently of luxU and was more active than a D47E variant (Fig. 5). However, by allowing these mutations to arise under natural selective pressures, our enrichment was biased toward mutations that provide a competitive advantage for the bacterium and may therefore serve as a more powerful tool for revealing strong effects on protein function than targeted mutagenesis alone.
A dearth of ainSR regulators.
We initially sought to identify novel controls of ainSR, and the fact that we did not identify any unknown regulators of ainS raises the question of whether there are any such regulators of ainSR beyond what is known for CRP and LitR (17, 22, 30), other than the possible regulation of ainSR by LuxR (22, 83). Our lack of success in identifying new regulators might reflect limitations of our original PainS-lacZ reporter-based screen. Moreover, the transposon library is not comprehensive and will also inherently miss any regulators of the system that are essential for V. fischeri's survival. In this regard, it is worth noting that while they are not essential, neither crp nor litR is among the genes disrupted in this transposon mutant library. Finally, regulators may become apparent only under different conditions (e.g., during growth in different media) or with other approaches. It seems likely that the regulation of ainSR is far less complex than that of luxI and luxR, which could reflect the function of the former being more general and the latter more context dependent.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cheryl Whistler and Randi Foxall for providing the transposon mutant library and for helpful discussions, Takahiko Sasaki and Zack Lewis for assistance in preparing and analyzing the genome resequencing data, and Julie Stoudenmire for providing pJLS27.
The National Science Foundation supported this research under grants IOS-0841480 and IOS-1121106. J.H.K. was partially supported with funds awarded by the University of Georgia Presidential Graduate Fellows Program.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00807-15.
REFERENCES
- 1.Fuqua WC, Winans SC, Greenberg EP. 1994. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol 176:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu J, Winans SC. 2001. The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A 98:1507–1512. doi: 10.1073/pnas.98.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaefer AL, Greenberg EP, Oliver CM, Oda Y, Huang JJ, Bittan-Banin G, Peres CM, Schmidt S, Juhaszova K, Sufrin JR, Harwood CS. 2008. A new class of homoserine lactone quorum-sensing signals. Nature 454:595–599. doi: 10.1038/nature07088. [DOI] [PubMed] [Google Scholar]
- 4.Lyell NL, Dunn AK, Bose JL, Stabb EV. 2010. Bright mutants of Vibrio fischeri ES114 reveal conditions and regulators that control bioluminescence and expression of the lux operon. J Bacteriol 192:5103–5114. doi: 10.1128/JB.00524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Septer AN, Stabb EV. 2012. Coordination of the Arc regulatory system and pheromone-mediated positive feedback in controlling the Vibrio fischeri lux operon. PLoS One 7:e49590. doi: 10.1371/journal.pone.0049590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunn AK, Stabb EV. 2007. Beyond quorum sensing: the complexities of prokaryotic parliamentary procedures. Anal Bioanal Chem 387:391–398. doi: 10.1007/s00216-006-0730-9. [DOI] [PubMed] [Google Scholar]
- 7.Bose JL, Rosenberg CS, Stabb EV. 2008. Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch Microbiol 190:169–183. doi: 10.1007/s00203-008-0387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stabb EV. 2005. Shedding light on the bioluminescence “paradox.” ASM News 71:223–229. [Google Scholar]
- 9.Sandoz KM, Mitzimberg SM, Schuster M. 2007. Social cheating in Pseudomonas aeruginosa quorum sensing. Proc Natl Acad Sci U S A 104:15876–15881. doi: 10.1073/pnas.0705653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velicer GJ, Kroos L, Lenski RE. 2000. Developmental cheating in the social bacterium Myxococcus xanthus. Nature 404:598–601. doi: 10.1038/35007066. [DOI] [PubMed] [Google Scholar]
