

Abstract
Vibrio parahaemolyticus is a bacterial pathogen that can cause illness after the consumption or handling of contaminated seafood. The primary virulence factors associated with V. parahaemolyticus illness are thermostable direct hemolysin (TDH) and Tdh-related hemolysin (TRH). However, clinical strains lacking tdh and trh have recently been isolated, and these clinical isolates are poorly understood. To help understand the emergence of clinical tdh- and trh-negative isolates, a genomic approach was used to comprehensively compare 4 clinical tdh- and trh-negative isolates with 16 environmental tdh- and trh-negative isolates and 34 clinical isolates positive for tdh or trh, or both, with the objective of identifying genomic features that are unique to clinical tdh- and trh-negative isolates. The prevalence of pathogenicity islands (PAIs) common to clinical isolates was thoroughly examined in each of the clinical tdh- and trh-negative isolates. The tdh PAI was not present in any clinical or environmental tdh- and trh-negative isolates. The trh PAI was not present in any environmental isolates; however, in clinical tdh- and trh-negative isolate 10-4238, the majority of the trh PAI including a partial trh1 gene was present, which resulted in reclassification of this isolate as a tdh-negative and trh-positive isolate. In the other clinical tdh- and trh-negative isolates, neither the trh gene nor the trh PAI was present. We identified 862 genes in clinical tdh- and trh-negative isolates but not in environmental tdh- and trh-negative isolates. Many of these genes are highly homologous to genes found in common enteric bacteria and included genes encoding a number of chemotaxis proteins and a novel putative type VI secretion system (T6SS) effector and immunity protein (T6SS1). The availability of genome sequences from clinical V. parahaemolyticus tdh- and trh-negative isolates and the comparative analysis may help provide an understanding of how this pathotype is able to survive in vivo during clinical illness.
INTRODUCTION
Vibrio parahaemolyticus is a Gram-negative, halophilic bacterium that is ubiquitous in marine and estuarine environments and is often found colonizing shellfish or shrimp. While most strains are nonpathogenic, many have acquired virulence factors that result in illness when individuals are exposed to V. parahaemolyticus strains carrying these virulence factors (1). V. parahaemolyticus is recognized to be a leading cause of foodborne illness worldwide and is transmitted via the handling and consumption of raw or undercooked contaminated seafood (1). Infections occur both sporadically and in very large outbreaks. The most common manifestation of V. parahaemolyticus infection is acute, watery diarrhea accompanied by abdominal pain and nausea, although symptoms can also be severe and include a dysentery-like illness or septicemia (2). Since most cases of illness caused by V. parahaemolyticus are self-limiting, rates of infection are probably underestimated due to underreporting.
Clinical V. parahaemolyticus isolates generally have at least one of two major toxigenic virulence factors, thermostable direct hemolysin (TDH) (3) and TDH-related hemolysin (TRH) (4). TDH has hemolytic activity on a blood-containing medium, Wagatsuma agar, and the process is referred to as the Kanagawa phenomenon (KP) (3). During infection, TDH is involved in cytotoxicity and hemolytic activity, and on the basis of the sequence similarity between TDH and TRH, TRH is believed to act similarly (5–7). The presence of tdh and/or trh is common in pathogenic isolates but relatively rare in environmental strains; therefore, the presence of these genes is used to assess the virulence potential of V. parahaemolyticus isolates (8, 9). In addition to tdh and trh, whole-genome sequencing (WGS) of V. parahaemolyticus led to the identification of two nonredundant type III secretion system (T3SS) gene clusters, dubbed T3SS1 and T3SS2, on chromosome 1 and chromosome 2, respectively, which are also involved in virulence (5, 10). T3SSs are needle-like apparatuses that inject bacterial effector proteins, such as toxins or hemolysins, directly through the membrane and into the cytoplasm of eukaryotic cells (11). On the basis of its G+C content and its high degree of sequence identity with the T3SSs of other Vibrio species, T3SS1 appears to have been ancestrally acquired and is present in all V. parahaemolyticus isolates, even nonpathogenic strains (10). Once it is in vivo, T3SS1 appears to inject effectors, such as VopQ, VopR, VopS, and VPA0450, directly into eukaryotic cells, resulting in cytotoxicity (5, 10). T3SS2 derives from two separate lineages: T3SS2α is typically found on a pathogenicity island (PAI) with the tdh gene, and T3SS2β is found with the trh gene. T3SS2 appears to inject VopA, VopC, VopL, and VopT into eukaryotic cells, resulting in both cytotoxicity and enterotoxicity (5, 12–14). T3SS2 has a lower G+C content than the genomic average, which is indicative of a recent acquisition of this region through lateral gene transfer (15).
Some V. parahaemolyticus strains also have two type VI secretion systems (T6SSs) (16). The T6SS was recently defined functionally in V. cholerae and is structurally similar to a contractile phage tail, but in the reverse orientation, that is fully assembled inside the bacterial cell and injects effectors directly into the recipient cell (17, 18). It is composed of 13 essential genes and a variable number of nonessential genes, including various effectors (19). In V. parahaemolyticus, T6SS2 is found in all strains, while T6SS1 is mostly associated with clinical isolates (20) and may play a role in virulence (21), although this has not yet been demonstrated conclusively. The T6SS1 also appears to have antibacterial activity in the environment, which may give isolates containing this system a competitive advantage (16).
Recent studies have reported the isolation of strains that lack both tdh and trh (tdh- and trh-negative strains) from clinical samples (22–27). However, the ability of tdh- and trh-negative strains to independently cause clinical illness is still controversial. For example, coinfection with multiple V. parahaemolyticus strains is known to occur, and if at least one infecting strain carries tdh or trh, it is possible that nonpathogenic tdh- and trh-negative strains could be isolated from a sick individual without having a direct involvement in illness (24). The coinfection model is supported by the finding that a single seafood sample is often contaminated by several different V. parahaemolyticus strains, some of which appear to be nonpathogenic (24). Alternatively, there are several lines of evidence that support the opinion that some tdh- and trh-negative isolates are able to induce clinical infection. During a coinfection study, three sick patients produced 30 tdh- and trh-negative isolates, and despite multiple culturing attempts, no other enteric pathogen or tdh- and trh-positive V. parahaemolyticus strain could be isolated from these patients (24). However, regardless of their independent pathogenicity, clinical tdh- and trh-negative isolates have a demonstrated ability to survive in vivo during illness, and we do not understand if the role of these isolates in human illness is as a causative agent, an innocent bystander, or an active participant in a multistrain infection. Therefore, this investigation was undertaken to better understand clinical tdh- and trh-negative isolates and how they compare on a genomic level to traditional pathogenic V. parahaemolyticus isolates and environmental tdh- and trh-negative isolates. Here we present a thorough comparative genomic analysis of multiple clinical tdh- and trh-negative isolates. This comprehensive approach has provided several insights into the pangenomics of clinical V. parahaemolyticus isolates and led to the identification of a novel putative T6SS effector.
MATERIALS AND METHODS
Genome sequencing, assembly, and annotation.
