

Abstract
Anti-angiogenic therapy is a promising therapeutic strategy for the highly vascular and malignant brain tumor, glioblastoma (GBM), although current clinical trials have failed to demonstrate an extension in overall survival. The small molecule tyrosine kinase inhibitor axitinib that targets vascular endothelial growth factor receptor, potently inhibits angiogenesis and has single-agent clinical activity in non-small cell lung, thyroid, and advanced renal cell cancer. Here we show that axitinib exerts direct cytotoxic activity against a number of patient-derived GBM stem cell (GSCs) and an endothelial cell line, and inhibits endothelial tube formation in vitro. Axitinib treatment of mice bearing hypervascular intracranial tumors generated from human U87 glioma cells, MGG4 GSCs and mouse 005 GSCs significantly extended survival that was associated with decreases in tumor-associated vascularity. We thus show for the first time the anti-angiogenic effect and survival prolongation provided by systemic single agent treatment with axitinib in preclinical orthotopic GBM models including clinically relevant GSC models. These results support further investigation of axitinib as an anti-angiogenic agent for GBM.
Keywords: Glioblastoma, Axitinib, Anti-angiogenic therapy, Glioblastoma stem cells, Molecular targeted therapy
Introduction
Glioblastoma (GBM) is a malignant brain tumor with a median survival time of 15 months with current multimodality treatment [1]. Despite increased molecular understanding of the tumor and extensive testing of novel investigational therapies, there has been no drastic improvement in the outcome for patients with this brain tumor [2, 3].
Angiogenesis plays a key role in the progression of glioma [4], and is mediated by signaling through a number of receptor tyrosine kinases including vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), epidermal growth factor receptor (EGFR), and Tie-2 receptor, as well as other cell surface receptors such as integrins [5, 6]. Anti-angiogenic therapy is considered a promising treatment option for GBM. Bevacizumab (Avastin®), a humanized monoclonal antibody against vascular endothelial growth factor (VEGF), was approved by the US Food and Drug Administration (FDA) in May 2009 for recurrent GBM [7]. In addition, several targeted drugs such as tyrosinekinase inhibitors (TKIs) have been tested in clinical trials either alone or in combination with other anti-cancer therapies [8, 9]. Clinical studies investigating agents that primarily target the VEGFR pathway have shown some clinical activity for GBM such as modest increases in progression-free survival (PFS) and lowering of corticosteroid use [10–14]. Unfortunately, recent phase III clinical trials with bevacizumab in newly diagnosed GBM did not show significant improvement in overall survival [15, 16]. While anti-angiogenic therapy remains promising for GBM, it is likely that other anti-angiogenic agents and combinations thereof will be necessary for efficacy improvement.
Axitinib (AG-013736) is an orally active small molecule TKI that selectively inhibits VEGFR-1, -2 and -3 at subnanomolar concentrations, amongst the lowest for any TKI, with additional inhibitory activity against PDGFRβ and c-KIT (CD117) [17–19]. Axitinib inhibits angiogenesis and vascular permeability, which further affects many interactions involved in tumor cell proliferation, tumor progression, and metastasis. In preclinical human xenograft models of breast, colon and lung cancer, melanoma, and neuroblastoma, axitinib exhibited anti-tumor activity that was associated with inhibition of angiogenesis and blood flow [18, 20]. In phase II studies, axitinib showed single-agent activity in a variety of tumor types, including thyroid cancer [21], advanced renal cell carcinoma [22] and non-small cell lung cancer [23]. In a Phase III randomized clinical trial for metastatic renal cell carcinoma, axitinib treatment resulted in longer PFS than sorafenib [24], and axitinib was approved for therapy-refractory renal cell carcinoma in 2012. However, the efficacy of axitinib for GBM has not yet been explored.
In the past decade cancer stem-like cells have been identified in various solid tumors, including GBM [25, 26]. GBM cancer stem cells (GSCs) are able to self-renew like neural stem cells, efficiently generate orthotopic tumors upon implantation into immunodeficient mice, and can differentiate to cells representative of the bulk of cancer cells. This suggests that GSCs may be responsible for GBM recurrence following conventional therapy, and that successful targeting of GSCs is necessary to cure GBM [27]. We and others previously reported that neurosphere cultures isolated from human GBM specimens enriches for GSCs [28, 29]. Orthotopic tumors derived from GSCs exhibit the phenotypic hallmarks of GBM such as hypervascularity and invasiveness, and maintain genetic aberrations such as EGFR amplification [30]. Thus, GSCs-based xenografts offer a clinically relevant disease model, superior to conventional cell lines, that is ideal for evaluating novel therapeutics for GBM and GSCs [31, 32]. On the other hand, genetically engineered mouse GSCs provide a brain tumor model in syngeneic mice with an intact immune system [33, 34].
