

Graphical abstract
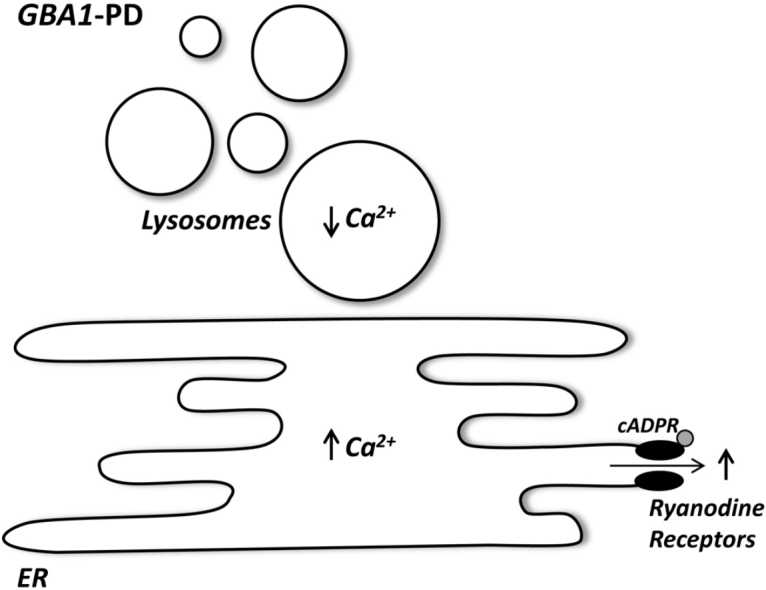
Abbreviations: ER, endoplasmic reticulum; cADPR, cyclic ADP-ribose; SERCA, sarco-endoplasmic reticulum Ca2+ ATPase; PD, Parkinson disease; GD, Gaucher disease; ASX, asymptomatic; CBE, conduritol B epoxide; GPN, glycyl-l-phenylalanine 2-naphthylamide; LAMP, lysosome associated membrane protein; LC3, microtubule-associated protein 1A/1B-light chain 3
Keywords: Ca2+, Endoplasmic reticulum, Lysosomes, Parkinson disease, Gaucher disease
Highlights
-
•
ER Ca2+ signalling is potentiated in PD patient fibroblasts.
-
•
Lysosomal Ca2+ signalling is inhibited in PD patient fibroblasts.
-
•
Remodelling of Ca2+ stores is age-dependent.
Abstract
Mutations in β-glucocerebrosidase (encoded by GBA1) cause Gaucher disease (GD), a lysosomal storage disorder, and increase the risk of developing Parkinson disease (PD). The pathogenetic relationship between the two disorders is unclear. Here, we characterised Ca2+ release in fibroblasts from type I GD and PD patients together with age-matched, asymptomatic carriers, all with the common N370S mutation in β-glucocerebrosidase. We show that endoplasmic reticulum (ER) Ca2+ release was potentiated in GD and PD patient fibroblasts but not in cells from asymptomatic carriers. ER Ca2+ signalling was also potentiated in fibroblasts from aged healthy subjects relative to younger individuals but not further increased in aged PD patient cells. Chemical or molecular inhibition of β-glucocerebrosidase in fibroblasts and a neuronal cell line did not affect ER Ca2+ signalling suggesting defects are independent of enzymatic activity loss. Conversely, lysosomal Ca2+ store content was reduced in PD fibroblasts and associated with age-dependent alterations in lysosomal morphology. Accelerated remodelling of Ca2+ stores by pathogenic GBA1 mutations may therefore feature in PD.