Each of the clinical isolates sequenced in this study were Canadian clinical strains originating from provincial public health laboratories and submitted to the National Microbiology Laboratory (Public Health Agency of Canada), the British Columbia Centre for Disease Control (BCCDC), or the Bureau of Microbial Hazards (BMH) (Health Canada). All isolates were routinely propagated on TSA-2N agar (Difco BD, NJ, USA). The environmental isolates were each isolated from seafood by the Vibrio Reference Laboratory in Canada. DNA for whole-genome sequencing was extracted using a Maxwell 16 SEV cell DNA purification kit (Promega, Madison, WI). The short-read sequence data were generated by preparing a paired-end library with a Nextera XT DNA sample preparation kit (Illumina, San Diego, CA) and sequencing the library on a MiSeq benchtop sequencer (Illumina) for 500 or 600 cycles. Previously, Banerjee et al. (26) performed PCR amplification of the tdh and trh genes, and this allowed us to select isolates for WGS and group isolates according to clinical pathotype (tdh and trh positive, tdh negative and trh positive, tdh positive and trh negative, or tdh and trh negative). This grouping scheme was used during the analysis in our study.
Genomes were assembled de novo using the SPAdes (version 3.1.1) program, and quality was assessed using QUAST, as outlined by Ronholm et al. (Table 1) (47). De novo assembly resulted in the PAIs of interest, including the tdh PAI and the trh PAI, being split between contigs. For analysis of pathogenicity islands, raw MiSeq reads were reassembled by reference-guided assembly using the Burrows-Wheeler aligner (BWA; version 07.05a) (29) and then visualized and inspected using the Tablet (version 1.14.10.20) program (30).
TABLE 1.
Sequencing and annotation results for V. parahaemolyticus strains analyzed during this studya
| Genotype and strain | Yr | Source | Serotype | BioProject accession no. | No. of contigs | Genome size (bp) | Coverage (fold) | No. of ORFs | Reference or source |
|---|---|---|---|---|---|---|---|---|---|
| tdh negative and trh negative | |||||||||
| 04-2548 | 2004 | Clinical | O1:KVI | JTGS00000000 | 101 | 5,549,224 | 68 | 5,001 | 44 |
| 09-5357 | 2009 | Clinical | O11:KUT | JTGT00000000 | 49 | 5,297,642 | 136 | 5,124 | 44 |
| 10-4238 | 2007 | Clinical | O1:KUT | JTGQ00000000 | 55 | 5,114,934 | 108 | 4,665 | 44 |
| 10-4239 | 2007 | Clinical | O4:KUT | JTGR00000000 | 63 | 5,082,538 | 133 | 4,780 | 44 |
| HS-22-14 | 2014 | Clam | Unknown | LIRV00000000 | 235 | 5,761,615 | 93 | 5,184 | This study |
| ISF-29-03 | 2011 | Shrimp | Unknown | LFYM00000000 | 94 | 5,368,163 | 175 | 4,956 | This study |
| ISF-54-12 | 2011 | Shrimp | Unknown | LIRR00000000 | 74 | 5,041,359 | 124 | 4,513 | This study |
| ISF-77-01 | 2011 | Shrimp | Unknown | LFZG00000000 | 95 | 5,042,958 | 178 | 4,605 | This study |
| RM-14-05 | 2014 | Mussel | Unknown | LFXK00000000 | 93 | 5,060,270 | 101 | 4,650 | This study |
| V110 | 2010 | Shrimp | Unknown | AQPJ00000000 | 167 | 5,505,021 | 100 | 5,084 | 45 |
| S171 | 2007 | Environment | Unknown | AWHJ00000000 | 342 | 5,146,548 | 81 | 4,505 | NCBI |
| VIP4-0444 | 2008 | Fish | Unknown | AXNR00000000 | 106 | 5,271,920 | 20 | 4,848 | NCBI |
| VIP4-0447 | 2008 | Oyster | Unknown | AXNS00000000 | 113 | 5,367,084 | 23 | 4,983 | NCBI |
| VPCR-2009 | 2009 | Water | Unknown | JDFL00000000 | 110 | 5,107,307 | 167 | 4,754 | NCBI |
| VPTS-2009 | 2009 | Water | Unknown | JDFM00000000 | 83 | 5,084,059 | 177 | 4,626 | NCBI |
| SG176 | 2006 | Water | Unknown | JMMQ00000000 | 48 | 4,952,407 | 100 | 4,543 | 27 |
| J-C2-34 | 1998 | Sediment | Unknown | JMMR00000000 | 91 | 5,150,449 | 100 | 4,814 | 27 |
| 22702 | 1998 | Sediment | Unknown | JMMT00000000 | 43 | 4,955,222 | 100 | 4,504 | 27 |
| NCKU-TN-S02 | 2008 | Shrimp | Unknown | JPKV00000000 | 97 | 5,410,371 | 100 | 4,969 | NCBI |
| VH3 | 2007 | Aquaculture | Unknown | LCVL00000000 | 67 | 4,955,051 | 89 | 4,453 | 46 |
| tdh positive and trh positive | |||||||||
| 04-1290 | 2004 | Clinical | O4:KII | JXVK00000000 | 97 | 5,143,304 | 111 | 4,767 | 47 |
| 09-3216 | 2009 | Clinical | O4:KII | JXVJ00000000 | 78 | 5,100,021 | 100 | 4,715 | 47 |
| 09-3217 | 2009 | Clinical | O4:K63 | JZAQ00000000 | 155 | 5,060,710 | 126 | 4,690 | This study |
| 10-4241 | 2006 | Clinical | O4:KII | JXVI00000000 | 57 | 5,104,503 | 44 | 4,719 | 47 |
| 10-4242 | 2006 | Clinical | O4:KII | JXVH00000000 | 74 | 5,126,748 | 55 | 4,758 | 47 |
| 10-4245 | 2006 | Clinical | O4:KII | JXVG00000000 | 70 | 5,097,053 | 66 | 4,697 | 47 |
| 10-4246 | 2006 | Clinical | O4:KII | JXVF00000000 | 74 | 5,098,537 | 80 | 4,704 | 47 |
| 10-4247 | 2006 | Clinical | O4:KII | JXVE00000000 | 84 | 5,124,180 | 107 | 4,745 | 47 |
| 10-4248 | 2006 | Clinical | O4:KII | JXVD00000000 | 117 | 5,112,922 | 101 | 4,737 | 47 |
| 10-4255 | 2006 | Clinical | O1:K56 | JYJT00000000 | 197 | 5,092,008 | 115 | 4,723 | This study |
| 10-4274 | 2005 | Clinical | O4:KII | JXVC00000000 | 96 | 5,115,101 | 73 | 4,751 | 47 |
| 10-4287 | 2003 | Clinical | O6:K18 | JYJU00000000 | 333 | 5,269,347 | 133 | 4,969 | This study |
| 10-4288 | 2003 | Clinical | O4:KII | JXVB00000000 | 61 | 5,109,523 | 70 | 4,717 | 47 |
| 10-4293 | 2002 | Clinical | O4:KII | JXVA00000000 | 58 | 5,202,165 | 50 | 4,841 | 47 |
| 10-4298 | 2001 | Clinical | O4:KII | JXUZ00000000 | 76 | 5,233,510 | 45 | 4,829 | 47 |
| 10-4303 | 2000 | Clinical | O4:KII | JXUY00000000 | 52 | 5,106,734 | 56 | 4,708 | 47 |
| 10-7197 | 2008 | Clinical | O4:KII | JXUX00000000 | 56 | 5,091,435 | 31 | 4,684 | 47 |
| tdh positive and trh negative | |||||||||
| 04-2549 | 2004 | Clinical | O11:KUT | JYNG00000000 | 138 | 5,129,592 | 99 | 4,750 | This study |
| 04-2551 | 2004 | Clinical | O3:K6 | LASF00000000 | 85 | 5,187,612 | 124 | 4,794 | This study |
| 07-1339 | 2007 | Clinical | O3:K6 | LASG00000000 | 88 | 5,251,833 | 65 | 4,849 | This study |
| 07-2965 | 2007 | Clinical | O5:KUT | JZAN00000000 | 85 | 5,216,789 | 113 | 4,817 | This study |
| 08-0278 | 2008 | Clinical | O2:K3 | JZAO00000000 | 188 | 5,206,818 | 111 | 4,922 | This study |
| 09-4435 | 2009 | Clinical | O3:K6 | JZAR00000000 | 115 | 5,120,353 | 87 | 4,725 | This study |
| 10-4251 | 2006 | Clinical | O3:K6 | JYJS00000000 | 156 | 5,220,324 | 105 | 4,815 | This study |
| tdh negative and trh positive | |||||||||
| 08-7626 | 2008 | Clinical | O1:K58 | JZAP00000000 | 121 | 5,207,542 | 101 | 4,841 | This study |
| 09-3218 | 2009 | Clinical | O1:KUT | LASH00000000 | 89 | 5,204,568 | 106 | 4,807 | This study |