In this study, we first used a number of GSCs and an endothelial cell line to test the effects of axitinib in vitro. We then investigated single agent efficacy of axitinib in three vascular GBM models (human U87 glioma cells and MGG4 GSCs, and mouse 005 GSCs) in vivo.
Materials and methods
Cell lines and reagents
Human U87 glioma cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and grown in complete Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal calf serum at 37 °C and 5 % CO2. Human GSCs MGG4, MGG8, MGG18, BT74 were isolated as previously described [28,30], and maintained as spheres in serum-free medium containing 20 ng/mL recombinant human EGF (R&D systems) and 20 ng/mL recombinant human FGF2 (Peprotech). GSCs were passaged by dissociating neurospheres using the Neuro-Cult Chemical Dissociation Kit (StemCell Technologies). Mouse 005 GSCs were provided by Dr. I. Verma (Salk Institute for Biological Studies, La Jolla, CA) [33, 34]. Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza. Human brain microvascular endothelial cells (HBMECs) were obtained from Dr. Ken Arai (MGH). Axitinib (Pfizer Inc) was provided by Pfizer and dissolved in DMSO as a 25 mM stock solution for in vitro studies. The final concentrations added to cells had less than 0.5 % DMSO, which was nontoxic to cells.
Cell viability/cytotoxicity assays
Cells were dissociated (GSCs) or trypsinized (HUVECs) and seeded into 96-well plates (5,500 GSCs, or 300 HUVECs/well). The next day, cells were treated with axitinib at varying doses. Five days after incubation, MTS assays (Promega) were performed following manufacturer’s instruction. Experiments were done in triplicate and repeated at least two times. Dose–response curves and IC50 values were calculated using Prism (GraphPad Software).
Endothelial tube formation assay
HUVECs or HBMECs were seeded at 4 × 104 cells/well on matrigel (Matrigel Matrix, BD Biosciences)-precoated 24-well culture plates and grown in EGM-2 medium (Lonza) with or without axitinib. Twelve (HUVECs) or 32 (HBMECs) hours later, microscopic pictures were captured and tube formation was assessed by counting branching points per field. Three to five fields per well were randomly chosen and each condition was tested in triplicate.
Secondary sphere formation assay
Single cell suspensions of dissociated GSCs were seeded into 96-well plates at 1, 3 or 10 cells/well, and exposed to either control or axitinib at the indicated concentrations. Sixteen days later, the number of wells containing tumor spheres (diameter >60 μm) was recorded.
Flow cytometric analysis
To detect apoptosis induction, GSCs were control or axitinib treated for 48 h and stained with Annexin V and propidium iodide using Annexin V apoptosis detection kit (eBioscience). Analysis was performed with an Accuri flow cytometer (BD Biosciences), and data were analyzed by FlowJo software (Tree Star).
Animal studies
Female athymic nu/nu and C57BL/6 mice aged 6–8 weeks were obtained from NCI Frederick (Frederick, MD). For intracranial tumor establishment, mice were injected stereotactically (2 mm lateral to the bregma at a depth of 3 mm) with 1 × 105 U87 (13 mice), 1 × 105 MGG4 cells (22 mice) or 2 × 104 005 cells (14 mice) in 2 μl DMEM. On day 10 (for U87), day 35 (for MGG4), or day 12 (for 005), mice were randomly divided into two groups, and axitinib (25 mg/kg, dissolved in polyethylene glycol 400 and acidified water) or vehicle solution (polyethylene glycol 400 and acidified water) was injected intraperitoneally daily for 4 weeks (for U87 model), 7 weeks (for MGG4 model, using a 5 days on and 2 days off cycle), or 23 days (for 005 model, 5 days on and 2 days off). Overall health status and body weight were recorded every 2–3 days, and animals were monitored for signs of discomfort or neurologic symptoms. Animals were sacrificed when they showed significant neurologic deficits or lost 15 % of body weight. All in vivo procedures were approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital.