- 11.Katzianer DS, Wang H, Carey RM, Zhu J. 2015. “Quorum non-sensing”: social cheating and deception in Vibrio cholerae. Appl Environ Microbiol 81:3856–3862. doi: 10.1128/AEM.00586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hastings JW, Greenberg EP. 1999. Quorum sensing: the explanation of a curious phenomenon reveals a common characteristic of bacteria. J Bacteriol 181:2667–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stabb EV, Schaefer A, Bose JL, Ruby EG. 2008. Quorum signaling in the marine luminous bacterium Vibrio fischeri p 233–250. In Winans SC, Bassler BL (ed), Chemical communication among bacteria, 2nd ed ASM Publishing, Washington, DC. [Google Scholar]
- 14.Stabb EV. 2006. The Vibrio fischeri-Euprymna scolopes light organ symbiosis, p 204–218. In Thompson FL, Austin B, Swings J (ed), The biology of vibrios. ASM Press, Washington, DC. [Google Scholar]
- 15.Kuo A, Blough NV, Dunlap PV. 1994. Multiple N-acyl-l-homoserine lactone autoinducers of luminescence in the marine symbiotic bacterium Vibrio fischeri. J Bacteriol 176:7558–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engebrecht J, Nealson K, Silverman M. 1983. Bacterial bioluminescence: isolation and genetic analysis of functions from Vibrio fischeri. Cell 32:773–781. doi: 10.1016/0092-8674(83)90063-6. [DOI] [PubMed] [Google Scholar]
- 17.Lupp C, Ruby EG. 2004. Vibrio fischeri LuxS and AinS: comparative study of two signal synthases. J Bacteriol 186:3873–3881. doi: 10.1128/JB.186.12.3873-3881.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lupp C, Urbanowski M, Greenberg EP, Ruby EG. 2003. The Vibrio fischeri quorum-sensing systems ain and lux sequentially induce luminescence gene expression and are important for persistence in the squid host. Mol Microbiol 50:319–331. doi: 10.1046/j.1365-2958.2003.t01-1-03585.x. [DOI] [PubMed] [Google Scholar]
- 19.Lupp C, Ruby EG. 2005. Vibrio fischeri uses two quorum-sensing systems for the regulation of early and late colonization factors. J Bacteriol 187:3620–3629. doi: 10.1128/JB.187.11.3620-3629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Federle MJ, Bassler BL. 2003. Interspecies communication in bacteria. J Clin Invest 112:1291–1299. doi: 10.1172/JCI20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pereira CS, Thompson JA, Xavier KB. 2013. AI-2-mediated signalling in bacteria. FEMS Microbiol Rev 37:156–181. doi: 10.1111/j.1574-6976.2012.00345.x. [DOI] [PubMed] [Google Scholar]
- 22.Kimbrough JH, Stabb EV. 2013. Substrate specificity and function of the pheromone receptor AinR in Vibrio fischeri ES114. J Bacteriol 195:5223–5232. doi: 10.1128/JB.00913-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fidopiastis PM, Miyamoto CM, Jobling MG, Meighen EA, Ruby EG. 2002. LitR, a new transcriptional activator in Vibrio fischeri, regulates luminescence and symbiotic light organ colonization. Mol Microbiol 45:131–143. doi: 10.1046/j.1365-2958.2002.02996.x. [DOI] [PubMed] [Google Scholar]
- 24.Miyashiro T, Wollenberg MS, Cao X, Oehlert D, Ruby EG. 2010. A single qrr gene is necessary and sufficient for LuxO-mediated regulation in Vibrio fischeri. Mol Microbiol 77:1556–1567. doi: 10.1111/j.1365-2958.2010.07309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray VA, Visick KL. 2012. LuxU connects quorum sensing to biofilm formation in Vibrio fischeri. Mol Microbiol 86:954–970. doi: 10.1111/mmi.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyamoto CM, Lin YH, Meighen EA. 2000. Control of bioluminescence in Vibrio fischeri by the LuxO signal response regulator. Mol Microbiol 36:594–607. [DOI] [PubMed] [Google Scholar]
- 27.Hammer BK, Bassler BL. 2003. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol 50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- 28.Enos-Berlage JL, Guvener ZT, Keenan CE, McCarter LL. 2005. Genetic determinants of biofilm development of opaque and translucent Vibrio parahaemolyticus. Mol Microbiol 55:1160–1182. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyell NL, Colton DM, Bose JL, Tumen-Velasquez MP, Kimbrough JH, Stabb EV. 2013. Cyclic AMP receptor protein regulates pheromone-mediated bioluminescence at multiple levels in Vibrio fischeri ES114. J Bacteriol 195:5051–5063. doi: 10.1128/JB.00751-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boettcher KJ, Ruby EG. 1990. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J Bacteriol 172:3701–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 33.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134. doi: 10.1016/j.plasmid.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 35.Stabb EV, Reich KA, Ruby EG. 2001. Vibrio fischeri genes hvnA and hvnB encode secreted NAD+-glycohydrolases. J Bacteriol 183:309–317. doi: 10.1128/JB.183.1.309-317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bose JL, Kim U, Bartkowski W, Gunsalus RP, Overley AM, Lyell NL, Visick KL, Stabb EV. 2007. Bioluminescence in Vibrio fischeri is controlled by the redox-responsive regulator ArcA. Mol Microbiol 65:538–553. doi: 10.1111/j.1365-2958.2007.05809.x. [DOI] [PubMed] [Google Scholar]
- 37.Dunn AK, Stabb EV. 2008. Genetic analysis of trimethylamine N-oxide reductases in the light organ symbiont Vibrio fischeri ES114. J Bacteriol 190:5814–5823. doi: 10.1128/JB.00227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colton DM, Stoudenmire JL, Stabb EV. 2015. Growth on glucose decreases cAMP-CRP activity while paradoxically increasing intracellular cAMP in the light-organ symbiont Vibrio fischeri. Mol Microbiol 97:1114–1127. doi: 10.1111/mmi.13087. [DOI] [PubMed] [Google Scholar]
- 39.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol 72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stabb EV, Ruby EG. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol 358:413–426. doi: 10.1016/S0076-6879(02)58106-4. [DOI] [PubMed] [Google Scholar]
- 41.Herrero M, de Lorenzo V, Timmis K. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in Gram-negative bacteria. J Bacteriol 172:6557–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pollack-Berti A, Wollenberg MS, Ruby EG. 2010. Natural transformation of Vibrio fischeri requires tfoX and tfoY. Environ Microbiol 12:2302–2311. doi: 10.1111/j.1462-2920.2010.02250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Visick KL, Quirke KP, McEwen SM. 2013. Arabinose induces pellicle formation by Vibrio fischeri. Appl Environ Microbiol 79:2069–2080. doi: 10.1128/AEM.03526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, Gunsalus R, Lostroh P, Lupp C, McCann J, Millikan D, Schaefer A, Stabb E, Stevens A, Visick K, Whistler C, Greenberg EP. 2005. Complete genome sequence of Vibrio fischeri: a symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A 102:3004–3009. doi: 10.1073/pnas.0409900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mandel MJ, Stabb EV, Ruby EG. 2008. Comparative genomics-based investigation of resequencing targets in Vibrio fischeri: focus on point miscalls and artefactual expansions. BMC Genomics 9:138. doi: 10.1186/1471-2164-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Septer AN, Wang Y, Ruby EG, Stabb EV, Dunn AK. 2011. The haem-uptake gene cluster in Vibrio fischeri is regulated by Fur and contributes to symbiotic colonization. Environ Microbiol 13:2855–2864. doi: 10.1111/j.1462-2920.2011.02558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adin DM, Engle JT, Goldman WE, McFall-Ngai MJ, Stabb EV. 2009. Mutations in ampG and lytic transglycosylase genes affect the net release of peptidoglycan monomers from Vibrio fischeri. J Bacteriol 191:2012–2022. doi: 10.1128/JB.01547-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colton DM, Stabb EV, Hagen SJ. 2015. Modeling analysis of signal sensitivity and specificity by Vibrio fischeri LuxR variants. PLoS One 10:e0126474. doi: 10.1371/journal.pone.0126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyell NL, Dunn AK, Bose JL, Vescovi SL, Stabb EV. 2008. Effective mutagenesis of Vibrio fischeri by using hyperactive mini-Tn5 derivatives. Appl Environ Microbiol 74:7059–7063. doi: 10.1128/AEM.01330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adin DM, Visick KL, Stabb EV. 2008. Identification of a cellobiose utilization gene cluster with cryptic β-galactosidase activity in Vibrio fischeri. Appl Environ Microbiol 74:4059–4069. doi: 10.1128/AEM.00190-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCann J, Stabb EV, Millikan DS, Ruby EG. 2003. Population dynamics of Vibrio fischeri during infection of Euprymna scolopes. Appl Environ Microbiol 69:5928–5934. doi: 10.1128/AEM.69.10.5928-5934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freeman JA, Bassler BL. 1999. A genetic analysis of the function of LuxO, a two-component response regulator involved in quorum sensing in Vibrio harveyi. Mol Microbiol 31:665–677. doi: 10.1046/j.1365-2958.1999.01208.x. [DOI] [PubMed] [Google Scholar]