1. Introduction
Changes in the concentration of cytosolic Ca2+ form the basis of a ubiquitous signalling pathway [1]. Ca2+ signals derive not only from the extracellular space, but also from Ca2+ stores, within the cell, through the opening of intracellular Ca2+-permeable channels [2]. The best characterised Ca2+ store is the ER which houses IP3- and ryanodine-sensitive Ca2+ channels. The latter are activated by the second messenger cyclic ADP-ribose [3]. Ca2+ pumps (such as SERCA), exchangers and buffers act to temper Ca2+ increases in a highly regulated Ca2+ network [2]. It is becoming increasingly clear that lysosomes and other acidic organelles such as lysosome-related organelles, endosomes, secretory granules and the Golgi complex are also integral sources of Ca2+ [4], [5]. Lysosomes are thought to drive global Ca2+ signals by providing a “trigger” release of Ca2+ which is then amplified by Ca2+ channels on the ER, possibly through recently described membrane-contact sites between the two organelles [6]. ER and lysosomal Ca2+ stores are thus functionally and physically coupled similar to coupling between the ER Ca2+ stores and mitochondria/plasma membrane [7].
Gaucher disease (GD) is the most common of the lysosomal storage disorders [8]. It results due to recessive mutations in GBA1 which encodes the lysosomal enzyme β-glucocerebrosidase responsible for hydrolysis of glucocerebroside to glucose and ceramide. Type I GD (often associated with the N370S mutation) is traditionally considered non-neuronopathic whereas types II and III are associated with neurodegeneration. But both type I GD sufferers and carriers of GBA1 mutations are up to 20 times more likely to develop Parkinson disease (PD). Mutations in GBA1 are therefore one of the highest known risk factors for this neurodegenerative disorder [9]. Genetic associations between PD and GD add to a body of literature implicating lysosomal dysfunction in the pathogenesis of PD [10], [11], which likely occurs upstream of established mitochondrial dysfunction [12]. The mechanism by which GBA1 mutations mediate PD pathogenesis remains undefined. It may involve the unfolded protein response and ER stress as a consequence of mutant protein trapping or interactions with α-synuclein metabolism leading to Lewy body formation [13]. However, not all GBA1 carriers develop PD suggesting additional pathogenic mechanisms are involved.
De-regulated Ca2+ signalling is established in a number of pathologies and has been implicated in both GD and PD as well as ageing, a major risk factor for neurodegenerative disease [7], [14]. ER Ca2+ stores appear to be hypersensitive to ryanodine receptor activation in a pharmacological neuronal model of GD resulting in sensitisation to cell death [15]. Whether lysosomal Ca2+ stores are affected in the disease is not known, although lysosomal Ca2+ content is reduced in Niemann–Pick type C1 disease [16], a distinct lysosomal storage disorder also potentially linked to PD [17]. In PD, attention has focussed mainly on Ca2+ influx since the affected dopaminergic neurons of the substantia nigra pars compacta exhibit unusual pace-making activity associated with influx of Ca2+ through L-type voltage-sensitive Ca2+ channels [18]. The resulting oscillations in cytosolic Ca2+ are thought to impose metabolic stress on the mitochondria [19], [20]. The role of ER and lysosomal Ca2+ stores in PD is largely unexplored.
In the present study, we identify age-dependent reciprocal changes in ER and lysosomal Ca2+ homeostasis in patient fibroblasts from GD and GBA1-linked PD sufferers. These data point to altered Ca2+ signalling in GBA1-disease and in ageing as possible contributors to PD pathology.
2. Methods
2.1. Patient fibroblasts
Primary fibroblast cultures were generated from skin biopsies as described in [21]. GD (type I) and PD patients carried the mutant allele encoding the N370S variant in β-glucocerebrosidase. The GD patient was a compound heterozygote with an additional 1263del55 mutation. For simplicity, these genotypes are referred to as GBA1mut/mut GD and GBA1wt/mut PD, respectively. Cultures were also established from asymptomatic (ASX) non-manifesting N370S GBA1 carriers (GBA1wt/mut ASX). Thus, all lines had the same mutant allele to facilitate comparison. For control purposes, fibroblasts were acquired from age-matched, apparently healthy individuals (GBA1wt/wt). The fibroblasts were categorised according to age. The “young” cohort were obtained from individuals under the age of 60 whereas the “aged” cohort were derived from individuals over 70 years old (exact age denoted in subscripts). A summary of fibroblasts used in this study is provided in Table S1.