Immunohistochemistry
Rodent brains were removed, frozen in OCT and 7 μm sections were obtained by cryostat. Tissue slides were dried and fixed in ice-cold acetone at −20 °C for immunohistochemistry and standard hematoxylin and eosin (H and E) staining. After blocking with serum, sections were incubated with rat anti-CD34 (MEC14.7, Abcam) or mouse anti-Ki67 (MIB-1, Dako) antibodies overnight at 4 °C. Immpress anti-rat Ig or Vectastain Elite kit (both from Vector) were used for CD34 and Ki67 staining, respectively, and color was developed with DAB (Dako). Sections were counterstained with hematoxylin, dehydrated, and mounted. Three slides (anterior, mid, and posterior regions of tumor) from each brain (n = 3/group) were selected and three microscopic tumor areas per slide were subjected to the measurement of CD34-immunopositive areas using NIH Image-J software. To quantify MIB-1 positivity, at least 500 cells were counted from randomly chosen fields per tumor (n = 3/group).
Statistical analysis
Tube formation assays were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. Microvascular densities and Ki67 labeling indices were compared using the unpaired student t test (two-tailed). Sphere forming efficiency was analyzed with Fisher’s exact test (two-tailed). Survival was analyzed by Kaplan–Meier curves, and comparisons were determined by Log-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test. All statistical analysis was conducted using Prism, and p values less than 0.05 were considered significant.
Results
Axitinib cytotoxicity of GBM cells, GSCs and endothelial cells in vitro
We first determined the sensitivity of GBM cells to axitinib (Fig. 1a). Three of four human GSC lines tested were sensitive to axitinib; IC50s for MGG4 and MGG18 were 2.1 and 6.4 μM, respectively, while MGG8, which has PDGFRA amplification, was highly sensitive with an IC50 of only 0.06 μM (Fig. 1a). BT74 GSCs were resistant to axitinib up to 300 μM. Established U87 glioma cell line was also mostly resistant to axitinib, with about a 25 % reduction in cell viability at 10 μM. As expected, HUVECs were very sensitive to axitinib with its IC50 about 0.3 μM (Fig. 1a). Furthermore, in vitro tube formation by HUVEC and HBMEC was inhibited by 65–70 % at 0.03 μM of axitinib (Fig. 1b, c, p < 0.0001).
Fig. 1.

Cytotoxic effect of axitinib in vitro. a Cell viability was measured after 5-day exposure to different concentrations of axitinib using MTS assay. Cell viability relative to untreated cells (± SD) is plotted for GSCs MGG4, MGG8, MGG18, and BT74, as well as U87 and HUVECs. b, c Endothelial tube formation assay. HUVECs (b) and HBMECs (c) were treated with indicated concentrations of axitinib or control, and tube formation was assessed by branching point counts. Representative microscopic pictures are shown. Scale bars, 100 μm. In b and c, p < 0.0001 by One-way ANOVA analysis. Lines over bars in the graph indicate which pairs of samples are statistically compared. *p < 0.05 comparing 0 versus 0.03, and 0 versus 0.3 μM (HUVECs) and 0 versus 0.003, 0 versus 0.03, 0 versus 0.3, 0.003 versus 0.03, and 0.003 versus 0.3 μM (HBMECs)
Axitinib inhibits sphere formation by GSCs and induces their apoptosis
We next wanted to determine whether axitinib treatment affects the stem-like properties of GSCs. As a surrogate of self-renewal, clonogenicity was assessed. Axitinib at the IC20 doses impaired secondary sphere formation by MGG4 and MGG8 seeded at clonogenic densities (Fig. 2a, b), indicating an inhibition of GSC self-renewal. At higher concentrations, axitinib induced apoptosis in MGG4 as shown by increases in fractions of cells positive for Annexin V and propidium iodide after 48-hour drug exposure (Fig. 2c).
Fig. 2.