| 09-4434 | 2009 | Clinical | O1:KUT | LASI00000000 | 80 | 5,194,240 | 148 | 4,789 | This study |
| 09-4660 | 2009 | Clinical | O1:KUT | LASJ00000000 | 73 | 5,191,811 | 118 | 4,784 | This study |
| 09-4663 | 2009 | Clinical | O1:KUT | JYJQ00000000 | 148 | 5,217,021 | 112 | 4,856 | This study |
| 09-4664 | 2009 | Clinical | O1:KUT | LASK00000000 | 89 | 5,196,673 | 69 | 4,810 | This study |
| 09-4681 | 2009 | Clinical | O3:KUT | LASL00000000 | 49 | 5,177,998 | 106 | 4,713 | This study |
| 10-4243 | 2006 | Clinical | OUT:KUT | LASM00000000 | 76 | 5,254,404 | 95 | 4,821 | This study |
| 10-4244 | 2006 | Clinical | O8:KUT | JYJR00000000 | 129 | 5,270,549 | 109 | 4,864 | This study |
| 10-7205 | 2008 | Clinical | O1:K58 | JYJV00000000 | 146 | 5,217,765 | 134 | 4,855 | This study |
Additional characterization of all clinical strains was completed by Banerjee et al. (26).
Annotation of the function of the protein-coding sequences was performed using the Basic Local Alignment Search Tool (BLAST), which was used to compare the sequences obtained against the sequences in the COG (Clusters of Orthologous Groups) of proteins databases and the NCBI nr protein database.
Comparative genomics.
An in-house Perl script was used to identify all orthologous, accessory, and unique genes between Vibrio genomes (the source code can be found at https://github.com/bfssi-nicholas-petronella/HARDCore.git.) Using the BLAST program, genes having 60% sequence identity over 80% of their length were considered orthologs (31, 32). The set of orthologous genes shared by all genomes was defined as the core genome, total genes identified within all genomes were defined as the pangenome, accessory genomes were defined as the set of genes possessed by a subset of genomes (pathotypes), and unique genomes were the subset of genes possessed only by a single strain's genome. To construct whole-genome rarefaction curves, an in-house script was also used in an iterative manner to obtain the number of genes contained in the pangenome relative to the number of genomes analyzed. For example, for the data points for the 16 genomes, the size of the pangenome was calculated for 16 randomly selected genomes (from the 38 possibilities). This was repeated 1,000 times using 16 genomes randomly selected from 38 genomes, and the average was taken. The purpose was to determine if V. parahaemolyticus had an open or closed pangenome and verify that the genomes from the number of strains selected for the experiment could adequately represent the core genome.
Phylogenetics.
To construct phylogenetic trees for the clinical isolates, the largest representative sequence of each core gene was retrieved from the pangenome. The homologue for each representative sequence was then retrieved from the whole genome of each strain. The Prokka software tool was used to generate the corresponding amino acid sequence (33). Trees were constructed from the core genome amino acid sequence. Each gene was aligned using the Muscle (version 3.8.31) program with default parameters (34), and then RAxML software was used to construct a maximum likelihood phylogeny from concatenated alignments with 100 bootstrap values (35).
To construct phylogenetic trees for each isolate used in the study, the multilocus sequence typing (MLST) sequences were retrieved from the WGS. Trees were constructed from the MLST alleles. Each gene was aligned using the Muscle (version 3.8.31) program with default parameters (34), and then RAxML was used to construct a maximum likelihood phylogeny from concatenated alignments with 1,000 bootstrap values (35).
Genetic islands and secretion systems.
The presence and absence of the tdh and trh pathogenicity islands and various secretion systems were investigated using BLASTn. The EasyFig (version 2.1) program was used for visualization of genomic islands (36). For larger genomic islands (tdh PAI and trh PAI), a Burrows-Wheeler transform reference-guided assembly was used on raw fastq data with the canonical sequence of the genomic island (29). The assembly was inspected for accuracy and validity using Tablet, prior to visualization (30). To possibly identify novel pathogenicity islands in the tdh- and trh-negative pathotype, the completed results of an IslandViewer analysis on strain CDC_K4557 were downloaded from the IslandViewer3 website (37). BLASTn was used to search each genome included in this study (Table 1) for each island that was identified.
Nucleotide sequence accession numbers.
All nucleotide sequence data referred to in this article have been deposited in the DDBJ/EMBL/GenBank database under the various BioProject accession numbers provided in Table 1. Additional important data are included in the supplemental material.
RESULTS AND DISCUSSION
Type III secretion systems and tdh and trh pathogenicity islands.
Traditionally, a pathogenic V. parahaemolyticus strain has been defined by the presence of tdh or trh, or both (5). These two virulence factors generally occur near a T3SS. Two T3SSs have been reported in V. parahaemolyticus, and two variants of T3SS2 (T3SS2α and T3SS2β) have been described (12). T3SS2α is associated with tdh, while T3SS2β is typically found with trh (14), though exceptions exist (27). T3SS1 is composed of 42 genes (VPA1656 to VAP1696 and VPA0450 in V. parahaemolyticus RIMD2210633 [15]). T3SS1 was present in each of the clinical and environmental isolates in this investigation. The tdh PAI (also known as VPAI-7) is composed of 87 coding sequences (in V. parahaemolyticus RIMD2210633) and includes tdhA, tdhS, and the T3SS2α genes (38). In our current work, due to multiple homologous areas in the tdh PAI and trh PAI areas, the use of de novo assembly led to this large PAI being split between multiple contigs. Therefore, to analyze this region properly, a reference-guided assembly based on the tdh PAI sequence (VPA1310 to VPA1396) from V. parahaemolyticus RIMD2210663 was used (39). Using this method, a complete tdh PAI including T3SS2α was consistently observed in all clinical tdh-positive and trh-negative isolates (Fig. 1A). Clinical tdh- and trh-positive isolates contained homologues to some coding sequences typically found in this island (VPA1310, VPA1311, VPA1313 to VPA1318, VPA1320, VPA1321, VPA1329, VPA1342, VPA1347, and VPA1382 to VPA1393), and their presence was consistent among all clinical tdh- and trh-positive isolates (see Fig. 3A). Isolates categorized as tdh negative and trh positive or tdh and trh negative did not contain a PAI with homology to tdh PAI genes (Fig. 1A).