2.2. Cell culture
Fibroblasts were maintained in DMEM. SH-SY5Y cells were maintained in a 1:1 mixture of DMEM:Ham's F12 media and 1% (v/v) non-essential amino acids (all from Invitrogen). SH-SY5Y cells with stable knock down of β-glucocerebrosidase were described in [22]. Media were supplemented with 10% (v/v) heat inactivated FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. Cells were cultured at 37 °C in a humidified atmosphere with 5% CO2. In some experiments, cells were cultured for 7–11 days with the irreversible β-glucocerebrosidase inhibitor, conduritol B epoxide (CBE, 10 μM; Sigma–Aldrich). Media, containing CBE, was replenished every 5 (fibroblasts) or 2–3 (SH-SY5Y) days. All cultures were analysed in parallel and fibroblast cultures differed by no more than 2 passages. Prior to experimentation, cells were plated onto glass coverslips (for Ca2+ imaging and immunocytochemistry) or directly into tissue culture flasks (for western blotting). For SH-SY5Y cells, glass coverslips were coated with 20 μg/mL poly-l-lysine.
2.3. Ca2+ imaging
Ca2+ imaging was performed using the fluorescent Ca2+ indicator Fura-2 as described in [6] using HEPES-buffered saline (HBS) consisting of 10 mM HEPES, 2 mM MgSO4, 156 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1.25 mM KH2PO4 and 10 mM glucose (pH 7.4). Cells were stimulated with thapsigargin (Merck), cADPR-AM, synthesised as described previously [23] and GPN (glycyl-l-phenylalanine 2-naphthylamide, SantaCruz Biotech). Where indicated, extracellular Ca2+ was replaced with 1 mM EGTA.
2.4. Western blotting
Western blotting was performed as described in [24]. Blots were incubated with either mouse anti-β-glucocerebrosidase (overnight at 4 °C, diluted 1:500, EMD Millipore), mouse anti-LAMP1 (1 h at RT, diluted 1:500, Santa Cruz Biotechnology) or rabbit-anti-LC3II (overnight at 4 °C, diluted 1:1000, Cell Signalling) primary antibodies. Blots were stripped and re-probed with a goat anti-actin (1 h at RT, diluted 1:500, Santa Cruz Biotechnology) primary antibody. Anti-mouse (Santa Cruz Biotechnology), anti-rabbit (Bio-Rad) or anti-goat (Santa Cruz Biotechnology) IgG conjugated to horse-radish peroxidase were used as the secondary antibodies (1 h at RT, 1:2000).
2.5. Other methods
β-Glucocerebrosidase and β-hexosaminidase enzyme activities were measured using 4-methylumbelliferyl-β-d-glucopyranoside and 4-methylumbelliferyl-N-acetyl-glucosaminide, respectively as described in [22]. Immunocytochemistry using primary antibodies raised to LAMP1 (mouse, 1 h at 37 °C; diluted 1:10, Developmental Studies Hybridoma Bank H4A3 clone supernatant) or LAMP2 (mouse, 1 h at 37 °C, diluted 1:100, Santa Cruz Biotechnology), Lysotracker™ Red staining and confocal microscopy were performed as described in [24], [25].
2.6. Data analysis
The magnitude of Ca2+ release was calculated by subtracting the basal Fura-2 fluorescence ratio prior to stimulation (60 s of data acquisition) from the peak response. The area under the curve was estimated by summating the increases in fluorescence ratio following stimulation over a given period. For thapsigargin, the periods were 750 s and 400 s for fibroblasts and SH-SY5Y cells, respectively. For GPN, the period was 400 s. These analyses were done at the individual cell level over the entire field of view (typically 15 cells). Data were derived from the number of passages stated in the figure legends, averaged over multiple fields of view (n, stated in the figure panels) and presented as mean ± standard error of the mean. Statistical analyses were performed using Minitab 17. Independent-samples t-tests were applied and in the case of multiple comparisons, ANOVA analysis followed by a post hoc Tukey test. p < 0.05 was considered statistically significant.