Effect of axitinib on GSC clonogenicity and apoptosis in vitro. a, b Secondary sphere formation assay with MGG4 (a) and MGG8 (b) GSCs. Axitinib dose was 1 μM (a) and 0.01 μM (b). Fraction of wells containing sphere(s) is shown for different cell densities (1, 3 and 10 cells per well). c MGG4 GSCs were treated with axitinib (3 μM) or control for 48 hours, stained for Annexin V and propidium iodide, and analyzed by flow cytometry. Numbers in each quadrant denote the percentage of each fraction
Anti-tumor and anti-angiogenic effects of axitinib in intracranial U87 glioma
The anti-tumor activity of axitinib for malignancies located in the CNS has not been investigated. We therefore tested whether daily systemic administration of axitinib was efficacious for established U87 intracranial tumors in mice. Since U87 is resistant to axitinib (Fig. 1a), the anti-tumor effects of axitinib in vivo are attributable to its impact on the tumor microenvironment. The median survival for the axitinib group was 34.5 days compared to 30 days for the control group (Fig. 3a, p = 0.002 with log-rank test, p = 0.004 with Gehan-Breslow-Wilcoxon test). Histopathology of tumor sections revealed that the axitinib-treated tumors had a 25 % decrease in tumor cellularity compared with the vehicle-treated tumors (Fig. 3b, p = 0.002). In line with this, axitinib significantly lowered the Ki67 labeling index of the treated tumors, revealing the ability of axitinib to suppress proliferation of neoplastic cells in the brain (Fig. 3c, p = 0.0003).
Fig. 3.

Axitinib treatment of U87 intracerebral tumors. a Kaplan–Meier survival analysis of mice bearing intracerebral U87 tumors after treatment with control (n = 7) or axitinib (n = 6). Red bracket indicates time of 4 weeks treatment. Axitinib treatment significantly extended survival over control treatment (p = 0.002, Log-rank (Mantel-Cox) test). b H and E staining of U87 xenografts treated with control (left) and axitinib (right). Original magnification, ×200. Scale bars, 50 μm. Quantification of cellular densities in control and axitinib groups is shown at right. c, d Immunohistochemistry for Ki67 (c, brown) and CD34 (d, brown) in U87 xenografts treated with control (left) or axitinib (middle). Original magnification ×200. Quantification of Ki67 positivity and CD34 + microvascular densities is shown at right as means with S.D. (for Ki67) or box plot (min–max; Spear) of the fraction of CD34 + area. p = 0.0003 (c) and 0.0002 (d), unpaired student t test
To analyze the effects of axitinib on tumor-associated vasculature, we performed immunohistochemisty for CD34, a commonly used endothelial marker. In vehicle-treated tumors, there was striking hypervascularity characterized by irregular-shaped and often tortuous or dilated blood vessels, a hallmark of GBM-associated aberrant angiogenesis (Fig. 3d). In contrast, axitinib-treated U87 tumors showed nearly complete disappearance of such features and displayed generally thin-walled, undilated vasculature that resembles the vasculature seen in the normal brain (Fig. 3d). Quantitative analysis of CD34 microvascular densities confirmed the anti-angiogenic effects as axitinib greatly decreased (>90 %, p = 0.0002) the microvascular density compared to vehicle treatment (Fig. 3d). Thus systemic axitinib treatment had anti-proliferative and potent anti-angiogenic effects in the orthotopic hypervascular U87 glioma model, which was associated with extended survival of the animals.
Axitinib efficacy in an orthotopic angiogenic human GSC-derived GBM model
Since the U87 glioma cell line model does not recapitulate the molecular pathology found in human GBM, we next used a more representative model generated from GSCs isolated from primary GBM specimens. We chose MGG4 because this GSC line generates hypervascular and hypoxic tumors [28, 30, 35], and thus provides a useful model to investigate anti-angiogenic strategies [36]. Athymic mice bearing intracranial MGG4 tumors were treated with either axitinib or vehicle solution, and animals followed for survival. Kaplan–Meier survival analysis showed that axitinib significantly extended survival (median survival: 80 days) compared to the control group (median survival: 75 days, p = 0.04 with log-rank test, p = 0.04 with Gehan-Breslow-Wilcoxon test) (Fig. 4a). Histopathologically, vehicle-treated MGG4 tumors exhibited frequent dilated and tortuous vasculature that was immunopositive for CD34, while axitinib-treated tumors had reduced vascular abnormalities (Fig. 4b). There was no difference in the weights of the vehicle- and axitinib-treated mice, indicating that axitinib at the administered dose was not toxic (Supplementary Fig. S1).
