


Abstract
Using a Systematic Evolution of Ligands by Exponential Enrichment (SELEX) protocol capable of selecting xeno-nucleic acid (XNA) aptamers, a 2′-deoxy-2′-fluoroarabinonucleotide (FANA) aptamer (referred to as FA1) to HIV-1 reverse transcriptase (HIV-1 RT) was selected. FA1 bound HIV-1 RT with KD,app values in the low pM range under different ionic conditions. Comparisons to published HIV-1 RT RNA and DNA aptamers indicated that FA1 bound at least as well as these aptamers. FA1 contained a 20 nucleotide 5′ DNA sequence followed by a 57 nucleotide region of FANA nucleotides. Removal of the fourteen 5′ DNA nucleotides did not affect binding. FA1's predicted structure was composed of four stems and four loops. All stem nucleotides could be modified to G-C base pairs (14 total changes) with a small effect on binding. Eliminating or altering most loop sequences reduced or abolished tight binding. Overall, results suggested that the structure and the sequence of FA1 were important for binding. FA1 showed strong inhibition of HIV-1 RT in extension assays while no specific binding to avian myeloblastosis or Moloney murine leukemia RTs was detected. A complete DNA version of FA1 showed low binding to HIV-1 RT, emphasizing the unique properties of FANA in HIV-1 RT binding.
INTRODUCTION
Human immunodeficiency virus (HIV) reverse transcriptase, the replicative polymerase for HIV, remains a major target for drug therapy. HIV-1 RT is a dimer composed of 66 (p66) and 51 (p51) kilodalton (kDa) subunits with the latter being derived from the former by cleavage of C-terminal amino acids. The enzyme possesses DNA polymerase activity that can be directed by either an RNA or DNA template and RNase H activity that cleaves RNA that is part of an RNA-DNA hybrid (1). Traditional small-molecule inhibitors to HIV RT, including nucleoside (NRTIs) and non-nucleoside (NNRTIs) inhibitors, have revolutionized our ability to treat HIV and Acquired Immune Deficiency Syndrome (AIDS). However, drug resistance and toxicity remain major concerns moving forward and highlight the need for alternative approaches.
Among the potential alternatives are aptamers, short nucleic acid molecules that bind with high affinity to targets (typically but not always proteins). Aptamers have many potential uses in diagnostic and therapeutic applications. These include, among others, replacement of antibodies in biochemical assays (e.g. ELISA), utilization as biosensors, as tools for studying the molecular biology of virus replication, and development of antiviral drugs (2–10). In addition to strongly inhibiting enzymatic activity in vitro (11–13), aptamers have shown potent antiviral activity and low toxicity in cell culture, and efficacy in animal models (6,14–25). Inhibitory aptamers to HIV-1 RT can bind with low nM KD values or even sub-nanomolar (11–13,26–28). An FDA approved aptamer (Macugen) for the treatment of macular degeneration is currently in use (7,29). Despite hurdles regarding effective delivery of aptamers to patients, understanding how these compounds interact so strongly with proteins could provide insights into the development of future inhibitors. Furthermore, there is great potential for developing aptamers that can overcome delivery problems and systems that can effectively transport aptamers and other drugs to target cells (30–33). This makes it all the more important to move forward on understanding aptamer-protein interactions and developing novel classes of aptamers.
Aptamer selection is usually performed using a method called Systematic Evolution of Ligands by Exponential Enrichment (SELEX), which was invented in the 1990s (34,35). In SELEX, repeated rounds of selection and amplification are used to select nucleic acid sequences that bind the target protein with high affinity from randomized starting pools with tremendous diversity (1014 or more different sequences). Aptamers to hundreds of proteins have been evolved over the years, including several classes of both RNA and DNA aptamers against HIV-1 RT. The RNA aptamers are structurally diverse, including some that form pseudoknots (27,36–38). Several DNA-based aptamers including those that form G-quadruplex structures (13), or loop-back primer-templates that resemble the HIV polypurine tract (11), as well as other unique classes (26,39) have been characterized. Some of these aptamers, including RNA pseudoknots (40) and more recently, a primer-template mimicking aptamer (41), have been crystalized with HIV-1 RT.
Potential recognition by host cell defense systems and degradation by host nucleases is an important concern for developing aptamers and other nucleic acid mediated drugs. These issues can be partially overcome by using aptamers constructed completely or in part from modified nucleotides that may evade recognition and are resistant to degradation. However, modifying DNA or RNA aptamers selected in SELEX protocols can significantly alter specificity and target binding. More recently, mixed aptamers consisting of ribonucleotide analogs (e.g. 2′-fluoro-2′-deoxyribonucleic acid) have been selected from starting pools using a T7 RNA polymerase optimized for incorporating 2′-F-dUTP and 2′-F-dCTP residues along with GTP and ATP. This approach allows direct selection of high affinity aptamers, which unlike aptamers composed solely of unmodified ribonucleotides, are RNase resistant (42–45).
Synthetic nucleotides (2′-deoxy-2′-fluoroarabinonucleotides, FANA) have also been used to modify the composition of thrombin aptamers containing G-quartets. Note that FANA is structurally distinct from the 2′-fluororibonucleotides described above as the sugar ring in FANA typically adopts a C2’/O4’-endo conformation as opposed to a C3’-endo conformation for 2′-fluoro-ribonucleotides, and the fluorine group in FANA is in the β as opposed to α conformation (46–48). The modified thrombin aptamers showed greater thermal stability and nuclease resistance, and tighter binding to thrombin (49,50). FANA is part of a group of synthetic nucleic acid analogs containing altered sugar backbones referred to as xeno-nucleic acid or XNA. XNAs are unique because the unnatural structure of the nucleoside is typically less susceptible to degradative enzymes (51–55). Like DNA and RNA, XNAs can form highly stable complex structures making them ideal for aptamer selection. A groundbreaking technique has enabled SELEX-based selection of aptamers directly from pools with all four nucleotides replaced by XNA nucleotides of several different types (51) (for commentary see (53–55)). The key breakthrough was the design by mutagenesis of thermostable polymerases that can transcribe DNA to XNA and the different types of XNA back to DNA.
In this report, we describe selection of FANA XNA aptamers to HIV-1 RT. These are the first FANA aptamers to any protein made by direct selection using all FANA nucleotides. These aptamers are structurally unique in comparison to other RNA and DNA aptamers and bind with pM affinity, comparable to the tightest binding HIV-1 RT aptamers currently known. They are also extremely potent inhibitors of HIV-1 RT in vitro. These represent the first XNA aptamers to HIV-1 RT and demonstrate that XNA is an excellent alternative for aptamer production against protein targets.
MATERIALS AND METHODS
Materials
All 2′-deoxy-2′-fluoro-arabino-nucleotides (faATP, faCTP, faGTP, faUTP) were obtained from Metkinen Chemistry (Kuusisto, Finland). Deoxyribonucleotide triphosphates (dNTPs) were from Roche. Taq polymerase, EcoRI HF® restriction enzyme, T4 polynucleotide kinase (PNK), calf intestinal alkaline phosphatase (CIP), murine leukemia virus reverse transcriptase (MuLV RT), 10X ThermoPol buffer (Mg2+-free), MgSO4, and 10× CutSmart buffer were from New England BioLabs. Avian myeloblastosis virus (AMV) RT was from Affymetrix. Radiolabeled ATP (γ-P32) was from Perkin-Elmer. G-25 spin columns were from Harvard Apparatus. Miniprep DNA preparation kits were purchased from Qiagen. Nitrocellulose filter disks (Protran BA 85, 0.45 μm pore size and 25-mm diameter) were from Whatman. All DNA oligonucleotides were from Integrated DNA Technologies. All other chemicals were from VWR, Fisher Scientific, or Sigma. The HIV-1 RT expression vector (derived from HXB2 proviral clone (56)) was a generous gift from Dr Michael Parniak (University of Pittsburgh). The enzyme was purified as described (57) and stored in aliquots at −80°C. Thermostable polymerase D4K was prepared as described and stored in aliquots at −20°C (51).