3. Results
3.1. ER Ca2+ release is disrupted in GD and PD fibroblasts
To examine whether GD and PD pathology is associated with impaired Ca2+ signalling, cytosolic Ca2+ levels were measured in age-segregated, passage-matched patient fibroblasts carrying the N370S mutation (see Section 2). In the first set of experiments, cultures from the younger cohort were used. We estimated ER Ca2+ content by challenging cells with the SERCA inhibitor thapsigargin (1 μM) in Ca2+-free medium. Thapsigargin-evoked Ca2+ release was significantly elevated in GD (GBA1mut/mut55GD) and PD (GBA1wt/mut55PD) cells when compared to cells from an age-matched (55 year old) healthy individual (GBA1wt/wt55) (Fig. 1A). These differences were quantified by measuring the magnitude of the response (Fig. 1B) or the area under the curve (Fig. S1A). To further examine ER Ca2+ release, fibroblasts were stimulated with a cell-permeable derivative of the intracellular Ca2+-mobilising messenger cyclic-ADP ribose (cADPR-AM) [23]. cADPR-AM (25 μM) evoked Ca2+ signals in a proportion of fibroblasts (Fig. 1C). The percentage of cells that responded to cADPR-AM was significantly increased in GBA1wt/mut55PD fibroblasts compared to fibroblasts from an age-matched healthy individual (Fig. 1D). These data identify defects in ER Ca2+ release in both GD and GBA1-linked PD.
Fig. 1.

Pathogenic GBA1 disrupts ER Ca2+ release. (A–D) ER Ca2+ release in GBA1wt/wt55, GBA1mut/mut55GD and GBA1wt/mut55PD cells (young cohort). (A) Cytosolic Ca2+ recordings from individual fibroblasts challenged with thapsigargin (1 μM) from the indicated representative populations. Experiments were performed in the absence of extracellular Ca2+. (B) Summary data (mean ± SEM) quantifying the magnitude of thapsigargin-evoked Ca2+ signals in the indicated number of fields of view. Results are from 5 to 9 independent passages analysing 154–367 cells. (C) Cytosolic Ca2+ recordings from individual fibroblasts stimulated with cADPR-AM (25 μM). Experiments were performed in the presence of extracellular Ca2+. (D) Summary data quantifying the percentage of cells responsive to cADPR. Results are from 2 to 3 independent passages analysing 39–75 cells. (E) Similar to A except thapsigargin-evoked Ca2+ release was assessed in GBA1wt/wt55, GBA1wt/mut58ASX and GBA1wt/mut55PD cells. (F) Summary data from 4 independent passages analysing 46–127 cells. (G) Similar to C except cADPR-evoked Ca2+ release was assessed in GBA1wt/wt55, GBA1wt/mut58ASX and GBA1wt/mut55PD cells. (H) Summary data from 3 to 6 independent passages analysing 73–257 cells. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
A significant number of individuals with heterozygous mutations in GBA1 never develop neurological conditions [9]. ER Ca2+ release was therefore assessed in asymptomatic individuals with heterozygotic mutations in GBA1. Although thapsigargin-evoked Ca2+ release appeared more heterogeneous in GBA1wt/mut58ASX when compared with control GBA1wt/wt55 fibroblasts (Fig. 1E), the mean amplitude of the Ca2+ elevations and the area under the curve did not differ between these cultures and those from an additional asymptomatic individual (GBA1wt/mut59ASX; Fig. 1F, Fig. S1A). Similarly, as shown in Fig. 1G-H, cADPR-AM-evoked Ca2+ release in GBA1wt/mut58ASX fibroblasts was not significantly different to control GBA1wt/wt55 fibroblasts. These data suggest that disrupted Ca2+ homeostasis correlates with PD in the same GBA1 genetic background.
3.2. ER Ca2+ defects are age-dependent
ER Ca2+ release in PD was further examined using fibroblasts from the aged cohort. Unlike the younger GBA1wt/mut55PD fibroblasts, thapsigargin-evoked Ca2+ release in GBA1wt/mut75PD fibroblasts was similar to fibroblasts from the age-matched healthy control (GBA1wt/wt78) (Fig. 2A and B, Fig. S1A). However, we noted that thapsigargin-evoked Ca2+ release in fibroblasts from both GBA1wt/wt78 and GBA1wt/mut75PD was kinetically irregular and larger than Ca2+ release evoked in fibroblasts from younger control subjects (compare with Fig. 1A). To investigate the effect of age on ER Ca2+ release, we examined the effects of thapsigargin in fibroblasts from healthy individuals of increasing age. As shown in Fig. 2E and F, thapsigargin-evoked Ca2+ release increased in an age-dependent manner in fibroblasts from control (GBA1wt/wt) individuals. Thapsigargin responses in the oldest fibroblasts examined (GBA1wt/wt82), closely resembled those from the younger GBA1wt/mut55PD fibroblasts (Fig. 2G). Such findings are consistent with the idea that some features of PD simulate an accelerated form of ageing [26].