Fig. 4.
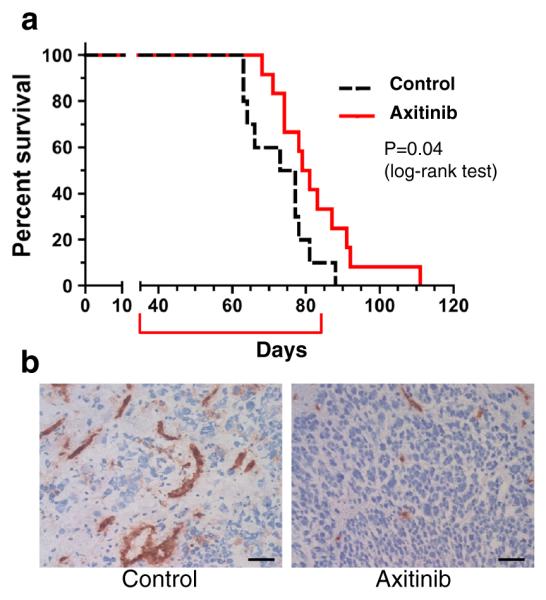
Axitinib treatment of MGG4 GSC-derived intracerebral tumors. a Survival of mice with established MGG4 intracranial tumors treated with control (n = 10) or axitinib (n = 12). Axitinib significantly increased survival (p = 0.04, Log-rank (Mantel-Cox) test). Red bracket indicates time of 7 weeks treatment. b Immunohistochemistry for CD34 (brown) in MGG4 xenografts treated with control (left) or axitinib (right). Original magnification, ×200. Scale bars, 50 μm
Axitinib efficacy in a mouse GSC-derived orthotopic GBM model
Finally, given reported roles for VEGFR signaling in regulating innate immune cell recruitment [37] and adaptive cellular immunity [38–40], we sought to determine axitinib efficacy in a GBM model with an intact immune system. To this end, we employed the mouse 005 GSC model that exhibits the pathological hallmarks of GBM, hypervascularity and invasiveness [34]. In this aggressive orthotopic GBM model, treatment with axitinib modestly but significantly prolonged the survival of tumor-bearing mice (Fig. 5a, median survival: 43 versus 38.5 days, p = 0.0001 with log-rank test, p = 0002 with Gehan-Breslow-Wilcoxon test), recapitulating the results we observed in the two human GBM xenografts. CD34 immunohistochemistry showed that axitinib efficacy was associated with decreased vascularity in axitinib-treated tumors compared to the control (Fig. 5b).
Fig. 5.
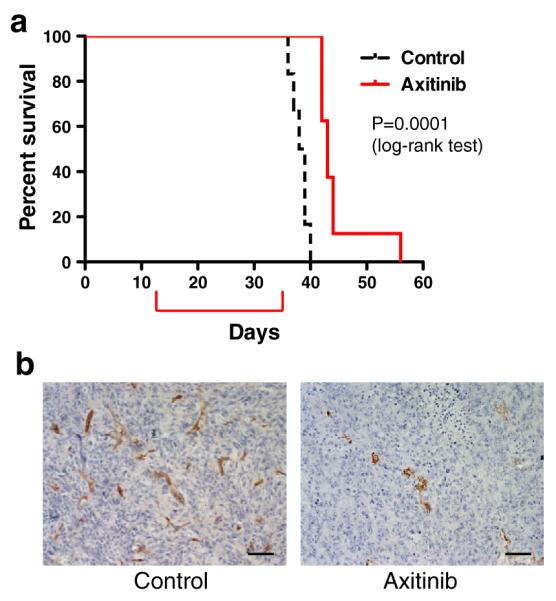
Axitinib treatment of 005 GSC-derived intracerebral tumors. a Survival of mice with syngeneic 005 intracranial tumors treated with control (n = 6) or axitinib (n = 8). Axitinib significantly increased survival (p = 0.0001, Log-rank (Mantel-Cox) test). Red bracket indicates time of 23 days treatment. b Immunohistochemistry for CD34 (brown) in intracerebral 005 tumors treated with control (left) or axitinib (right). Original magnification, ×200. Scale bars, 50 μm
Discussion
In this study, we show for the first time that the VEGFR TKI axitinib exhibits anti-angiogenic activity and prolongs survival of mice bearing orthotopic GBMs. The therapeutic activity of axitinib was demonstrated in three hypervascular intracerebral GBM models: conventional U87 glioma, MGG4 GSCs and mouse 005 GSCs.