- 54.Gode-Potratz CJ, McCarter LL. 2011. Quorum sensing and silencing in Vibrio parahaemolyticus. J Bacteriol 193:4224–4237. doi: 10.1128/JB.00432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bose JL, Wollenberg MS, Colton DM, Mandel MJ, Septer AN, Dunn AK, Stabb EV. 2011. Contribution of rapid evolution of the luxR-luxI intergenic region to the diverse bioluminescence outputs of Vibrio fischeri strains isolated from different environments. Appl Environ Microbiol 77:2445–2457. doi: 10.1128/AEM.02643-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrun B, Lostroh CP. 2013. Vibrio fischeri exhibit the growth advantage in stationary-phase phenotype. Can J Microbiol 59:130–135. doi: 10.1139/cjm-2012-0439. [DOI] [PubMed] [Google Scholar]
- 57.Raychaudhuri S, Jain V, Dongre M. 2006. Identification of a constitutively active variant of LuxO that affects production of HA/protease and biofilm development in a non-O1, non-O139 Vibrio cholerae O110. Gene 369:126–133. doi: 10.1016/j.gene.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 58.Jung SA, Chapman CA, Ng W-L. 2015. Quadruple quorum-sensing inputs control Vibrio cholerae virulence and maintain system robustness. PLoS Pathog 11:e1004837. doi: 10.1371/journal.ppat.1004837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. 2002. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110:303–314. doi: 10.1016/S0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 60.Vance RE, Zhu J, Mekalanos JJ. 2003. A constitutively active variant of the quorum-sensing regulator LuxO affects protease production and biofilm formation in Vibrio cholerae. Infect Immun 71:2571–2576. doi: 10.1128/IAI.71.5.2571-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thungapathra AM, Sinha KK, Chaudhuri SR, Garg P, Ramamurthy T, Nair GB, Ghosh A. 2002. Occurrence of antibiotic resistance gene cassettes aac(6′)-Ib, dfrA5, dfrA12, and ereA2 in class I integrons in non-O1, Non-O139 Vibrio cholerae strains in India. Antimicrob Agents Chemother 46:2948–2955. doi: 10.1128/AAC.46.9.2948-2955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bortner SR. 1988. Biochemical and genetic studies of a Vibrio cholerae extracellular protease. Ph.D. thesis. Harvard University, Cambridge, MA. [Google Scholar]
- 63.Belas R, Simon M, Silverman M. 1986. Regulation of lateral flagella gene transcription in Vibrio parahaemolyticus. J Bacteriol 167:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keynan A, Hastings JW. 1961. The isolation and characterization of dark mutants of luminescent bacteria. Biol Bull 121:375. [Google Scholar]
- 65.Silverman M, Martin M, Engebrecht J. 1989. Regulation of luminescence in marine bacteria, p 71–86. In Hopwood DA, Chater KF (ed), Genetics of bacterial diversity. Academic Press, Waltham, MA. [Google Scholar]
- 66.Bassler BL, Wright M, Silverman MR. 1994. Sequence and function of LuxO, a negative regulator of luminescence in Vibrio harveyi. Mol Microbiol 12:403–412. doi: 10.1111/j.1365-2958.1994.tb01029.x. [DOI] [PubMed] [Google Scholar]
- 67.McCarter LL. 1998. OpaR, a homolog of Vibrio harveyi LuxR, controls opacity of Vibrio parahaemolyticus. J Bacteriol 180:3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaques S, McCarter LL. 2006. Three new regulators of swarming in Vibrio parahaemolyticus. J Bacteriol 188:2625–2635. doi: 10.1128/JB.188.7.2625-2635.