FIG 1.

Pathogenicity islands and T3SS2. tdh PAI (A) and trh PAI (B) are the two canonical genomic islands present in clinical isolates of V. parahaemolyticus. The presence or absence of these islands is strongly correlated with the presence of the tdh and trh genes. In three of the four tdh- and trh-negative isolates, these genes are entirely absent. (C) In the fourth tdh- and trh-negative isolate, 10-4328, a trh PAI including a partial trh1 gene is mostly present. Abbreviations: neg, negative; pos, positive.
FIG 3.

COG profiles of the core (A), accessory (B), and unique (C) genomes of clinical tdh- and trh-negative isolates. The numbers at the top of each column denote the number of genes in the unique genome of the corresponding strain listed at the bottom of the column. Abbreviations: neg, negative; pos, positive.
The trh PAI, composed of 81 coding sequences, was also independently assembled using reference-guided assembly and V. parahaemolyticus VIPARAQ4037 residues 1748 to 1830 as a reference and was consistently observed in isolates of both the tdh-negative and trh-positive pathotype and the tdh- and trh-positive pathotype and in all instances included trh1 and the T3SS2β genes (Fig. 1B). The association between the trh PAI and T3SS2β with the tdh-negative and trh-positive pathotype and the tdh- and trh-positive pathotype agrees with the findings of previous studies (12, 27). Three clinical tdh- and trh-negative isolates, isolates 04-2548, 09-5357, and 10-4239, did not contain the tdh PAI, the trh PAI, or a T3SS2; however, an almost complete trh PAI including a partial trh gene, urease gene cluster, and the T3SS2β genes was identified in isolate 10-4238, which was categorized as tdh and trh negative by PCR analysis by Banerjee et al. (26) (Fig. 1C). In addition to the TRH hemolysins, the T3SSβ present in this PAI also contains effectors thought to be involved in enterotoxicity and cytotoxicity (5). T3SS2β appears to be a recent acquisition by V. parahaemolyticus and is sometimes found in non-O1, non-O139, and CTX V. cholerae strains (12). The presence of the trh PAI in 10-4238 made us question whether this strain should indeed be classified as a clinical tdh- and trh-negative isolate or if it would be more accurate, on the basis of its WGS, to classify it as a tdh-negative and trh-positive strain. Therefore, this strain was removed from the remaining analysis specific to clinical tdh- and trh-negative isolates.
General genomic features of V. parahaemolyticus clinical isolates.
To depict the genetic diversity of pathogenic V. parahaemolyticus strains, 38 clinical isolates (Table 1) representing each of the four previously described genotypes (tdh and trh positive, tdh negative and trh positive, tdh positive and trh negative, and tdh and trh negative) (40, 41) were extensively compared. The pangenome of clinical V. parahaemolyticus isolates was calculated and consisted of 8,399 protein-coding genes (Fig. 2A). To assess the accuracy of computing of a pangenome using draft genomes, three clinical isolates with closed genomes (see Table S1 in the supplemental material) were added to our data set and the pangenome size was recalculated. The addition of three closed genomes caused the size of the pangenome of the clinical isolates to increase to 8,609 genes. This increase would also have been expected after the addition of three draft genomes, and therefore, we concluded that our calculations based on the draft genomes were accurate.
FIG 2.
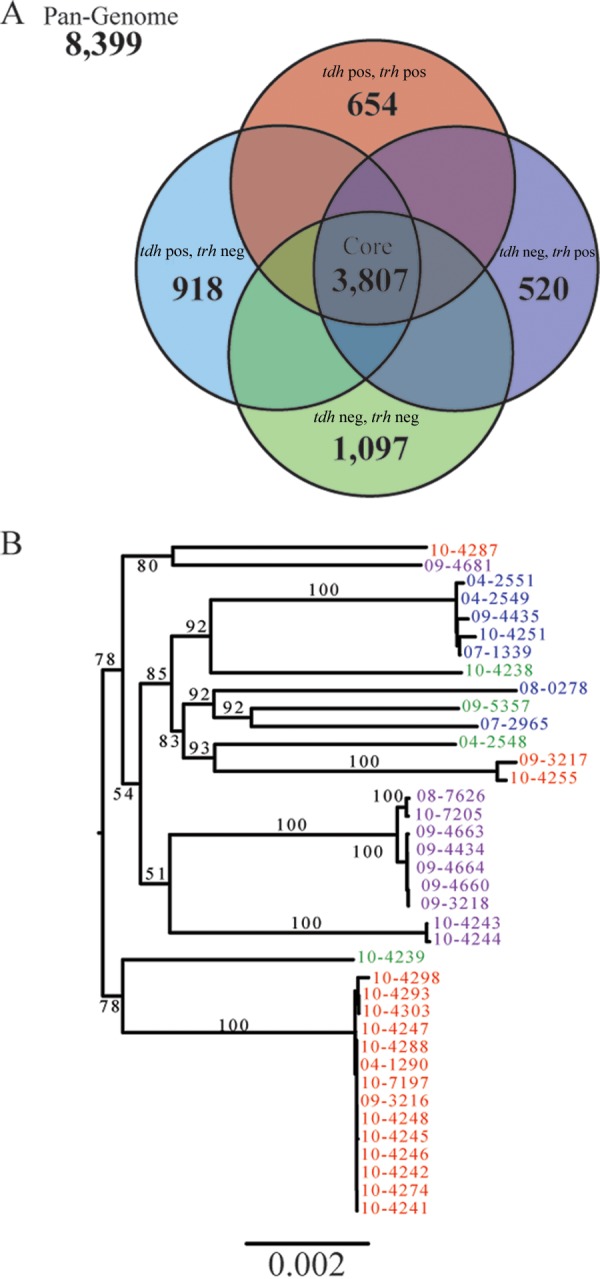
General genomic features of clinical V. parahaemolyticus isolates. (A) The pangenome of clinical V. parahaemolyticus isolates was constructed using the de novo assembly of 38 clinical isolates and contained 8,399 genes. Pangenomes were also assembled for each pathotype and compared. (B) A phylogenetic tree, constructed from concatenated core genes, shows the phylogeny of clinical isolates and demonstrates that each of the pathotypes is polyphyletic.
A gene intersection analysis of the accessory genomes was performed. From this analysis, we defined a core genome of orthologous genes that were shared by all clinical V. parahaemolyticus isolates. The core genome contained 3,807 protein-coding genes, which represented between 76 to 81% of each isolate's genome (Fig. 2A). The size of the core genome remained relatively stable for each additional genome added after the first four. When the closed genomes of three additional clinical isolates (see Table S1 in the supplemental material) were added to the data set and the core genome was recalculated, it decreased to a size of 3,803 protein-coding genes. This indicated that the 38 draft genomes sequenced here provided an excellent estimation of the true core genome of clinical V. parahaemolyticus isolates.