Our in vitro investigation indicated that axitinib not only suppresses the viability of endothelial cells but also GSCs isolated from three of four patients, with sensitivities ranging from IC50s of 0.06–6 μM. Axitinib was previously shown to inhibit the growth of 9L rat gliosarcoma cells in vitro [41]. Although traditionally VEGFR2 was regarded as an endothelial specific protein, recent research described an autocrine VEGF–VEGFR2–Neuropilin-1 signaling pathway that promotes GSC viability and tumor growth [42]. Thus, the observed cytotoxic effect mediated by axitinib, including sphere forming suppression and apoptosis induction, may be through its inhibition of VEGFRs. Notably, MGG8 GSC was the most sensitive to axitinib, which may have been due to inhibition of PDGFR since MGG8 has PDGFRA gene amplification [30]. In contrast, U87 was relatively resistant to axitinib. It will be of interest to study whether undifferentiated GSCs are more sensitive to the direct impact of axitinib compared to more differentiated GBM cells.
In subcutaneous tumor models, axitinib treatment inhibited growth of rat 9L [41], and human U87 [18] and U251 gliomas [37], although tumor growth resumed after cessation of axitinib treatment [37]. In order to evaluate the anti-angiogenic and anti-tumor effects of axitinib on tumors located in the brain, we selected three orthotopic GBM models that are highly vascular. In the commonly used U87 model, daily axitinib treatment prominently reduced tumor-associated microvascular density and prolonged survival. The MGG4 GSC-derived tumor model displays frequent abnormally dilated and tortious blood vessels and intratumoral hemorrhages, a vascular hallmark of human GBM [28, 30, 36]. CD34 endothelium staining revealed axitinib-mediated anti-angiogenic effects in the MGG4 and 005 models as well. While survival was significantly extended in all three orthotopic GBM models by axitinib treatment, the effects were modest. This suggests that despite robust inhibition of angiogenesis in these vascular tumors, tumor growth was not dependent upon neovascularization. In the MGG4 model, where MGG4 GSCs are sensitive to axitinib in vitro, both direct impacts on tumor cells and inhibition on tumor vasculature may contribute to the survival benefit.
Cediranib and axitinib share a very similar tyrosine kinase inhibition profile, blocking VEGFR1-3, PDGFRβ, and c-Kit [18, 43]. Preclinically, cediranib extended survival of mice bearing intracerebral GBMs, including U87, without altering the growth kinetics of tumors [44]. A randomized phase 3 clinical trial testing cediranib with and without lomustine for recurrent GBM showed that the use of cediranib delayed the time to neurological deterioration and had corticosteroid sparing effect, however it failed to demonstrate an effect on PFS [13]. In a phase 2 trial of cediranib for newly diagnosed GBM patients, cediranib induced an increase of tumor perfusion in a subset of patients and these responders had an overall survival benefit [14]. This improved blood perfusion and alleviated GBM-associated edema indicates some clinical activity of cediranib for at least a subset of GBM patients. In this study, we observed a notable disappearance of dilated CD34 + vessels in the U87 and MGG4 models, which is consistent with the induction of vascular normalization. Analysis of molecular interactions of several TKIs and VEGFR2 identified a positive correlation between in vitro ligand efficiency (LipE) and PFS in renal cell carcinoma trials [45]. Axitinib is superior to cediranib in LipE, kinase Ki, as well as HUVEC IC50 for both VEGFR2 phosphorylation and survival [45], suggesting that axitinib may perform better in the clinic than cediranib, as was the case for axitinib and sorafenib [24]. These targeting properties, ability to penetrate the brain, and possible inhibitory effects on GBM cells could confer axitinib with an advantage over anti-angiogenic antibodies, such as the VEGF antibody bevacizumab, which was recently shown to provide no overall survival benefit for newly diagnosed GBMs [15, 16].