Methods
End-labeling of oligonucleotides with T4 PNK
The DNA oligonucleotides were 5′ end-labeled in a 50 μl volume containing 10–50 pmol of the oligonucleotide of interest, 1× T4 PNK reaction buffer, 10 U of T4 PNK and 10 μl of (γ-32P) ATP (3000 Ci/mmol, 10 μCi/μl). The labeling reaction was done at 37°C for 30 min according to manufacturer's protocol. PNK enzyme was heat inactivated by incubating the reaction at 75°C for 15 min. Excess radiolabeled nucleotides were then removed by centrifugation using a Sephadex G-25 column.
Synthesis of the FANA starting pool
FANA synthesis was performed using a 97 nucleotide DNA template (5′-AGGCCAACTGGATAGCGAA(N)40CGAATTCAGCACTACCTTTTGGCAAACGCTAATAAGGG-3′) with a randomized 40 nucleotide central region (Primer 1 region underlined and EcoRI restriction site is in bold italics). The 25 μl reaction contained 1.6 μM of DNA template, 1 μM of 5′ end-labeled DNA primer 1 (5′-AAAAGGTAGTGCTGAATTCG-3′, see Figure 1), 250 μM of each faNTP, 1X ThermoPol buffer (20 mM Tris–HCl, 10 mM (NH4)2SO4 10 mM KCl, 0.1% Triton® X-100, pH 8.8@25°C) and 2 mM MgSO4. Reactions were denatured and annealed by heating to 94°C for 2 min and cooling to 4°C at rate of 0.1°C/s. D4K enzyme 0.3 μM (final concentration) was added and FANA synthesis was carried out in 4 cycles of 50°C (5 min), 65°C (5 min), and 94°C (1 min). Upon completion, an equal volume of 2× gel-loading buffer (90% formamide, 10 mM EDTA pH 8, 0.25% each bromophenol blue and xylene cyanol) was added and the material was run on an 8% denaturing polyacrylamide-7M urea gel (19:1 (w:w) acrylamide:bisacrylamide) in Tris–boric acid-EDTA (TBE) buffer (58). The 79 nucleotide fully extended product (20 nucleotides 5′ DNA-59 nucleotide FANA), which was 18 nucleotides shorter than the DNA template, was excised from the gel and eluted by the crush and soak method in 10 mM Tris–HCl (pH 7.0) buffer overnight (58). Material was passed through a syringe filter and the nucleic acid was recovered by ethanol precipitated using 50 μg of glycogen as a carrier. Each reaction yielded ∼5 pmol of 79 nucleotide material. Several reactions were performed to obtain enough FANA to start the first round of selection.
Figure 1.

SELEX approach used to select FANA aptamers with tight binding to HIV-1 RT. The protocol used a DNA template containing a 40 nucleotide random region flanked by two fixed sequences at the 5′ end (19 nucleotide) and 3′ end (38 nucleotides). The FANA random pool produced using primer 1 was 79 nucleotides long, composed of 20 DNA nucleotides on the 5′ end and 59 FANA nucleotides. DNA template and FANA random pool were separated based on size on a denaturing polyacrylamide gel. Refer to Material and Methods for further details.
Selection of FANA aptamers with HIV-1 RT using gel-shift
About 200 pmol of FANA random pool was heated to 90°C for 3 min (buffer described below) then quickly cooled on ice. This material was incubated with 12.5 pmol of HIV-1 RT in 25 μl of buffer containing 50 mM Tris–HCl (pH 8), 80 mM KCl, 2 mM MgCl2 and 1 mM DTT. The incubation was at room temperature for 1 h. After incubation, 6 μl of 6× gel loading buffer (30% glycerol, 0.25% each (w/v) bromophenol blue and xylene cyanol) were added and the material was run on a 6% native polyacrylamide gel (29:1 (w:w) acrylamide–bisacrylamide) in TBE buffer at 150 V and 10°C until the blue dye marker was ∼2–3 in. below the wells. A control reaction containing FANA random pool without HIV-1 RT was also run. Shifted FANA material was excised from the gel and eluted as described above. The recovered material was reverse transcribed to DNA in a 20 μl reaction containing 500 μM of each dNTP, 1× ThermoPol buffer (see above) containing 1 mM of MgSO4, rather than 2 mM, 0.5 μM of DNA primer 2 (5′-AGGCCAACTGGATAGCGAA-3′, see Figure 1) and 0.3 μM of polymerase D4K. The enzyme was added after the denaturation and hybridization step, as described above. Reverse transcription was performed in a single cycle for 2 h at 65°C. Three different reactions were performed following reverse transcription to obtain the FANA substrate for the next round of selection: (i) All material from the reverse transcription was used for a 100 μl PCR reaction that contained 1 μM 5′-P32 labeled primer 2, 1 μM DNA primer 3 (5′-CCCTTATTAGCGTTTGCCAAAAGGTAGTGCTGAATTCG-3′, see Figure 1), 200 μM of each dNTP, 1× Taq buffer, and 5 U of Taq polymerase. The PCR was performed at 94°C (2 min), followed by cycles at 94°C (30 s), 55°C (30 s) and 72°C (30 s). Thirty-three microliters were removed at cycles 15, 18 and 21. The material was run on 12% native polyacrylamide gel. Products corresponding to the correct size 97 base pair dsDNA were excised and processed as described above. If less than ∼10 pmol of product was recovered, another PCR reactions was performed using 0.1 pmol of recovered dsDNA and the above conditions for 8, 10 and 12 cycles. (ii) The second reaction was performed to produce single-stranded DNA template to regenerate FANA. An 800 μl reaction volume contained about 8 pmol of material from PCR 1, 1 μM of 5′-P32 labeled primer 2, 200 μM of each dNTP, 1× Taq buffer, and 40 U of Taq polymerase. The reaction was divided into eight tubes (100 μl each) and asymmetric PCR was carried out as described above for 20 cycles. Reactions were combined and the DNA was recovered by ethanol precipitation. The material was run on an 8% denaturing polyacrylamide-7M urea gel as described above. Single strand DNA of the correct length (97 nucleotides) was excised and recovered as described above. (iii) The last reaction was performed to synthesize FANA from the single-stranded DNA as described above under ‘Synthesis of FANA starting pool’ with the following changes: the amount of 5′-P32 labeled DNA primer 1 used was equal to the amount of recovered single stranded DNA. Reactions were split into several tubes with ∼40 pmol of single stranded DNA template in each reaction. This approach typically yielded ∼10–20 total pmol of FANA for the next selection round. In round 2, the amount of HIV-1 RT was decreased to 5 pmol then 2 pmol in round 3 and 1 pmol thereafter. Selection was continued for a total of seven rounds. After the second round, some material from PCR 1 was saved as a source to regenerate the selected material from these rounds.
Sequences analysis of FANA products recovered from round 5
Sequences from FANA selected material from round 5 were cloned using a TOPO TA cloning kit from Life Technologies. DNA mini-preps were prepared and the products were sequenced by Macrogen (Rockville, Maryland). The sequences were analyzed using BioEdit and folded structures were generated using the online mfold program and the default settings for RNA (59). The appropriate DNA oligonucleotide templates for some recovered sequences were synthesized and generation of FANA material was performed as described above.
Apparent equilibrium dissociation constant (KD,app) and dissociation rate constant (koff) determinations using nitrocellulose filter binding assay and gel-shift
Standard reactions for KD,app determinations were performed in 1 ml of buffer containing (final concentrations): 2 pM 5′ end-labeled aptamer, 50 mM Tris–HCl (pH 8), 80 mM KCl, 1 mM DTT, 2 mM MgCl2 and 0.1 mg/ml BSA (for some determinations with the FANA FA1 aptamer, other buffer conditions were employed as indicated in Table 2). Increasing amounts of HIV-1 RT (or AMV and MuLV RTs as indicated) diluted in the above reaction buffer was added in amounts that approximately flanked the KD,app value (estimated from initial experiments) for the aptamer. For aptamers with very low KD,app values (e.g. FA1 (Table 1), KD,app = 4 ± 3 pM) the amount of RT used was 0, 1, 2, 4, 8, 16, 32, 64, 128, 10 000 pM. HIV-1 RT concentrations below 1 pM were not used as they could not be reproducibly differentiated from the no enzyme sample background. After 10 min at room temperature, the reactions were applied to a 25 mm nitrocellulose disk (0.45 μm pore, Protran BA 85, Whatman™) pre-soaked in filter wash buffer (25 mM Tris–HCl pH 7.5, 10 mM KCl). The filter was washed three times under vacuum with 1 ml of wash buffer at a flow rate of ∼0.25 ml/s. Filters were then counted in a scintillation counter. The concentration of bound aptamer was determined for each RT concentration using a saturating RT level as a reference for the maximum amount of material that could be bound in the assay. A plot of bound aptamer vs. RT concentration was fit to the following equation using KaleidaGraph in order to determine the KD,app: [ED] = 0.5([E]t + [D]t + KD,app) – 0.5(([E]t + [D]t + KD,app)2 – 4[E]t[D]t)1/2, where [E]t is the total enzyme concentration and [D]t is the total aptamer concentration (60). For some constructs with KD,app values in the nM range, the above assay was modified to include 100 pM nucleic acid in 50 μl assay volume. This modification allowed the use of higher concentrations of RT while using less total RT.