Accessory genes that were unique to each pathotype were also identified, and 654, 520, 918, and 1,097 genes were specific to the tdh- and trh-positive, tdh-negative and trh-positive, tdh-positive and trh-negative, and tdh- and trh-negative pathotypes of clinical isolates, respectively (Fig. 2A). Strain-specific unique genes were also identified, and the sizes of the unique genome varied between strains. For example, seven strains (09-3216, 10-4303, 10-7197, 09-3218, 09-4434, 09-4660, and 09-4664) possessed no unique genes, while one strain, 04-2548 (tdh and trh negative), had 405 unique genes. Clinical isolates of the tdh- and trh-negative pathotype consistently had large unique genomes.
To determine if clinical tdh- and trh-negative isolates are monophyletic and possibly the result of a single loss of a pathogenicity island, an unrooted phylogenetic tree was constructed from the concatenated amino acid sequences of the 3,899 core genes (Fig. 2B). Use of the core genes provides a high-resolution view of phylogeny. This revealed that isolates of each of the pathotypes, tdh- and trh-negative isolates, are polyphyletic. This may indicate a high degree of mobility of pathogenicity elements between V. parahaemolyticus isolates, rather than a single PAI deletion event.
The distribution of clusters of orthologous groups (COGs) of proteins was determined for the core genome, the accessory genome of each pathotype, and the unique genome of each of the tdh- and trh-negative isolates, to determine if there were differences in the proportion of the genome attributable to particular cellular processes in clinical isolates (see Table S2 in the supplemental material). Almost 20% of the core genome was classified as having an unknown function; this proportion dropped to less than 10% in each of the pathotype-specific unique genomes (Fig. 3A and B). The tdh- and trh-negative isolates had a large proportion of genes involved in cell motility relative to the proportion for the other pathotypes (Fig. 3B). Individual tdh- and trh-negative isolates also had large and functionally consistent unique genomes (Fig. 3C). Within this functional category of cell motility, each of the three clinical tdh- and trh-negative isolates contained a methyl-accepting chemotaxis protein not observed in the other pathotypes or in environmental isolates. Cell motility is generally considered to be a factor associated with the ability of V. parahaemolyticus to survive in vivo (5); therefore, the finding of an increase in the number of genes involved in motility in clinical isolates is logical.
General genomic features of V. parahaemolyticus environmental isolates.
To provide a basis for comparison of the clinical tdh- and trh-negative isolates, we sequenced the genomes of 5 environmental tdh- and trh-negative isolates and collected the genomes of an additional 11 from the NCBI database (Table 1). To demonstrate the wide diversity and the relationships of the strains used in this study, a rooted phylogenetic tree was constructed from the concatenated nucleotide sequences of seven housekeeping genes (recA, gyrB, dnaE, dtdS, pntA, pyrC, and tnaA) traditionally used in V. parahaemolyticus MLST analysis (Fig. 4A). Clinical and environmental isolates shared several common lineages, again demonstrating the dynamic nature of virulence factors in this species.
FIG 4.

General genomic features of environmental V. parahaemolyticus isolates. (A) A phylogenetic tree, constructed from concatenated MLST sequences, demonstrates the diversity of the strains used in this study, as well as the relationships between each clinical and environmental strain included in this study. (B) A rarefaction curve of the genetic diversity of clinical and environmental V. parahaemolyticus strains was created. Environmental isolates have a much greater genetic diversity than clinical isolates. In addition, these curves demonstrate that the pangenome of V. parahaemolyticus is open.
The sizes of the core genome and pangenome were calculated for the 16 environmental isolates. The core genome of the environmental tdh- and trh-negative isolates was composed of 2,773 protein-coding genes, and though the pangenome was constructed from fewer genomes, it was much larger than that of the clinical isolates at 11,669 protein-coding genes (Fig. 4B). For both the clinical and environmental isolates, the pangenome size increased, in terms of protein-coding gene number, after the addition of each genome, which indicates an open pangenome (42), and this is in agreement with the findings of earlier studies (43) (Fig. 4B). The increased size of the pangenome in environmental isolates is likely due to their ability to survive in diverse niches in situ. For example, environmental isolates were collected from colonized shrimp, muscle, oyster, and clams as well as from marine water and sediments, while clinical isolates have been preselected by use of a very narrow criterion, which is the ability to colonize humans and cause illness. This likely leads to the low level of genetic diversity observed in clinical isolates.
Whole-genome comparison of clinical and environmental tdh- and trh-negative isolates.
The genomes of the 3 clinical tdh- and trh-negative isolates were compared with the genomes of 16 environmental tdh- and trh-negative isolates, resulting in the identification of 862 protein-coding genes unique to clinical isolates (Fig. 5; see also Table S3 in the supplemental material). From these genes, 529 were annotated as hypothetical proteins. A large portion of both hypothetical proteins and annotated proteins had high sequence similarity with other genes from other enteric bacteria, such as V. cholerae, Listeria monocytogenes, Campylobacter jejuni, Salmonella enterica, Escherichia coli, and Enterobacter. While this finding is not conclusive, it may indicate that the acquisition of genes from other enteric bacteria may contribute to the ability of clinical tdh- and trh-negative isolates to colonize humans during a gastrointestinal illness.
FIG 5.
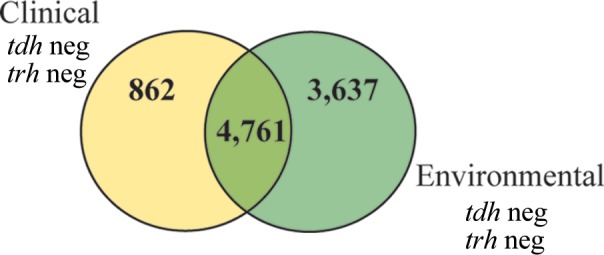
Genomic comparison of environmental and clinical tdh- and trh-negative isolates. On the basis of a comparison of protein-coding genes, 862 protein-coding genes that are unique to clinical tdh- and trh-negative isolates were identified. neg, negative.
Pathogenicity islands.
By examining the complete genome of V. parahaemolyticus RIMD2210633, Hurley et al. (2006) identified seven genomic islands which occur in pathogenic V. parahaemolyticus isolates (38). VPAI-1, VPAI-4, VPAI-5, and VPAI-6 appeared to represent DNA acquired by pandemic V. parahaemolyticus isolates (38). The presence of these islands was variable in our clinical and environmental isolates (Tables 2 and 3). VPAI-1 was present in most clinical tdh-positive and trh-negative isolates but was largely absent from isolates of the other pathotypes. The entire VPAI-2 was present in most tdh-positive and trh-negative isolates and tdh-negative and trh-positive isolates but was only partially present in tdh- and trh-positive and tdh- and trh-negative strains. VPAI-6 was consistently present in each of the clinical isolates. Strains 07-2965 and 08-0278 did not typically have the same PAI profile as the other tdh-positive and trh-negative strains, and this was also reflected in the phylogenetic tree (Fig. 2B). The 10-4238 tdh- and trh-negative strain carried VPAIs more similar to those carried by the tdh-negative and trh-positive isolates, agreeing with findings presented earlier in this paper that this strain is likely a tdh-negative and trh-positive strain and was misidentified by PCR analysis. The environmental tdh- and trh-negative isolates carried pathogenicity island profiles similar to those of the clinical isolates (Table 3), this observation raises several questions about the true roles of these islands and whether their inclusion in pandemic strains is associated more with fitness in the environment than with pathogenicity.
TABLE 2.