Although axitinib extended survival, long-term disease control or cures were not obtained in any model tested, despite large decreases in neovascularization. This suggests that clinical use of axitinib as monotherapy for GBM would not be efficacious in prolonging overall survival. Combinatory approaches with other therapeutic modalities may be able to overcome limitations with anti-angiogenic strategies and enhance efficacy. However, selection of combinatorial partners with VEGFR TKI needs caution as subcutaneous glioma tumor regression induced by metronomic cyclophosphamide chemotherapy was inhibited by axitinib due to inhibition of innate anti-tumor immune cell recruitment [37]. We previously showed that bevacizumab-mediated reduced vascular permeability enhances distribution of intratumorally injected therapeutic oncolytic herpes simplex virus (oHSV) [46]. When systemic bevacizumab was combined with oHSV expressing anti-angiogenic angiostatin (G47Δ-mAngio), there was marked increase in survival of mice bearing intracerebral U87 GBM [46]. It will be of interest to investigate whether axitinib and oHSV have similar synergistic effects in GSC-derived orthotopic GBM models.
In summary, we show for the first time that systemic daily treatment with axitinib markedly inhibited GBM-associated angiogenesis and modestly extended survival in three preclinical orthotopic GBM models. These results support further investigation of axitinib as an anti-angiogenic agent for GBM.
Supplementary Material
Acknowledgments
We thank Pfizer for providing axitinib, Drs. Giulia Fulci, Brent J. Passer, and David J. Waxman for technical advice, and Melissa R. Marinelli, Andrea Murphy, Tooba Cheema, and Shinichi Esaki for their experimental help. This work was supported by a grant from the National Institute of Health (Grant number R01NS032677).
Footnotes
Conflict of interest The authors declare no conflict of interest.
Electronic supplementary material The online version of this article (doi:10.1007/s11060-014-1612-1) contains supplementary material, which is available to authorized users.
Contributor Information
Lei Lu, Department of Neurosurgery, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA; Department of Neurosurgery, Huashan Hospital, Fudan University, Shanghai 200040, China.
Dipongkor Saha, Department of Neurosurgery, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Robert L. Martuza, Department of Neurosurgery, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA
Samuel D. Rabkin, Department of Neurosurgery, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA
Hiroaki Wakimoto, Department of Neurosurgery, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5 year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013;331:139–146. doi: 10.1016/j.canlet.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 4.Das S, Marsden PA. Angiogenesis in glioblastoma. N Engl J Med. 2013;369:1561–1563. doi: 10.1056/NEJMcibr1309402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuettenberg J, Friedel C, Vajkoczy P. Angiogenesis in malignant glioma––a target for antitumor therapy? Crit Rev Oncol Hematol. 2006;59:181–193. doi: 10.1016/j.critrevonc.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Wong ML, Prawira A, Kaye AH, Hovens CM. Tumour angiogenesis: its mechanism and therapeutic implications in malignant gliomas. J Clin Neurosci. 2009;16:1119–1130. doi: 10.1016/j.jocn.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 8.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerstner ER, Batchelor TT. Antiangiogenic therapy for glioblastoma. Cancer J. 2012;18:45–50. doi: 10.1097/PPO.0b013e3182431c6f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 11.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 12.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–3218. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, Ancukiewicz M, Polaskova P, Pinho MC, Jennings D, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci USA. 2013;110:19059–19064. doi: 10.1073/pnas.1318022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 17.Kelly RJ, Rixe O. Axitinib (AG-013736) Recent Results Cancer Res. 2010;184:33–44. doi: 10.1007/978-3-642-01222-8_3. [DOI] [PubMed] [Google Scholar]
- 18.Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14:7272–7283. doi: 10.1158/1078-0432.CCR-08-0652. [DOI] [PubMed] [Google Scholar]
- 19.Solowiej J, Bergqvist S, McTigue MA, Marrone T, Quenzer T, Cobbs M, Ryan K, Kania RS, Diehl W, Murray BW. Characterizing the effects of the juxtamembrane domain on vascular endothelial growth factor receptor-2 enzymatic activity, autophosphorylation, and inhibition by axitinib. Biochemistry. 2009;48:7019–7031. doi: 10.1021/bi900522y. [DOI] [PubMed] [Google Scholar]