Table 2. KD,app measurements for FANA binding to HIV-1 RT under different conditions.
| Buffer conditions used to measure KD,app | 1Approximate ionic strength (mM) | 2KD,app (pM) |
|---|---|---|
| Gel shift assay: 50 mM Tris–HCl pH 8.0, 80 mM KCl, 1 mM DTT, 2 mM MgCl2, 0.1 mg/ml BSA | 99 | 8±1 |
| Filter binding assay: | ||
| 50 mM Tris–HCl pH 8.0, 80 mM KCl, 1 mM DTT, 2 mM MgCl2, 0.1 mg/ml BSA | 99 | 4±3 |
| 50 mM Tris–HCl pH 8.0, 50 mM KCl, 1 mM DTT, 10 mM MgCl2, 0.1 mg/ml BSA | 93 | 16±8 |
| 1xPBS + Mg2+: 1.7 mM KH2PO4, 5 mM Na2HPO4, 150 mM NaCl, 2 mM MgCl2 | 170 | 22±4 |
| 50 mM Tris–HCl pH 8.0, 150 mM KCl, 1 mM DTT, 2 mM MgCl2, 0.1 mg/ml BSA | 190 | 74±20 |
1—Ionic strength was calculated assuming 50% ionization for Tris buffer as the pH was very close to the buffer pKa. An online calculator was used for the determinations (http://www.lenntech.com/calculators/activity/activity-coefficient.htm).
2—KD,app measurements were performed using nitrocellulose filter binding or gel shift assay as described in Materials and Methods. Results are an average of three or more independent experiment ± standard deviations.
Table 1. KD,app and koff measurements for FANA and DNA aptamers.
| 1FANA aptamers and various DNA aptamers | 2KD,app values (pM) | 2koff (s−1) |
|---|---|---|
| FANA aptamers | ||
| FANA random pool | 6100±700 | |
| FA1 | 4±3 | 0.00011±0.00005 |
| FA2 | 16±6 | |
| FA3 | 270±20 | |
| FA1Δ1–14 | 5±1 | 0.00009±0.00002 |
| FA1GCstems | 12±5 | 0.00013±0.00006 |
| FA1GCstemsΔL4/S4 | 1500±400 | |
| FA1GCstemsΔL2 | 260±40 | |
| FA1GCstemsΔL1 | 18±6 | |
| FA1GCstemsL1→stem | 19±5 | |
| MuLV (random pool)a | 2200±400 | |
| MuLV (FA1)a | 1300±400 | |
| AMV (random pool)a | 3200±300 | |
| AMV (FA1)a | 5000±2100 | |
| DNA aptamers | ||
| 38 NT SELEX | 160±20 | |
| 38NT2,4-methyl | 14±2 | 0.00010±0.00004 |
| R1T G-quadruplex | 6±1 | 0.00014±0.00003 |
| FA1→DNAa | 23 000±9000 | |
| Random pool DNAa | 24 000±4000 | |
| RNA aptamer | ||
| 26 nt RNApseudoknot | 210±10 | |
1—Refer to text and Figure 4 for information on the various FANA aptamers. All experiments were with HIV-1 RT except those designated as MuLV and AMV.
2—KD,app and dissociation rate constant (koff) measurements shown in this table were performed using nitrocellulose filter bind as described in Materials and Methods. Results are an average of three or more independent experiment ± standard deviations.
aDetermined using 100 pM alternative KD assay (see Material and Methods).
A gel-shift assay was also performed for the FA1 aptamer. The 50 μl total reaction volume contained the same buffer described above for filter binding and 2 pM aptamer. HIV-1 RT dilutions used in this assay were 0, 1, 2, 4, 8, 16, 32, 64, 128, 256, 512, 1024 and 2048 pM. After 10 min of incubation at room temperature, 10 μl of 6× loading gel buffer was added to the reaction and the material was run on a 6% native polyacrylamide gel in TBE buffer at 150 V and 10°C until the blue dye marker was ∼2–3 in. below the wells. The gel was dried, exposed to an imager screen for about 60 hours, and quantified using a phosphorimager (Fuji FLA7000). KD,app values were determined by plotting the total amount of gel-shifted aptamer vs. the RT concentrations and fitting the data to an equation for ligand binding one-site saturation in SigmaPlot. The equation was y = Bmax(x)/KD + x where x is the concentration of RT and y is the amount of gel-shifted aptamer. The experiment was performed three times and the KD,app value in Table 2 is an average of those experiments ± standard deviation.
Dissociation rate constant (koff) determinations were performed in the same buffer with 5 nM (final concentration) of 5′ end-labeled aptamer and 5 nM of HIV-1 RT in 98 μl. The reaction was incubated at room temperature for 5 min. Two microliters of non-radioactive aptamer (1 μM final concentration) was added to the reaction at time ‘0’. This was used as a ‘trap’ to sequester RT molecules as they dissociated from the labeled aptamer. Aliquots of 10 μl were vacuum filtered over nitrocellulose filters as above at times 0, 15, 30, 60, 90, 120, 180 and 240 minutes. For some aptamers that bound less stably to RT, a shorter time scale was used. The nitrocellulose filter disks were washed, and processed as described above. Off-rates (koff) were determined by plotting the retained counts versus time, and fitting the data to an equation for single, two parameter exponential decay using SigmaPlot. The equation was y = ae−bx, where y is the amount of labeled aptamer bound at time x and a is the amount bound at time 0. Experiments were repeated at least two times and averages ± standard deviations are reported.
Preparation of substrate for reverse transcriptase inhibition assays
Fifty pmol of 50 nucleotide template (5′-TTGTAATACGACTCACTATAGGGCGAATTCGAGCTCGGTACCCGGGGATC-3′) and 50 pmol of 33 nucleotide 5′ 32P end-labeled primer (5′-GATCCCCGGGTACCGAGCTCGAATTCGCCCTAT-3′) were mixed in 20 μl containing 50 mM Tris–HCl (pH 8), 1 mM DTT, and 80 mM KCl. The mixture was heated to 80°C for 2 min, and then cooled at a rate of 1°C per minute to 30°C. This material was used directly in the assays.
Preparation of ddG-terminated 38 NT SELEX substrate
Fifty pmol of 38 NT SELEX DNA was incubated with 2 units of Klenow DNA polymerase in 50 μl of buffer containing 50 mM Tris–HCl (pH 8), 1 mM DTT, 50 mM KCl, 6 mM MgCl2, and 50 μM ddGTP for 30 min at 37°C. The material was extracted with phenol–chloroform and precipitated with ethanol. The precipitated material was run through a Sephadex G-25 spin column to remove any remaining unused ddGTP. The recovered material was not extendable by HIV-1 RT in the presence of dNTPs (determined by 5′ end-labeling a portion of the material with γ 32P ATP), indicating that it was 3′ terminated with ddG.
Reverse transcriptase inhibition assay
Inhibition assays were performed essentially as described previously (12). Reactions contained substrate (1:1 primer:template, final concentration in reactions was 50 nM in 5′ 32P end-labeled primer) in 30 μl of buffer containing 50 mM Tris–HCl (pH 8), 1 mM DTT, 80 mM KCl, 6 mM MgCl2, and 0.1 μg/μl BSA. Aptamer inhibitors (1 nM final concentration) were included in some assays. Five μl of a supplement containing 800 μM dNTPs (100 μM final) in the above buffer were added to the reactions. The mixture was placed at 37°C for 2 min. Primer extension was initiated by adding 5 μl of HIV-1 RT (0.25 nM final concentration in reactions) in the above buffer. Five microliters aliquots were removed at 2, 5, 10, 15 and 20 min and add to 5 μl of 2× formamide gel loading buffer. Samples were run on a 10% denaturing polyacrylamide–7M urea gel (19:1 acrylamide:bisacrylamide), dried, then quantified using a phosphorimager (Fuji FLA7000). A graph of the number of phosphorimager counts (photo-stimulated luminescence (PSL) radiation units) versus time was plotted. Experiments were repeated at least twice. Some experiments with AMV and MuLV RTs were also conducted (see Supplemental Data).