Distribution of pathogenicity islands in clinical isolates
| Genotype and strain | Gene(s) in the following V. parahaemolyticus PAI or presence or absence of a PAI: |
|||||
|---|---|---|---|---|---|---|
| VPAI-1 (VP0380 to VP0403) | VPAI-2 (VP0635 to VP0643) | VPAI-3 (VP1071 to VP1094) | VPAI-4 (VP2131 to VP2144) | VPAI-5 (VP2900 to VP2910) | VPAI-6 (VPA1253 to VPA1270) | |
| tdh negative and trh negative | ||||||
| 04-2548 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 09-5357 | VP0380, VP0398, VP0400 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| (T12739) 10-4238 | − | + | VP1077 to VP1094 | − | − | + |
| (T9109) 10-4239 | VP0380, VP0397 to VP0403 | VP0635, VP0636 | VP1071, VP1088 to VP1094 | − | − | + |
| tdh positive and trh positive | ||||||
| 04-1290 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 09-3216 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 09-3217 | − | VP0635, VP0636 | VP1088 to VP1094 | VP2144 | − | + |
| 10-4241 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4242 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4245 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4246 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4247 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4248 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4255 | VP0380 to VP0384, VP0386 to VP0392 | VP0635, VP0636 | VP1088 to VP1094 | VP2144 | − | + |
| VP0396 to VP0400, VP0402, VP0403 | ||||||
| 10-4274 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4287 | VP0380, VP0397 to VP0400 VP0402, VP0403 | VP0635, VP0636 | VP1071, VP1076, VP1088 to VP1094 | VP2144 | − | + |
| 10-4288 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4293 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4298 | VP0380, VP0382, VP0386, VP0395, VP0402 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4303 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-7197 | − | VP0635, VP0636 | VP1088−VP1094 | − | − | + |
| tdh positive and trh negative | ||||||
| 04-2549 | + | + | + | − | + | + |
| 04-2551 | + | + | + | + | + | + |
| 07-1339 | + | + | + | + | + | + |
| 07-2965 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 08-0278 | VP0380, VP0398, VP0400, VP0402, VP0403 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 09-4435 | + | + | + | + | + | + |
| 10-4251 | + | + | + | + | + | + |
| tdh negative and trh positive | ||||||
| 08-7626 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
| 09-3218 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
| 09-4434 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
| 09-4660 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
| 09-4663 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
| 09-4664 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
| 09-4681 | VP0380 to VP0388, VP0395 to VP0400 VP0402, VP0403 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4243 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-4244 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 10-7205 | VP0380, VP0398 to VP0400, VP0402, VP0403 | + | VP1088 to VP1094 | − | − | + |
TABLE 3.
Distribution of pathogenicity islands in environmental isolates
| tdh- and trh-negative strain | Gene(s) in the following V. parahaemolyticus PAI or presence or absence of a PAI: |
|||||
|---|---|---|---|---|---|---|
| VPAI-1 (VP0380 to VP0403) | VPAI-2 (VP0635 to VP0643) | VPAI3 (VP1071 to VP1094) | VPAI-4 (VP2131 to VP2144) | VPAI-5 (VP2900 to VP2910) | VPAI-6 (VPA1253 to VPA1270) | |
| HS-22-14 | VP0387 to VP0395 | − | − | − | − | VPA1254, VPA1256, VPA1258 to VPA1259, VPA1262 to VPA1265, VPA1267 |
| ISF-29-3 | VP0380, VP0398 to VP0400, VP0402, VP0403 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| ISF-54-12 | − | VP0635 to VP0636, VP0638 to VP0643 | VP1088 to VP1094 | − | − | + |
| ISF-77-01 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| RM-14-5 | − | VP0635, VP0636 | VP1088 to VP1094 | VP2131 to VP2133 VP2136 to VP2144 | − | + |
| V110 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| S171 | VP0381 to VP0384 | VP0635, VP0636 | VP1088 to VP1094 | VP2144 | − | + |
| VIP4-0444 | − | + | VP1071, VP1088 to VP1094 | − | − | + |
| VIP4-0447 | − | VP0635, VP0636 | VP1088 to VP1094 | VP2131 to VP2133 VP2136 to VP2144 | − | + |
| VPCR-2009 | − | + | VP1071, VP1073, VP1074, VP1075, VP1088 to VP1094 | − | − | + |
| VPTS-2009 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| SG176 | VP0381 to VP0384 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| J-C2-34 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| 22702 | − | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| NCKU-TN-S02 | VP0381 to VP0383, VP0388, VP0395 | VP0635, VP0636 | VP1088 to VP1094 | − | − | + |
| VH3 | VP0635, VP0636 | VP1071, VP1076, VP1088 to VP1094 | − | − | + | |
IslandViewer3 was used to search the closed genome of CDC_K4557, which is also a clinical V. parahaemolyticus tdh- and trh-negative isolate, for genomic islands that are common to clinical isolates but that are not found in environmental tdh- and trh-negative isolates (37). We reasoned that if an island was present in at least some of the clinical tdh- and trh-negative isolates but absent from all of the environmental tdh- and trh-negative isolates, it would be a reasonable candidate for evaluation as a novel pathogenicity island. IslandViewer3 was used to identify 29 genomic islands on chromosome 1 and 8 genomic islands on chromosome 2 (Fig. 6). These genomic islands were assessed for their presence or absence across our 43 genomes (see Table S4 in the supplemental material). We found that islands 1, 3, 33, 35, and 36 (as denoted in Fig. 6) were present in almost every strain of V. parahaemolyticus. An island that was present in clinical tdh- and trh-negative strains but not in environmental tdh- and trh-negative strains was not identified.
FIG 6.