- 20.Rossler J, Monnet Y, Farace F, Opolon P, Daudigeos-Dubus E, Bourredjem A, Vassal G, Geoerger B. The selective VEGFR1-3 inhibitor axitinib (AG-013736) shows antitumor activity in human neuroblastoma xenografts. Int J Cancer. 2011;128:2748–2758. doi: 10.1002/ijc.25611. [DOI] [PubMed] [Google Scholar]
- 21.Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, Kane MA, Sherman E, Kim S, Bycott P, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26:4708–4713. doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, Motzer RJ, Bycott P, Liau KF, Freddo J, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8:975–984. doi: 10.1016/S1470-2045(07)70285-1. [DOI] [PubMed] [Google Scholar]
- 23.Schiller JH, Larson T, Ou SH, Limentani S, Sandler A, Vokes E, Kim S, Liau K, Bycott P, Olszanski AJ, et al. Efficacy and safety of axitinib in patients with advanced non-small-cell lung cancer: results from a phase II study. J Clin Oncol. 2009;27:3836–3841. doi: 10.1200/JCO.2008.20.8355. [DOI] [PubMed] [Google Scholar]
- 24.Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, Oudard S, Gore ME, Tarazi J, Hariharan S, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552–562. doi: 10.1016/S1470-2045(13)70093-7. [DOI] [PubMed] [Google Scholar]
- 25.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 26.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 27.Nduom EK, Hadjipanayis CG, Van Meir EG. Glioblastoma cancer stem-like cells: implications for pathogenesis and treatment. Cancer J. 2012;18:100–106. doi: 10.1097/PPO.0b013e3182452e0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakimoto H, Kesari S, Farrell CJ, Curry WT, Jr, Zaupa C, Aghi M, Kuroda T, Stemmer-Rachamimov A, Shah K, Liu TC, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, Modrusan Z, Meissner H, Westphal M, Lamszus K. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2008;27:2897–2909. doi: 10.1038/sj.onc.1210949. [DOI] [PubMed] [Google Scholar]
- 30.Wakimoto H, Mohapatra G, Kanai R, Curry WT, Jr, Yip S, Nitta M, Patel AP, Barnard ZR, Stemmer-Rachamimov AO, Louis DN, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14:132–144. doi: 10.1093/neuonc/nor195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 32.Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, Rosenblum M, Mikkelsen T, Zenklusen JC, Fine HA. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res. 2008;6:21–30. doi: 10.1158/1541-7786.MCR-07-0280. [DOI] [PubMed] [Google Scholar]
- 33.Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, Martuza RL, Rabkin SD. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci USA. 2013;110:12006–12011. doi: 10.1073/pnas.1307935110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sgubin D, Wakimoto H, Kanai R, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus counteracts the hypoxia-induced modulation of glioblastoma stem-like cells. Stem Cells Transl Med. 2012;1:322–332. doi: 10.5966/sctm.2011-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Fulci G, Wakimoto H, Cheema TA, Buhrman JS, Jeyaretna DS, Stemmer-Rachamimov AO, Rabkin SD, Martuza RL. Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia. 2013;15:591–599. doi: 10.1593/neo.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doloff JC, Waxman DJ. VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer Res. 2012;72:1103–1115. doi: 10.1158/0008-5472.CAN-11-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, Hurwitz HI, Dev I, Nixon AB, Lyerly HK, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother. 2008;57:1115–1124. doi: 10.1007/s00262-007-0441-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. 2001;23:263–272. doi: 10.1385/IR:23:2-3:263. [DOI] [PubMed] [Google Scholar]
- 40.Mimura K, Kono K, Takahashi A, Kawaguchi Y, Fujii H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother. 2007;56:761–770. doi: 10.1007/s00262-006-0234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma J, Waxman DJ. Modulation of the antitumor activity of metronomic cyclophosphamide by the angiogenesis inhibitor axitinib. Mol Cancer Ther. 2008;7:79–89. doi: 10.1158/1535-7163.MCT-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, Moudry P, Bartek J, Jr, Fischer W, Lukas J, et al. Autocrine VEGF–VEGFR2–Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209:507–520. doi: 10.1084/jem.20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M, Curwen JO, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 44.Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA, Batchelor TT, di Tomaso E, Duda DG, Munn LL, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009;27:2542–2552. doi: 10.1200/JCO.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci USA. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Fulci G, Buhrman JS, Stemmer-Rachamimov AO, Chen JW, Wojtkiewicz GR, Weissleder R, Rabkin SD, Martuza RL. Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther. 2012;20:37–45. doi: 10.1038/mt.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.