RESULTS
Protocol for FANA aptamer selection
Aptamers were selected from a starting pool of approximately 1014 different sequences by the method shown in Figure 1 and described in Materials and Methods. The random pool contained a 20 nucleotide fixed DNA sequence at the 5′ end followed by a 40 nucleotide region of random FANA nucleotides, then 19 nucleotides of FANA fixed sequence. Since FANA is not completely resistant to DNases (50), selection was performed without the removal of the 20 nucleotide 5′ end DNA. Therefore the selected aptamers were chimeric and contained a relatively short fixed DNA region along with a longer FANA region. Although this introduces the possibility of a DNA–FANA hybrid region that could be important for HIV-1 RT selection, results with the tightest binding aptamer (FA1, Table 1, and Figure 4) indicated that at least the first 14 DNA nucleotides could be deleted from the selected aptamer without a loss of binding affinity.
Figure 4.

Structure of FANA aptamers (denoted FA) recovered from round 5 of FANA SELEX (see Figure 1) and 38 NT SELEX aptamer. For FANA aptamers, red indicates DNA nucleotides that were derived from the SELEX 5′ primer. Blue nucleotides are FANA nucleotides corresponding to the SELEX 3′ primer while sequence derived from the 40 nucleotide random region are in black. Various derivatives of FA1 and FA1GCstems are also shown (see text for a description). Folding was performed with mfold using standard conditions for RNA. Biochemical analysis of the various aptamers is reported in Table 1 and the Result section. *38 NT SELEX is a previously reported DNA aptamer (11).
Selection was performed by gel-shift. A titration of the random pool with HIV-1 RT is shown in Figure 2 (right panel). Shifting was observed only in the far right lane where 500 nM HIV-1 RT was added. This concentration of RT was used in the first round of the selection process. By round 5 a discrete band was observed even at the lowest concentration of RT used in the experiment shown in Figure 2 (left panel). In contrast, the random pool shifted only at the highest concentrations and no prominent band was observed. No further increase in binding was observed in two additional rounds of SELEX conducted after round 5 so the selection was terminated at this point.
Figure 2.

Titration of FANA with HIV-1 RT. A gel-shift experiment performed using 10 nM of selected material from round 5 of the SELEX protocol (Figure 1) or random pool, and different amounts of HIV-1 RT is shown. The concentrations of RT used were from left to right 0, 50, 100, 200, 300, 400 and 500 nM. Positions of unshifted and shifted material are indicated.
Sequences recovered from the FANA SELEX
Material from round 5 of the SELEX was cloned and 31 sequences were recovered (see Supplemental Data, Figure S1). Several sequences had lost one or more bases from the random region (initially 40 nucleotides) during the selection process. No sequence appeared more than once, although two sequences differed by just a single nucleotide (sequences 5 and 10), and others differed by just two nucleotides (sequences 6 and 8). This is quite different from other SELEX experiments performed in this lab where late rounds typically showed several repeats of the same sequence (39,61,62). The sequences were folded using the mfold program for RNA (59). Since FANA is different from RNA or DNA and no programs for folding XNAs are available, structures produced by mfold may not be highly accurate, although FANA is known to bind strongly to DNA and RNA sequences (46) and probably forms structures that are highly related to these nucleic acids. Nine different sequences were chosen for further analysis (sequences 1–9 in Supplemental Data, Figure S1). Both structure and sequence were considered in choosing which recovered sequences to further analyze. Sequences which showed considerable levels of identity (e.g. 1, 2 and 3; 5 and 10; and 6 and 8) to other sequences in the pool were analyzed. Also, representative examples of those with common folding trends (based on mfold predications) were tested. This was the basis for choosing 4, 7 and 9. FANA sequences were produced from chemically synthesized DNA as described in Materials and Methods. Of the nine, only three sequences (1, 2 and 3) bound significantly better than the random pool, with one and two showing low pM binding to HIV-1 RT (Table 1).
The sequences 1, 2 and 3 (denoted FA1, FA2 and FA3, Figure 3) were related to each other at the sequence level but showed much less identity to any of the other recovered sequences. FA1 and FA2 showed 75% identity (pairwise alignment using BioEdit) in the random region while FA1 and FA3, and FA2 and FA3 each showed 63% identity in this region (Figure 3). FA1 and FA2 produced similar folded structures in mfold (Figure 4). A notable difference was the unpaired 3′ terminal nucleotide in FA2. In contrast, despite relatively high identity to the other sequences, FA3 produced a different structure (Figure 4) in mfold. As mfold is not designed to fold FANA, it remains possible that FA3 could form a similar but perhaps less stable version of the FA1 and FA2 structures, and this could help explain its modestly enhanced affinity (see below) for HIV-1 RT. Based on KD,app values, FA3 bound about 23 times better than random pool while FA1 and FA2 bound about 1500 and 400 times more tightly, respectively (Table 1, see Supplemental Data Figure S2 for examples of graphs used for filter binding KD,app determinations).
Figure 3.

(A) Full length sequences of FA1, FA2 and FA3 aptamers recovered from Round 5 of SELEX. FA1 and FA3 lost two nucleotides and FA2 lost one nucleotide from the 40 nucleotide random starting pool region (non-underlined nucleotides) during the SELEX process. The 5′ and 3′ underlined nucleotides were derived from fixed sequence primers and were composed of DNA and FANA, respectively (see Figure 1). (B) Pairwise (using BioEdit) alignment of nucleotides derived from the 40 nucleotide random starting pool region of FA1, FA2 and FA3 aptamers. FA1/FA2 showed 75% identity while FA1/FA3 and FA2/FA3 each showed 63% identity in this region.
The aptamer structure and the sequence are important to tight binding and the DNA sequence at the 5′ end can be mostly truncated without a significant effect on binding
To test the importance of the DNA region at the 5′ end of the aptamer for tight binding to HIV-1 RT, an EcoRI restriction site present in the DNA was used to truncate 14 nucleotides on the 5′ end of the FA1 aptamer (FA1 was used because it bound HIV-1 RT modestly more tightly than FA2). This produced a new aptamer that retained just 6 nucleotides of DNA (FA1Δ1–14, Figure 4). This aptamer bound HIV-1 RT with essentially the same affinity as FA1 (Table 1), suggesting that at least the first 14 nucleotides of the DNA portion of the aptamer is not required for tight binding.
It was notable that the DNA portion of FA1 and FA2 was predicted to form a 5′ overhang with the 3′ recessed FANA (Figure 4), which mimics the natural substrate of a polymerase. Other HIV-1 RT DNA aptamers also generated recessed 3′ termini which could be extended by HIV-1 RT in the presence of nucleotides (26). Experiments were conducted with dNTPs and faNTPs and several polymerases to test extension from the 3′ end of FA1 (see Supplemental Figure S3). No extension was observed with HIV-1 RT or any other polymerase. Although this may suggest that HIV-1 RT is not binding to the 3′ terminus in the proper orientation for extension, it could also be due to an inability to extend from a FANA nucleotide.
To test the binding of the aptamer to HIV-1 RT several modifications of the sequence and underlying structure were examined. A version of FA1 in which all the stem structures were converted to G-C base pairs was constructed (Figure 4, FA1GCstems). This construct differed from FA1 by 14 nucleotides, but maintained the basic structure of FA1. FA1GCstems presumably takes advantage of the stability of G-C versus A-T or G-U base pairs in stems, and mfold predicted a ΔG of −32.1 kcal/mol for an RNA with the sequence of FA1GCstems versus −16.5 kcal/mol for FA1. The KD,app for binding to RT was ∼3 times higher for FA1GCstems than FA1 (Table 1). This small decrease in binding affinity, despite changing several nucleotides, emphasizes the importance of the secondary structure for binding RT.