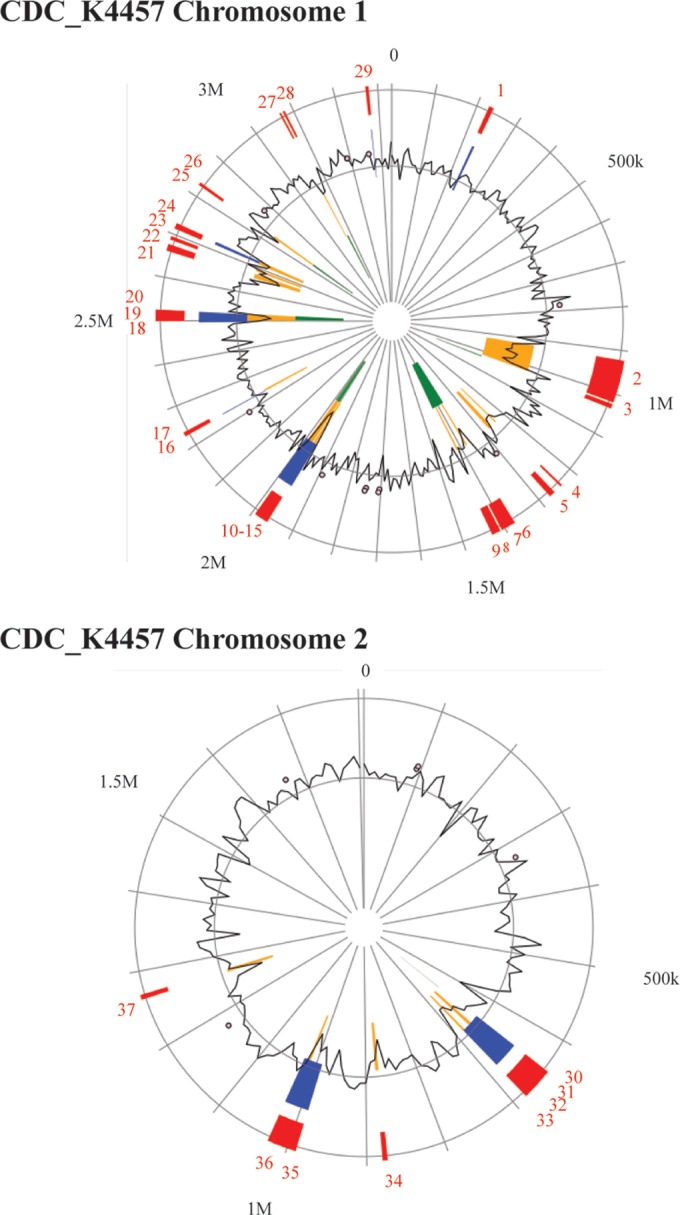
Genomic islands identified in CDC_K4557. A closed genome is required to identify novel genomic islands. To determine if a novel pathogenicity island is responsible for virulence in the tdh- and trh-negative pathotype, the closed genome of V. parahaemolyticus CDC_K4557, another clinical tdh- and trh-negative isolate, was searched for genomic islands using IslandViewer3, and the images shown above were modified from the IslandViewer3 (37) website (http://www.pathogenomics.sfu.ca/islandviewer/accession/NC_021848.1/ [top panel] and http://www.pathogenomics.sfu.ca/islandviewer/accession/NC_021822.1/ [bottom panel]). BLASTn was used to search for each of the islands in the clinical and environmental isolates from Table 1. IslandViewer3 uses multiple algorithms (IslandPick, SIGI-HMM, and IslandPath-DIMOB) to predict the presence of genomic islands. Red, islands identified by an integrative algorithm incorporating multiple mechanisms of island prediction; blue, islands predicted by the IslandPath algorithm, yellow, islands predicted by the SIGI-HMM algorithm; green, islands predicted by the IslandPick algorithm. M, millions; k, thousands.
Type VI secretion systems.
Two T6SSs have previously been found in the pangenome of V. parahaemolyticus. T6SS1 (VP1387 to VP1414) is found on chromosome 1 and is commonly associated with clinical isolates, while T6SS2 (VPA1025 to VPA1046) is found on chromosome 2 and has been found in all tested strains (16, 20). In the current study, six clinical isolates (10-4238, 08-0278, 09-5357, 07-2965, 09-3217, and 10-4255) and eight environmental isolates (ISF-54-12, S171, VPCR-2009, VPTS-2009, SG176, 22720, J-C2-34, and VH3) did not have a T6SS1. Of the isolates that had a T6SS1, variance in this gene cluster was observed between isolates, although variance occurred only in the VP1388, VP1389, and VP1390 genes (Fig. 7). There were five different alleles of these genes, and a phylogenetic tree is shown to demonstrate which isolates contained which alleles (Fig. 7). The 10-4239 isolate had unique alleles for each of these genes which were not observed in any of the other isolates. The N terminus of VP1388 was conserved in all isolates that had a T6SS1, while variance was observed in the C terminus. There is biological significance underlying the variation observed in VP1388 to VP1390. VP1388 has previously been identified to be an antibacterial effector, and VP1389 is its associated immunity protein (21). Changes in the effector must be accompanied by changes in the immunity protein to maintain self-protection. The putative functionality of the third larger gene downstream (VP1390) is still unknown, but on the basis of its association with VP1388 and VP1390, it may have a role in antimicrobial activity. The finding of a novel putative effector in a V. parahaemolyticus clinical tdh- and trh-negative isolate indicates that this protein should be further investigated for roles during infection.
FIG 7.

T6SS1. The gene cluster homologous to T6SS1 is present in 32 of our clinical isolates; however, variance in the VP1388 to VP1390 genes was observed between isolates.
Conclusion.
The ability of tdh- and trh-negative strains to cause clinical illness is still controversial, and several theories have been proposed to explain why tdh- and trh-negative strains are sometimes isolated from clinical cases, including coinfection with pathogenic V. parahaemolyticus strains, the loss of virulence genes during infection, the presence of novel and uncharacterized virulence factors, or the fact that they play a role in a multistrain infection. However, in this investigation we have identified 862 genes that are present in clinical tdh- and trh-negative isolates but that are not present in environmental isolates. Several of these genes are highly homologous to genes from other enteric bacteria, indicating that horizontal gene transfer may play an important role in the ability of tdh- and trh-negative isolates to survive in the human gastrointestinal tract. In addition, tdh- and trh-negative isolate 10-4239 contains a unique T6SS1 effector/immunity gene combination that should be investigated further.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to Robyn Kenwell for technical assistance with the sequencing of the genomes analyzed in this paper. We also thank John Austin and Franco Pagotto of the BMH Research Division of Health Canada for reviewing the manuscript and offering helpful comments.
This work was funded (A-base) by Health Canada in support of Canada's Food Safety Programs. J.R. is supported by the Visiting Fellow in a Government Laboratory program.
J.R., N.P., C.C.L., and S.K.B. conceived of and designed the experiments. J.R. and C.C.L. performed all wet laboratory experimental, molecular, and next-generation sequencing work. J.R. and N.P. conducted the in silico analysis, including the use of novel tools contributed by A.W.P.; J.R. wrote the paper with input from all other authors.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03465-15.