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Showalter RE, Martin MO, Silverman MR. 1990. Cloning and nucleotide sequence of luxR, a regulatory gene controlling bioluminescence in Vibrio harveyi. J Bacteriol 172:2946–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang M, Schaefer AL, Dandekar AA, Greenberg EP. 2015. Quorum sensing and policing of Pseudomonas aeruginosa social cheaters. Proc Natl Acad Sci U S A 112:2187–2191. doi: 10.1073/pnas.1500704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dandekar AA, Chugani S, Greenberg EP. 2012. Bacterial quorum sensing and metabolic incentives to cooperate. Science 338:264–266. doi: 10.1126/science.1227289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shao Y, Feng L, Rutherford ST, Papenfort K, Bassler BL. 2013. Functional determinants of the quorum-sensing non-coding RNAs and their roles in target regulation. EMBO J 32:2158–2171. doi: 10.1038/emboj.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teng S-W, Schaffer JN, Tu KC, Mehta P, Lu W, Ong NP, Bassler BL, Wingreen NS. 2011. Active regulation of receptor ratios controls integration of quorum-sensing signals in Vibrio harveyi. Mol Syst Biol 7:491–491. doi: 10.1038/msb.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Henares BM, Higgins KE, Boon EM. 2012. Discovery of a nitric oxide responsive quorum sensing circuit in Vibrio harveyi. ACS Chem Biol 7:1331–1336. doi: 10.1021/cb300215t. [DOI] [PubMed] [Google Scholar]
- 75.Shikuma NJ, Fong JCN, Odell LS, Perchuk BS, Laub MT, Yildiz FH. 2009. Overexpression of VpsS, a hybrid sensor kinase, enhances biofilm formation in Vibrio cholerae. J Bacteriol 191:5147–5158. doi: 10.1128/JB.00401-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee K-H, Ruby EG. 1992. Detection of the light organ symbiont, Vibrio fischeri, in Hawaiian seawater by using lux gene probes Appl Environ Microbiol 58:942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee KH, Ruby EG. 1994. Effect of the squid host on the abundance and distribution of symbiotic Vibrio fischeri in nature. Appl Environ Microbiol 60:1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fazil MH, Kumar S, Rao NS, Selvaraj C, Singh SK, Pandey HP, Singh DV. 2012. Comparative structural analysis of two proteins belonging to quorum sensing system in Vibrio cholerae. J Biomol Struct Dyn 30:574–584. doi: 10.1080/07391102.2012.687523. [DOI] [PubMed] [Google Scholar]
- 79.Meyer M, Park S, Zeringue L, Staley M, McKinstry M, Kaufman RI, Zhang H, Yan D, Yennawar NH, Yennawar HP, Farber GK, Nixon BT. 2001. A dimeric two-component receiver domain inhibits the σ54-dependent ATPase in DctD. FASEB J 15:1326–1328. [DOI] [PubMed] [Google Scholar]
- 80.Park S, Meyer M, Jones AD, Yennawar HP, Yennawar NH, Nixon BT. 2002. Two-component signaling in the AAA+ ATPase DctD: binding Mg2+ and BeF3− selects between alternative dimeric states of the receiver domain. FASEB J 16:1964–1966. [DOI] [PubMed] [Google Scholar]
- 81.Doucleff M, Chen B, Maris AE, Wemmer DE, Kondrashkina E, Nixon BT. 2005. Negative regulation of AAA+ ATPase assembly by two-component receiver domains: a transcription activation mechanism that is conserved in mesophilic and extremely hyperthermophilic bacteria. J Mol Biol 353:242–255. doi: 10.1016/j.jmb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 82.Lee S-Y, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE. 2003. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev 17:2552–2563. doi: 10.1101/gad.1125603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gilson L, Kuo A, Dunlap PV. 1995. AinS and a new family of autoinducer synthesis proteins. J Bacteriol 177:6946–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee K-H. 1994. Ecology of Vibrio fischeri, the light organ symbiont of the Hawaiian sepiolid squid Euprymna scolopes. Ph.D. thesis. University of Southern California, Los Angeles, CA. [Google Scholar]
- 85.Ruby EG, Lee K-H. 1998. The Vibrio fischeri-Euprymna scolopes light organ association: current ecological paradigms. Appl Environ Microbiol 64:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boettcher KJ, Ruby EG. 1994. Occurrence of plasmid DNA in the sepiolid squid symbiont Vibrio fischeri. Curr Microbiol 29:279–286. doi: 10.1007/BF01577441. [DOI] [Google Scholar]
- 87.Nishiguchi MK, Ruby EG, McFall-Ngai MJ. 1998. Competitive dominance among strains of luminous bacteria provides an unusual form of evidence for parallel evolution in sepiolid squid-Vibrio symbioses. Appl Environ Microbiol 64:3209–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.