To investigate the importance of various regions of the aptamer for HIV-1 RT binding, truncations were made in FA1GCstems and binding was measured. FA1GCstems was used instead of FA1 to take advantage of the expected stabilization of G-C pairs, where truncations could be made without significantly disturbing structural motifs distal to the truncated region. For clarity, the four loop regions in the folded structure of FA1GCstems are denoted in Figure 4 as L1–L4 with L1 most proximal to the 5′ and 3′ ends. The four stem regions were denoted S1–S4 with S1 most proximal to the 5′ and 3′ ends. Deletion of nucleotides 47–56 resulted in removal of L4 and S4 (Figure 4, FA1GCstemsΔL4/S4). Though folding indicated that the general structure of FA1GCstemsΔL4/S4 outside of the deleted region was unchanged, binding to HIV-1 RT was severely reduced to near the level of the random pool (Table 1). Removal of L1 (FA1GCstemsΔL1) or changing L1 to a stem (FA1GCstemsL1→stem) did not significantly change binding (Table 1). Removal of the asymmetric L2 loop (FA1GCstemsΔL2) resulted in a much greater loss in binding affinity of approximately 23-fold (Table 1). From these experiments we conclude that the secondary structure conformation of the aptamer as well as L2 and the L4/S4 regions are pivotal for tight binding to HIV-1 RT. These experiments do not rule out the possibility that some specific sequences in the loop regions are important for binding. They also did not test for the possibility that small changes in the lengths of specific stems or size of loop regions could affect binding. However, they do indicate that the general structure of the aptamer must be maintained for high affinity binding to HIV-1 RT.
Results above indicated that FA1 binds very tightly to HIV-1 RT when filter binding is used to assess affinity. To confirm this result, binding was examined by a gel-shift method (Figure 5). Results were in good agreement with filter binding with a KD,app of 8 ± 1 pM versus 4±2 pM for filter binding under the same assay conditions (Table 2). In gel-shifts, the level of shifted material appeared to saturate at ∼32 pM RT and a portion of the FA1 material (∼1/3) was not shifted, even at very high RT concentrations above 1 nM (Figure 5). The unshifted material may represent mis-folded FA1 or an alternative folding form with lower affinity for HIV-1 RT. Since the KD,app value for these experiments was determined by fitting the amount of shifted material versus [RT] to an equation for ligand binding one-site saturation (see Materials and Methods), the value only reflects the form of FA1 that was able to shift under the experimental conditions.
Figure 5.

Gel-shift analysis of FA1 with HIV-1 RT. (A) Radiolabeled FANA aptamer FA1 (2 pM final concentration, see Figure 4 for a depiction of FA1) was incubated at room temperature for 10 minutes with increasing concentrations of HIV-1 RT (as indicated) in 50 μl of buffer containing 50 mM Tris–HCl pH 8, 80 mM KCl, 2 mM MgCl2, 1 mM DTT, and 0.1 mg/ml BSA. Ten μl of 6× native gel loading buffer was added and the samples were loaded on a 6% native PAGE gel and electrophoresed and processed as described in Material and Methods. The positions of unshifted and shifted FA1 aptamer are indicated. (B) A graph of shifted material versus [HIV-1 RT] is shown. RT concentrations beyond 64 pM are not shown on the graph to emphasize the area where an increase in shifted material was observed. Beyond 32 pM the values for shifted material were essentially constant. The results are an average of 3 exp. ± standard deviations (error bars). The curve was generated by fitting the data to an equation for ligand binding with one-site saturation using SigmaPlot. The r2 value for this fit was 0.96. The KD,app value for gel-shift experiments in Table 2 of 8 ± 1 pM was determined from the mean of the individual experiments ± the standard deviation.
Comparison of FANA aptamers with other HIV-1 RT aptamers
Several DNA and RNA aptamers to HIV-1 RT have been selected (see Introduction). Binding constants for other aptamers that are known to bind tightly to HIV-1 RT were also determined using the same filter binding assay that was used for FANA aptamers, and the KDapp values are shown in Table 1. The primer-template mimicking aptamers 38 NT SELEX (11) and 38NT2,4-methyl were developed before by our group and the latter aptamer was recently crystalized with HIV-1 RT (41). The G quadruplex aptamer R1T (13) and the 26 nucleotide RNA pseudoknot aptamer have been previously described (27). This RNA pseudoknot constitutes of the minimum domain required for high affinity binding to HIV-1 RT as described for aptamer ‘1.1’ in Tuerk et al. (27). The R1T aptamer bound HIV-1 RT about as stably as FA1 while 38NT2,4-methyl also bound very tightly with a KD,app approximately equal to FA1GCstems and FA2. The koff values for these aptamers were also comparable to values for FA1, FA1Δ1–14 and FA1GCstems. Aptamers 38 NT SELEX and the RNA pseudoknot bound considerably less tightly, though still with high affinity. A FANA mimic of 38 NT SELEX was also constructed and bound essentially like FANA random pool to RT (Figure 4, FA38 NT SELEX). A complete DNA version of FA1 also bound with low affinity to RT (FA1→DNA) (Table 1). In this case, the affinity was even lower than the binding of HIV-1 RT to the FANA random pool. This is probably because HIV-1 RT binds FANA modestly more tightly than DNA. Overall, these results show that the tight binding of the FANA aptamers requires properties that are unique to FANA and cannot be completely mimicked by DNA.
The effect of ionic strength and buffer conditions on binding of FANA aptamer FA1 to HIV-1 RT
Binding of nucleic acids to proteins is highly dependent on the assay conditions including ionic strength and the concentration of divalent cations. The standard buffer conditions used above to measure KD,app were the same as those used in the SELEX selection process, and this buffer had an approximate ionic strength of 100 mM (assuming about 50% ionization of the Tris buffer). In cells the ionic strength is higher, and this is likely to weaken binding (63). Because of the complexities of the cellular environment, it would be difficult to directly correlate in vitro assays to binding in the cell. However, a buffer with ionic strength nearer to what exists in cells may more closely represent binding of the aptamer to HIV-1 RT in cells. Phosphate buffered saline (PBS) is commonly used for cell suspension and is isotonic for many cells. The ionic strength of this buffer (with 2 mM MgCl2) is approximately 170 mM. Binding of FA1 was tested in PBS with 2 mM MgCl2 and the results are shown in Table 2. The KD,app value rose about 5-fold in this buffer to 22 ± 4 pM, consistent with the increase in ionic strength. A version of the standard buffer with 150 mM KCl had an ionic strength of about 190 mM and resulted in a KD,app of 74 ± 20 pM. Finally, others have shown that a 33 nucleotide RNA pseudoknot aptamer bound HIV-1 RT with a low pM KD, similar to what was observed for FA1 (28). The Tris buffer used in those experiments contained 10 mM MgCl2 and 50 mM KCl and had approximately the same ionic strength as the standard buffer used here. With FA1, this buffer resulted in a KD,app of 16 ± 8 pM. Overall the results show that the binding of FA1, like other nucleic acids, is affected by ionic strength and buffer composition. However, binding to HIV-1 RT is very tight, even in buffers with ionic strengths that resemble the environment in cells.
FANA aptamers are potent inhibitors of HIV-1 RT
Competition binding experiments have previously shown that many aptamers to HIV-1 RT are probably bound in the nucleic acid binding pocket, or at least overlap with that pocket (12). The FA1 aptamer also binds in this pocket as in competition experiments (Supplementary Data Figure S4), it can displace 38NT2,4-methyl, which has been shown to bind in a primer-template configuration to HIV-1 RT in crystal structures (41). Binding in this pocket leads to competition for primer-template binding and inhibition of enzyme activity. To test the potency of FA1 in HIV-1 RT inhibition, the experiment shown in Figure 6 was performed. A primer-template was included at 50 nM in the assay along with 0.25 nM HIV-1 RT and 1 nM of various aptamers. Extension of the radiolabeled primer was monitored over time by gel electrophoresis and autoradiography (see Supplemental Data Figure S5). FANA random pool showed essentially no inhibition, while 38 NT SELEX (with a 3′ terminal ddG residue added to prevent extension of the aptamer by RT) inhibited the reaction by about 40%. Both FA1 and the R1T G-quadruplex aptamers showed nearly complete inhibition of RT extension. Note that this occurred despite the aptamer being present at only 1/50th the concentration of the primer-template. The inhibition is consistent with the extremely tight and stable binding observed for FA1 and R1T.
Figure 6.