Fig. 2.

ER Ca2+ defects are age-dependent. (A) Cytosolic Ca2+ recordings from individual fibroblasts challenged with thapsigargin (1 μM) from representative populations of GBA1wt/wt78 and GBA1wt/mut75PD cells (aged cohort). (B) Summary data 3 independent passages analysing 112–117 cells. (C) Similar to A except, ER Ca2+ release was assessed in GBA1wt/wt82 and GBA1wt/mut80ASX cells. (D) Summary data from 3 independent passages analysing 131–134 cells. (E) ER Ca2+ release from GBA1wt/wt fibroblasts with increasing age. (F) Summary data from 1 to 14 independent passages analysing 30–483 cells. (G) Magnitude of ER Ca2+ release versus age for both the young and aged cohort. All experiments were performed in the absence of extracellular Ca2+.
3.3. ER Ca2+ defects are independent of β-glucocerebrosidase activity loss
Whether pathogenic effects of GBA1 are due to loss of enzymatic function or gain of toxic function is debated [27]. To probe the mechanism of how mutant GBA1 disrupts ER Ca2+ release, the effects of thapsigargin were examined in fibroblasts from healthy controls by reducing the activity of β-glucocerebrosidase using pharmacological and molecular means. Fibroblasts were chronically treated with conduritol B epoxide (CBE, 10 μM), an inhibitor of β-glucocerebrosidase, which reduced β-glucocerebrosidase activity to 6 ± 0.03%. Thapsigargin-induced Ca2+ release after exposure to CBE was unchanged (Fig. 3A and B, Fig. S1B). To extend these studies to a more neuronal context, we examined the effect of CBE on dopaminergic SH-SY5Y cells. As in fibroblasts, thapsigargin-evoked Ca2+ release was not different following CBE treatment (Fig. 3C and D, Fig. S1B) despite substantial reduction in β-glucocerebrosidase enzyme activity to 8 ± 0.4%. To probe further the role of β-glucocerebrosidase, we examined the effect of thapsigargin upon stable knockdown of GBA1 [22]. Reducing the levels of β-glucocerebrosidase did not affect thapsigargin-evoked Ca2+ release (Fig. 3E and F, Fig. S1B). Taken together, these data show that reducing β-glucocerebrosidase enzyme activity, under our experimental conditions, appears not to induce ER Ca2+ dysfunction.
Fig. 3.

Inhibition of β-glucocerebrosidase does not affect ER Ca2+ release. (A) Cytosolic Ca2+ recordings from individual control GBA1wt/wt fibroblasts challenged with thapsigargin (1 μM) from a representative population treated with 10 μM CBE for 8 days. (B) Summary data from 2 independent treatments analysing 87–90 cells. (C–F) Cytosolic Ca2+ recordings from individual SH-SY5Y cells challenged with thapsigargin (1 μM) from a representative population treated with 10 μM CBE for 10–11 days (C) or stably expressing either scrambled shRNA (GBA1+/+) or shRNA targeting GBA1 (GBA1−/−) (E). Summary data from 3 independent treatments analysing 117–204 cells (D) and 3 independent passages analysing 150–143 cells (F). All experiments were performed in the absence of extracellular Ca2+. ns, not significant. Inset (F) is a Western blot using antibodies to β-glucocerebrosidase (top) or actin (bottom) and homogenates (14 μg) from SH-SY5Y cells treated with the indicated shRNA.