REFERENCES
- 1.Ceccarelli D, Hasan NA, Huq A, Colwell RR. 2013. Distribution and dynamics of epidemic and pandemic Vibrio parahaemolyticus virulence factors. Front Cell Infect Microbiol 3:97. doi: 10.3389/fcimb.2013.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joseph SW, Colwell RR, Kaper JB. 1982. Vibrio parahaemolyticus and related halophilic vibrios. Crit Rev Microbiol 10:77–124. doi: 10.3109/10408418209113506. [DOI] [PubMed] [Google Scholar]
- 3.Nishibuchi M, Fasano A, Russell RG, Kaper JB. 1992. Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect Immun 60:3539–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Honda T, Ni Y, Miwatani T, Adachi T, Kim J. 1992. The thermostable direct hemolysin of Vibrio parahaemolyticus is a pore-forming toxin. Can J Microbiol 38:1175–1180. doi: 10.1139/m92-192. [DOI] [PubMed] [Google Scholar]
- 5.Broberg CA, Calder TJ, Orth K. 2011. Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect 13:992–1001. doi: 10.1016/j.micinf.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hiyoshi H, Kodama T, Iida T, Honda T. 2010. Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect Immun 78:1772–1780. doi: 10.1128/IAI.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuda S, Kodama T, Okada N, Okayama K, Honda T, Iida T. 2010. Association of Vibrio parahaemolyticus thermostable direct hemolysin with lipid rafts is essential for cytotoxicity but not hemolytic activity. Infect Immun 78:603–610. doi: 10.1128/IAI.00946-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Letchumanan V, Chan K-G, Lee L-H. 2014. Vibrio parahaemolyticus: a review on the pathogenesis, prevalence, and advance molecular identification techniques. Front Microbiol 5:705. doi: 10.3389/fmicb.2014.00705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyamoto Y, Kato T, Obara Y, Akiyama S, Takizawa K, Yamai S. 1969. In vitro hemolytic characteristic of Vibrio parahaemolyticus: its close correlation with human pathogenicity. J Bacteriol 100:1147–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. 2003. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 11.Izoré T, Perdu C, Job V, Attree I, Faudry E, Dessen A. 2011. Structural characterization and membrane localization of ExsB from the type III secretion system (T3SS) of Pseudomonas aeruginosa. J Mol Biol 413:236–246. doi: 10.1016/j.jmb.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 12.Okada N, Iida T, Park K-S, Goto N, Yasunaga T, Hiyoshi H, Matsuda S, Kodama T, Honda T. 2009. Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect Immun 77:904–913. doi: 10.1128/IAI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park K-S, Ono T, Rokuda M, Jang M-H, Okada K, Iida T, Honda T. 2004. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun 72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noriea NF III, Johnson CN, Griffitt KJ, Grimes DJ. 2010. Distribution of type III secretion systems in Vibrio parahaemolyticus from the northern Gulf of Mexico. J Appl Microbiol 109:953–962. doi: 10.1111/j.1365-2672.2010.04722.x. [DOI] [PubMed] [Google Scholar]
- 15.Ham H, Orth K. 2012. The role of type III secretion system 2 in Vibrio parahaemolyticus pathogenicity. J Microbiol 50:719–725. doi: 10.1007/s12275-012-2550-2. [DOI] [PubMed] [Google Scholar]
- 16.Salomon D, Gonzalez H, Updegraff BL, Orth K. 2013. Vibrio parahaemolyticus type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PLoS One 8:e61086. doi: 10.1371/journal.pone.0061086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salomon D, Orth K. 2015. Type VI secretion system. Curr Biol 25:R265–R266. doi: 10.1016/j.cub.2015.02.031. [DOI] [PubMed] [Google Scholar]
- 18.Pukatzki S, McAuley SB, Miyata ST. 2009. The type VI secretion system: translocation of effectors and effector-domains. Curr Opin Microbiol 12:11–17. doi: 10.1016/j.mib.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I. 2009. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics 10:104. doi: 10.1186/1471-2164-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu Y, Yang H, Li J, Zhang P, Wu B, Zhu B, Zhang Y, Fang W. 2012. Putative type VI secretion systems of Vibrio parahaemolyticus contribute to adhesion to cultured cell monolayers. Arch Microbiol 194:827–835. doi: 10.1007/s00203-012-0816-z. [DOI] [PubMed] [Google Scholar]
- 21.Salomon D, Kinch LN, Trudgian DC, Guo X, Klimko JA, Grishin NV, Mirzaei H, Orth K. 2014. Marker for type VI secretion system effectors. Proc Natl Acad Sci U S A 111:9271–9276. doi: 10.1073/pnas.1406110111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lüdeke CHM, Kong N, Weimer BC, Fischer M, Jones JL. 2015. Complete genome sequences of a clinical isolate and an environmental isolate of Vibrio parahaemolyticus. Genome Announc 3(2):e00216-15. doi: 10.1128/genomeA.00216-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meador CE, Parsons MM, Bopp CA, Gerner-Smidt P, Painter JA, Vora GJ. 2007. Virulence gene- and pandemic group-specific marker profiling of clinical Vibrio parahaemolyticus isolates. J Clin Microbiol 45:1133–1139. doi: 10.1128/JCM.00042-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhoopong P, Palittapongarnpim P, Pomwised R, Kiatkittipong A, Kamruzzaman M, Nakaguchi Y, Nishibuchi M, Ishibashi M, Vuddhakul V. 2007. Variability of properties of Vibrio parahaemolyticus strains isolated from individual patients. J Clin Microbiol 45:1544–1550. doi: 10.1128/JCM.02371-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones JL, Lüdeke CHM, Bowers JC, Garrett N, Fischer M, Parsons MB, Bopp CA, DePaola A. 2012. Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J Clin Microbiol 50:2343–2352. doi: 10.1128/JCM.00196-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banerjee SK, Kearney AK, Nadon CA, Peterson C-L, Tyler K, Bakouche L, Clark CG, Hoang L, Gilmour MW, Farber JM. 2014. Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J Clin Microbiol 52:1081–1088. doi: 10.1128/JCM.03047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hazen TH, Lafon PC, Garrett NM, Lowe TM, Silberger DJ, Rowe LA, Frace M, Parsons MB, Bopp CA, Rasko DA, Sobecky PA. 2015. Insights into the environmental reservoir of pathogenic Vibrio parahaemolyticus using comparative genomics. Front Microbiol 6:204. doi: 10.3389/fmicb.2015.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 31.Marchesi JR. 2012. Metagenomics: current innovations and future trends. Future Microbiol 7:813–814. doi: 10.2217/fmb.12.41. [DOI] [Google Scholar]
- 32.Chen JC, Sun S, Li W, Wooley JC. 2011. A community cyberinfrastructure for metagenomics: CAMERA 2.0. In Metagenomics: current innovations and future trends. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 33.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 34.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan MJ, Petty NK, Beatson SA. 2011. EasyFig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, Pereira SK, Waglechner N, McArthur AG, Langille MGI, Brinkman FSL. 2015. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res 43(W1):W104–W108. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurley CC, Quirke A, Reen FJ, Boyd EF. 2006. Four genomic islands that mark post-1995 pandemic Vibrio parahaemolyticus isolates. BMC Genomics 7:104. doi: 10.1186/1471-2164-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyd EF, Cohen ALV, Naughton LM, Ussery DW, Binnewies TT, Stine OC, Parent MA. 2008. Molecular analysis of the emergence of pandemic Vibrio parahaemolyticus. BMC Microbiol 8:110. doi: 10.1186/1471-2180-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishibuchi M, Kaper JB. 1995. Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect Immun 63:2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Honda T, Ni Y, Miwatani T. 1988. Purification and characterization of a hemolysin produced by a clinical isolate of Kanagawa phenomenon-negative Vibrio parahaemolyticus and related to the thermostable direct hemolysin. Infect Immun 56:961–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tettelin H, Riley D, Cattuto C, Medini D. 2008. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 11:472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Wong H, Nong W, Cheung MK, Law PTW, Kam KM, Kwan HS. 2014. Comparative genomic analysis of clinical and environmental strains provides insight into the pathogenicity and evolution of Vibrio parahaemolyticus. BMC Genomics 15:1135. doi: 10.1186/1471-2164-15-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banerjee S, Petronella N, Chew Leung C, Farber J. 2015. Draft genome sequences of four Vibrio parahaemolyticus isolates from clinical cases in Canada. Genome Announc 3:e01482-14. doi: 10.1128/genomeA.01482-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Chen S. 2013. Draft genome sequence of Vibrio parahaemolyticus V110, isolated from shrimp in Hong Kong. Genome Announc 1:e00300-13. doi: 10.1128/genomeA.00300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castillo D, Jun JW, D'Alvise P, Middelboe M, Gram L, Liu S, Katharios P. 2015. Draft genome sequence of Vibrio parahaemolyticus VH3, isolated from an aquaculture environment in Greece. Genome Announc 3:e00731-15. doi: 10.1128/genomeA.00731-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ronholm J, Petronella N, Kenwell R, Banerjee S. 2015. Draft whole-genome sequences of 14 Vibrio parahaemolyticus clinical isolates with an ambiguous K serogroup. Genome Announc 3:e00217-15. doi: 10.1128/genomeA.00217-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.