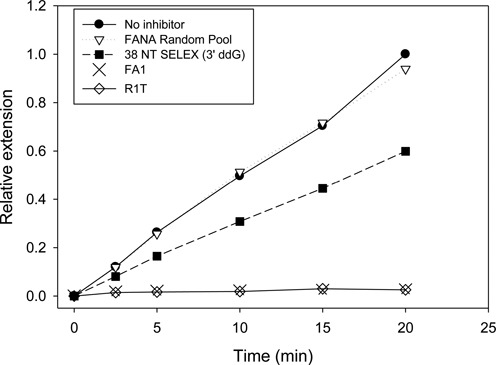
Inhibition of HIV-1 RT primer extension by selected aptamers. Assays included 50 nM primer-template (33 nucleotide primer: 50 nucleotide template) and 0.25 nM HIV-1 RT. One nM of the indicated aptamer was included. Reactions were initiated by adding dNTPs (50 μM final) and aliquots were removed at the indicated time points. Primer extension was monitored on denaturing polyacrylamide gels and quantified using a phosphorimager. All values for ‘Relative Extension’ are relative to the level of extension at 20 minutes in the ‘No inhibitor’ sample. The 3′ nucleotide of 38 NT SELEX was replaced with ddG using Klenow for this assay. FANA random pool was used for selection in round 1 of SELEX and has random nucleotides in the 40 base random region (Figure 1). Refer to Table 1 for information on the DNA aptamers, 38 NT SELEX and R1T. The structures of FA1 and 38 NT SELEX are shown in Figure 4.
FA1 does not bind other RTs with enhanced affinity
The binding of FA1 to two commonly employed RTs was tested. Although MuLV RT bound tighter to the FANA random pool than HIV-1 RT, the binding affinity for FA1 was only about twice that for binding to the random pool (Table 1). A similar result was obtained with AMV RT although this enzyme bound less strongly to both FANA random pool and FA1. AMV and MuLV RTs are structurally related to HIV-1 RT and all three enzymes use very similar poly-purine tracts to initiate second strand DNA synthesis. In addition, primer-template mimicking aptamers for all 3 enzymes show strong homology (62). Despite this, MuLV and AMV RTs showed no significant preference for binding FA1 over FANA random pool. In addition, FA1 did not strongly inhibit either AMV or MuLV RT (Supplemental Data, Figure S6).
DISCUSSION
In this report, a method to directly select aptamers to HIV-1 RT containing only FANA nucleotides in the randomized region is demonstrated. These are the first such aptamer selected for binding to HIV-1 RT or any other protein. The methodology employed could be used in theory to produce FANA aptamers to any protein. In addition, several other classes of XNA aptamers (LNA (locked nucleic acid), HNA (1,5-anhydrohexitol nucleic acid), ANA (arabinonucleic acid), TNA (α-L-threofuranosyl nucleic acid), CeNA (cyclohexenyl nucleic acid)) can potentially be selected using the modified enzymes used here and others (51). The FANA aptamers produced here bound extremely tightly to HIV RT, equivalent to the levels of the tightest binding DNA aptamer (Table 1). They also competed for binding with the primer-template mimicking 38NT2,4-methyl aptamer, suggesting that like other HIV-1 RT aptamers (12), FA1 binds in the substrate binding pocket or at a site that overlaps the binding pocket.
Strong binding to target proteins by aptamers containing unnatural nucleic acid moieties has been demonstrated by others (42,49,50,64–67). In these cases, the aptamers were produced by replacement of specific nucleotides in DNA or RNA aptamers, or direct selection of aptamers containing both unnatural and natural nucleotides. Some of these ‘mixed aptamers’ bind much more tightly to the target protein than the homogeneous RNA or DNA aptamers, and may also be more resistant to RNases in comparison to RNA aptamers (see Introduction). A commonly used approach for producing mixed aptamers is to use 2′-F-dCTP and 2′-F-dUTP along with natural ATP and GTP (see Introduction). This approach led to mixed aptamers to HIV Env that were RNase resistant and capable of inhibiting virus infection in tissue culture (42,44,68). Another approach involved direct selection of aptamers that contained dNTPs and a limited number of 7-(2-thienyl)imidazo[4,5-b]pyridine (Ds) residues (69). This approach took advantage of the ability of a thermostable polymerase to incorporate an unnatural nucleotide complementary to Ds that could be copied back to the Ds nucleotide by the polymerase. Aptamers to vascular endothelial cell growth factor 165 (VEGF-165) and interferon γ (INF γ) bound these target proteins with affinities over 100 times greater than those containing only natural bases. VEGF-165 is the target protein of the macular degeneration drug Macugen (Pegaptanib Sodium), the only current FDA approved aptamer therapeutic (29). The Ds modified VEGF-165 aptamers bind several-fold more tightly to the protein target than the current therapeutic which was produced by SELEX with 2′-F-dUTP and 2′-F-dCTP as described above, followed by subsequent modification (43). Unlike the above approaches, the approach we used to produce HIV-1 RT FANA aptamers allows selection of aptamers that are composed completely of the unnatural nucleotides in the randomized region of the aptamer. Mixed aptamers can also be produced by this approach since the modified enzyme used to construct the aptamer starting pool and amplify the selected material in each round can incorporate both natural and unnatural nucleotides (51). It remains to be determined if aptamers composed completely of unnatural nucleotides or those of a mixed composition will bind more tightly to target proteins and it may depend on the particular target.
The FA1 aptamer produced here bound very tightly to HIV-1 RT with a KD,app in the low pM range (Tables 1 and 2). Although this was a clear improvement over some RNA aptamers and DNA aptamers, binding affinity and dissociation rates were essentially the same as the R1T G-quadruplex forming aptamer produced by others (13) and an RNA pseudoknot aptamer previously characterized (28). Also, although the 5′ DNA tail on FA1 could be mostly removed without loss of binding affinity, attempts to truncate other regions (using FA1GCstems) reduced binding affinity (Figure 4 and Table 1). This resulted in FANA aptamers that were relatively large (63 nucleotides for FA1Δ1–14, the smallest high affinity aptamer tested). Performing aptamer selections with a shorter randomized region or testing more post-selection modifications could potentially produce shorter FANA aptamers.
In conclusion, we have produced FANA aptamers to HIV-1 RT that bind the protein with pM affinity and potently inhibit polymerase activity. The protocol used allows the production of aptamers that have only unnatural bases in the randomized region (51). FANA aptamers produced by this approach show greater resistance to nucleases (50) and the protocol can also be used with several other unnatural XNAs (51). FANA was used here because FANA triphosphates are readily available and FANA represents a good proof-of-principle model for XNA aptamers. Future work will focus on developing aptamers with other XNAs and testing their ability to inhibit HIV replication in cell culture.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Medical Research Council UK [program no. U105178804 to P.H. and C.C.]; National Institute of Allergy and Infectious Diseases [R03AI116380 to J.D.]. Funding for open access charge: Medical Research Council UK [program no. U105178804 to P.H. and C.C.]; National Institute of Allergy and Infectious Diseases [R03AI116380 to J.D.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Telesnitsky A., Goff S.P. In: Retroviruses. Coffin JM, Hughes SH, Varmus HE, editors. NY: Cold Spring Harbor Laboratory Press; 1997. pp. 121–160. [PubMed] [Google Scholar]
- 2.Brody E.N., Gold L. Aptamers as therapeutic and diagnostic agents. J. Biotechnol. 2000;74:5–13. doi: 10.1016/s1389-0352(99)00004-5. [DOI] [PubMed] [Google Scholar]
- 3.Deisingh A.K. Aptamer-based biosensors: biomedical applications. Handb. Exp. Pharmacol. 2006:341–357. doi: 10.1007/3-540-27262-3_17. [DOI] [PubMed] [Google Scholar]
- 4.Gold L. The SELEX process: a surprising source of therapeutic and diagnostic compounds. Harvey Lect. 1995;91:47–57. [PubMed] [Google Scholar]
- 5.James W. Aptamers in the virologists’ toolkit. J. Gen. Virol. 2007;88:351–364. doi: 10.1099/vir.0.82442-0. [DOI] [PubMed] [Google Scholar]
- 6.Joshi P.J., Fisher T.S., Prasad V.R. Anti-HIV inhibitors based on nucleic acids: emergence of aptamers as potent antivirals. Curr. Drug Targets Infect. Disord. 2003;3:383–400. doi: 10.2174/1568005033481060. [DOI] [PubMed] [Google Scholar]
- 7.Nimjee S.M., Rusconi C.P., Sullenger B.A. Aptamers: an emerging class of therapeutics. Annu. Rev. Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 8.Porschewski P., Grattinger M.A., Klenzke K., Erpenbach A., Blind M.R., Schafer F. Using aptamers as capture reagents in bead-based assay systems for diagnostics and hit identification. J. Biomol. Screen. 2006;11:773–781. doi: 10.1177/1087057106292138. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z., Blank M., Schluesener H.J. Nucleic acid aptamers in human viral disease. Arch. Immunol. Ther. Exp. (Warsz) 2004;52:307–315. [PubMed] [Google Scholar]
- 10.Mok W., Li Y. Recent progress in nucleic acid aptamer-based biosensors and bioassays. Sensors. 2008;8:7050–7084. doi: 10.3390/s8117050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeStefano J.J., Nair G.R. Novel aptamer inhibitors of human immunodeficiency virus reverse transcriptase. Oligonucleotides. 2008;18:133–144. doi: 10.1089/oli.2008.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai Y.T., DeStefano J.J. DNA aptamers to human immunodeficiency virus reverse transcriptase selected by a primer-free SELEX method: characterization and comparison to other aptamers. Nucleic Acid Ther. 2012;22:162–176. doi: 10.1089/nat.2011.0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michalowski D., Chitima-Matsiga R., Held D.M., Burke D.H. Novel bimodular DNA aptamers with guanosine quadruplexes inhibit phylogenetically diverse HIV-1 reverse transcriptases. Nucleic Acids Res. 2008;36:7124–7135. doi: 10.1093/nar/gkn891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreola M.L., Pileur F., Calmels C., Ventura M., Tarrago-Litvak L., Toulme J.J., Litvak S. DNA aptamers selected against the HIV-1 RNase H display in vitro antiviral activity. Biochemistry. 2001;40:10087–10094. doi: 10.1021/bi0108599. [DOI] [PubMed] [Google Scholar]
- 15.de Soultrait V.R., Lozach P.Y., Altmeyer R., Tarrago-Litvak L., Litvak S., Andreola M.L. DNA aptamers derived from HIV-1 RNase H inhibitors are strong anti-integrase agents. J. Mol. Biol. 2002;324:195–203. doi: 10.1016/s0022-2836(02)01064-1. [DOI] [PubMed] [Google Scholar]
- 16.Hotoda H., Koizumi M., Koga R., Kaneko M., Momota K., Ohmine T., Furukawa H., Agatsuma T., Nishigaki T., Sone J., et al. Biologically active oligodeoxyribonucleotides. 5. 5′-End-substituted d(TGGGAG) possesses anti-human immunodeficiency virus type 1 activity by forming a G-quadruplex structure. J. Med. Chem. 1998;41:3655–3663. doi: 10.1021/jm970658w. [DOI] [PubMed] [Google Scholar]
- 17.Kolb G., Reigadas S., Castanotto D., Faure A., Ventura M., Rossi J.J., Toulme J.J. Endogenous expression of an anti-TAR aptamer reduces HIV-1 replication. RNA Biol. 2006;3:150–156. doi: 10.4161/rna.3.4.3811. [DOI] [PubMed] [Google Scholar]
- 18.Neff C.P., Zhou J., Remling L., Kuruvilla J., Zhang J., Li H., Smith D.D., Swiderski P., Rossi J.J., Akkina R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Science translational medicine. 2011;3:66ra66. doi: 10.1126/scitranslmed.3001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J., Neff C.P., Liu X., Zhang J., Li H., Smith D.D., Swiderski P., Aboellail T., Huang Y., Du Q., et al. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol. Ther. 2011;19:2228–2238. doi: 10.1038/mt.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferguson M.R., Rojo D.R., Somasunderam A., Thiviyanathan V., Ridley B.D., Yang X., Gorenstein D.G. Delivery of double-stranded DNA thioaptamers into HIV-1 infected cells for antiviral activity. Biochem. Biophys. Res. Commun. 2006;344:792–797. doi: 10.1016/j.bbrc.2006.03.201. [DOI] [PubMed] [Google Scholar]
- 21.Fisher T.S., Joshi P., Prasad V.R. HIV-1 reverse transcriptase mutations that confer decreased in vitro susceptibility to anti-RT DNA aptamer RT1t49 confer cross resistance to other anti-RT aptamers but not to standard RT inhibitors. AIDS Res. Ther. 2005;2:8. doi: 10.1186/1742-6405-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi P.J., North T.W., Prasad V.R. Aptamers directed to HIV-1 reverse transcriptase display greater efficacy over small hairpin RNAs targeted to viral RNA in blocking HIV-1 replication. Mol. Ther. 2005;11:677–686. doi: 10.1016/j.ymthe.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Matzen K., Elzaouk L., Matskevich A.A., Nitzsche A., Heinrich J., Moelling K. RNase H-mediated retrovirus destruction in vivo triggered by oligodeoxynucleotides. Nat. Biotechnol. 2007;25:669–674. doi: 10.1038/nbt1311. [DOI] [PubMed] [Google Scholar]
- 24.Moelling K., Abels S., Jendis J., Matskevich A., Heinrich J. Silencing of HIV by hairpin-loop-structured DNA oligonucleotide. FEBS Lett. 2006;580:3545–3550. doi: 10.1016/j.febslet.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 25.Somasunderam A., Ferguson M.R., Rojo D.R., Thiviyanathan V., Li X., O'Brien W.A., Gorenstein D.G. Combinatorial selection, inhibition, and antiviral activity of DNA thioaptamers targeting the RNase H domain of HIV-1 reverse transcriptase. Biochemistry. 2005;44:10388–10395. doi: 10.1021/bi0507074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider D.J., Feigon J., Hostomsky Z., Gold L. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus. Biochemistry. 1995;34:9599–9610. doi: 10.1021/bi00029a037. [DOI] [PubMed] [Google Scholar]
- 27.Tuerk C., MacDougal S., Gold L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 1992;89:6988–6992. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kensch O., Connolly B.A., Steinhoff H.J., McGregor A., Goody R.S., Restle T. HIV-1 reverse transcriptase-pseudoknot RNA aptamer interaction has a binding affinity in the low picomolar range coupled with high specificity. J. Biol. Chem. 2000;275:18271–18278. doi: 10.1074/jbc.M001309200. [DOI] [PubMed] [Google Scholar]
- 29.Macugen Food and Drug Administration. [Google Scholar]
- 30.Pangburn T.O., Petersen M.A., Waybrant B., Adil M.M., Kokkoli E. Peptide- and aptamer-functionalized nanovectors for targeted delivery of therapeutics. J. Biomech. Eng. 2009;131:074005. doi: 10.1115/1.3160763. [DOI] [PubMed] [Google Scholar]
- 31.De Rosa G., La Rotonda M.I. Nano and microtechnologies for the delivery of oligonucleotides with gene silencing properties. Molecules. 2009;14:2801–2823. doi: 10.3390/molecules14082801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou J., Bobbin M.L., Burnett J.C., Rossi J.J. Current progress of RNA aptamer-based therapeutics. Front. Genet. 2012;3:234. doi: 10.3389/fgene.2012.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tripathi S., Chaubey B., Barton B.E., Pandey V.N. Anti HIV-1 virucidal activity of polyamide nucleic acid-membrane transducing peptide conjugates targeted to primer binding site of HIV-1 genome. Virology. 2007;363:91–103. doi: 10.1016/j.virol.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 35.Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 36.Whatley A.S., Ditzler M.A., Lange M.J., Biondi E., Sawyer A.W., Chang J.L., Franken J.D., Burke D.H. Potent Inhibition of HIV-1 Reverse Transcriptase and Replication by Nonpseudoknot, ‘UCAA-motif’ RNA Aptamers. Mol. Ther. Nucleic Acids. 2013;2:e71. doi: 10.1038/mtna.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burke D.H., Scates L., Andrews K., Gold L. Bent Pseudoknots and Novel RNA Inhibitors of Type 1 Human Immunodeficiency Virus (HIV-1) Reverse Transcriptase. J. Mol. Biol. 1996;264:650–666. doi: 10.1006/jmbi.1996.0667. [DOI] [PubMed] [Google Scholar]
- 38.Ditzler M.A., Lange M.J., Bose D., Bottoms C.A., Virkler K.F., Sawyer A.W., Whatley A.S., Spollen W., Givan S.A., Burke D.H. High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of HIV reverse transcriptase. Nucleic Acids Res. 2013;41:1873–1884. doi: 10.1093/nar/gks1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai Y.T., DeStefano J.J. A primer-free method that selects high-affinity single-stranded DNA aptamers using thermostable RNA ligase. Anal. Biochem. 2011;414:246–253. doi: 10.1016/j.ab.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaeger J., Restle T., Steitz T.A. The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J. 1998;17:4535–4542. doi: 10.1093/emboj/17.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller M.T., Tuske S., Das K., DeStefano J.J., Arnold E. Structure of HIV-1 reverse transcriptase bound to a novel 38-mer hairpin template-primer DNA aptamer. Protein Sci. 2015 doi: 10.1002/pro.2776. doi:10.1002/pro.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou J., Swiderski P., Li H., Zhang J., Neff C.P., Akkina R., Rossi J.J. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009;37:3094–3109. doi: 10.1093/nar/gkp185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruckman J., Green L.S., Beeson J., Waugh S., Gillette W.L., Henninger D.D., Claesson-Welsh L., Janjic N. 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998;273:20556–20567. doi: 10.1074/jbc.273.32.20556. [DOI] [PubMed] [Google Scholar]
- 44.London G.M., Mayosi B.M., Khati M. Isolation and characterization of 2′-F-RNA aptamers against whole HIV-1 subtype C envelope pseudovirus. Biochem. Biophys. Res. Commun. 2015;456:428–433. doi: 10.1016/j.bbrc.2014.11.101. [DOI] [PubMed] [Google Scholar]
- 45.Tawaraya Y., Hyodo M., Ara M.N., Yamada Y., Harashima H. RNA aptamers for targeting mitochondria using a mitochondria-based SELEX method. Biol. Pharm. Bull. 2014;37:1411–1415. doi: 10.1248/bpb.b14-00112. [DOI] [PubMed] [Google Scholar]
- 46.Martin-Pintado N., Deleavey G.F., Portella G., Campos-Olivas R., Orozco M., Damha M.J., Gonzalez C. Backbone FC-H…O hydrogen bonds in 2′ F-substituted nucleic acids. Angew. Chem. Int. Ed. Engl. 2013;52:12065–12068. doi: 10.1002/anie.201305710. [DOI] [PubMed] [Google Scholar]
- 47.Berger I., Tereshko V., Ikeda H., Marquez V.E., Egli M. Crystal structures of B-DNA with incorporated 2′-deoxy-2′-fluoro-arabino-furanosyl thymines: implications of conformational preorganization for duplex stability. Nucleic Acids Res. 1998;26:2473–2480. doi: 10.1093/nar/26.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trempe J.F., Wilds C.J., Denisov A.Y., Pon R.T., Damha M.J., Gehring K. NMR solution structure of an oligonucleotide hairpin with a 2′ F-ANA/RNA stem: implications for RNase H specificity toward DNA/RNA hybrid duplexes. J. Am. Chem. Soc. 2001;123:4896–4903. doi: 10.1021/ja003859p. [DOI] [PubMed] [Google Scholar]
- 49.Peng C.G., Damha M.J. G-quadruplex induced stabilization by 2′-deoxy-2′-fluoro-D-arabinonucleic acids (2′ F-ANA) Nucleic Acids Res. 2007;35:4977–4988. doi: 10.1093/nar/gkm520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watts J.K., Damha M.J. 2′F-Arabinonucleic acids (2′F-ANA) — History, properties, and new frontiers. Can. J. Chem. 2008;86:641–656. [Google Scholar]
- 51.Pinheiro V.B., Taylor A.I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J.C., Wengel J., Peak-Chew S.Y., McLaughlin S.H., et al. Synthetic genetic polymers capable of heredity and evolution. Science. 2012;336:341–344. doi: 10.1126/science.1217622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinheiro V.B., Holliger P. The XNA world: progress towards replication and evolution of synthetic genetic polymers. Curr. Opin. Chem. Biol. 2012;16:245–252. doi: 10.1016/j.cbpa.2012.05.198. [DOI] [PubMed] [Google Scholar]
- 53.Shen H. Enzymes grow artificial DNA. Nature. 2012 doi:10.1038/nature.2012.10487. [Google Scholar]
- 54.Steele F.R., Gold L. The sweet allure of XNA. Nat Biotech. 2012;30:624–625. doi: 10.1038/nbt.2298. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt M. Xenobiology: a new form of life as the ultimate biosafety tool. Bioessays. 2010;32:322–331. doi: 10.1002/bies.200900147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fletcher R.S., Holleschak G., Nagy E., Arion D., Borkow G., Gu Z., Wainberg M.A., Parniak M.A. Single-step purification of recombinant wild-type and mutant HIV-1 reverse transcriptase. Protein Expr. Purif. 1996;7:27–32. doi: 10.1006/prep.1996.0004. [DOI] [PubMed] [Google Scholar]
- 57.Hou E.W., Prasad R., Beard W.A., Wilson S.H. High-level expression and purification of untagged and histidine-tagged HIV-1 reverse transcriptase. Protein Expr. Purif. 2004;34:75–86. doi: 10.1016/j.pep.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. 3rd edn. NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 59.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsieh J.C., Zinnen S., Modrich P. Kinetic mechanism of the DNA-dependent DNA polymerase activity of human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 1993;268:24607–24613. [PubMed] [Google Scholar]
- 61.DeStefano J.J., Cristofaro J.V. Selection of primer-template sequences that bind human immunodeficiency virus reverse transcriptase with high affinity. Nucleic Acids Res. 2006;34:130–139. doi: 10.1093/nar/gkj426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nair G.R., Dash C., Le Grice S.F., DeStefano J.J. Viral reverse transcriptases show selective high affinity binding to DNA-DNA primer-templates that resemble the polypurine tract. PloS One. 2012;7:e41712. doi: 10.1371/journal.pone.0041712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hart D.J., Speight R.E., Cooper M.A., Sutherland J.D., Blackburn J.M. The salt dependence of DNA recognition by NF-kappaB p50: a detailed kinetic analysis of the effects on affinityand specificity. Nucleic Acids Res. 1999;27:1063–1069. doi: 10.1093/nar/27.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuwahara M., Obika S. In vitro selection of BNA (LNA) aptamers. Artif. DNA PNA XNA. 2013;4:39–48. doi: 10.4161/adna.25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kasahara Y., Irisawa Y., Fujita H., Yahara A., Ozaki H., Obika S., Kuwahara M. Capillary electrophoresis-systematic evolution of ligands by exponential enrichment selection of base- and sugar-modified DNA aptamers: target binding dominated by 2′-O,4′-C-methylene-bridged/locked nucleic acid primer. Anal. Chem. 2013;85:4961–4967. doi: 10.1021/ac400058z. [DOI] [PubMed] [Google Scholar]
- 66.Kasahara Y., Irisawa Y., Ozaki H., Obika S., Kuwahara M. 2′,4′-BNA/LNA aptamers: CE-SELEX using a DNA-based library of full-length 2′-O,4′-C-methylene-bridged/linked bicyclic ribonucleotides. Bioorg. Med. Chem. 2013;23:1288–1292. doi: 10.1016/j.bmcl.2012.12.093. [DOI] [PubMed] [Google Scholar]
- 67.Elle I.C., Karlsen K.K., Terp M.G., Larsen N., Nielsen R., Derbyshire N., Mandrup S., Ditzel H.J., Wengel J. Selection of LNA-containing DNA aptamers against recombinant human CD73. Mol. Biosyst. 2015;11:1260–1270. doi: 10.1039/c5mb00045a. [DOI] [PubMed] [Google Scholar]
- 68.Khati M., Schuman M., Ibrahim J., Sattentau Q., Gordon S., James W. Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2′ F-RNA aptamers. J. Virol. 2003;77:12692–12698. doi: 10.1128/JVI.77.23.12692-12698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kimoto M., Yamashige R., Matsunaga K.-i., Yokoyama S., Hirao I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotech. 2013;31:453–457. doi: 10.1038/nbt.2556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.