3.4. Lysosomal morphology and Ca2+ content is disrupted in GD and PD fibroblasts
Lysosomes are increasingly implicated in PD pathogenesis [10], [11]. We recently identified lysosome morphology defects in LRRK2-PD fibroblasts which we correlated with lysosomal Ca2+ defects [24]. We therefore probed potential physical and functional lysosome alterations in GBA1-PD fibroblasts. Using an antibody raised to the late endosome/lysosome marker LAMP1, lysosome morphology was compared in the fibroblasts from the young and aged cohorts (Fig. 4). Lysosome morphology was altered in the GBA1mut/mut55GD fibroblasts (Fig. 4B) compared to age-matched control fibroblasts (GBA1wt/wt55; Fig. 4A). Lysosome morphology was also altered in GBA1wt/mut55PD fibroblasts (Fig. 4C). In both cases, lysosomes appeared enlarged and clustered. Similar morphological alterations were apparent in the GD and PD cells using an antibody raised to LAMP2 (Fig. S2A–C and F) and in live cells labelled with the acidotrope, Lysotracker (Fig. S2D–F). There was little change in LAMP1 protein levels quantified by Western blotting in either GD or PD fibroblasts consistent with our previous analysis [21], although levels of the autophagic marker LC3II were increased (Fig. S2G).
Fig. 4.

Pathogenic GBA1 disrupts lysosomal morphology. (A–H) Representative confocal fluorescence images of LAMP1 staining (white) in the indicated fibroblasts from the young (A–D) and aged (E–H) cohort. Nuclei were stained with DAPI (blue). Zoomed images are displayed in the right panels. Scale bars, 10 μm. (I) Summary data quantifying LAMP1 intensity as a percentage of the indicated age-matched control (82–654 cells). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Morphological alterations to the lysosomal system were also found in asymptomatic GBA1 carriers (GBA1wt/mut58ASX and GBA1wt/mut59ASX) but to a lesser extent than in GD and PD fibroblasts (Fig. 4D and data not shown). Importantly, lysosome morphology did not differ in healthy, PD and asymptomatic carriers from the aged cohort (Fig. 4E–H). These data are summarised in Fig. 4I. Thus, similar to ER Ca2+ defects, lysosome morphology defects are age-dependent.
To estimate lysosomal Ca2+ content, we challenged cells with the lysosomotropic agent GPN (200 μM) which induces leak of low molecular weight solutes (<10 kDa) in fibroblasts upon hydrolysis by the lysosomal protease, cathepsin C [28]. GPN stimulated complex cytosolic Ca2+ increases, as reported previously [6], and no differences were observed across the GBA1wt/wt55, GBA1mut/mut55GD and GBA1wt/mut55PD fibroblast cultures (Fig. S3A and B). Potential differences in lysosomal Ca2+ content may have been masked due to recruitment of ER-localised receptors upon lysosomal destabilisation [6]. Indeed, in human fibroblasts we have previously shown that lysosomal Ca2+ release triggers Ca2+ responses through IP3, but not ryanodine, receptors [6]. We therefore isolated lysosomal Ca2+ release by blocking IP3 receptors with 2-APB prior to GPN challenge. Under these conditions, GPN-evoked Ca2+ release was largely monotonic and reduced in GBA1wt/mut55PD fibroblasts relative to controls (Fig. 5A and B, Fig. S1C). This reduction was not due to differences in cathepsin C activity/lysosomal permeabilisation because the rate of fluorescence loss in cells loaded with Lysotracker in response to GPN, was similar between fibroblasts cultures (Fig. S3C and D). Activity of β-hexosaminidase was also unchanged in GD and PD cells (106 ± 10% and 103 ± 3% of control, respectively). Similar to our ER Ca2+ estimates, reducing the activity of β-glucocerebrosidase with CBE had little effect on GPN-evoked Ca2+ release in fibroblasts from healthy controls (Fig. 5C and D). We therefore identify Ca2+ defects at the lysosomal level in PD that are likely independent of β-glucocerebrosidase activity loss.
Fig. 5.

Pathogenic GBA1 disrupts lysosomal Ca2+ content. (A–D) Cytosolic Ca2+ measurements from individual fibroblasts stimulated with GPN (200 μM). Experiments were performed in the presence of extracellular Ca2+ and following a 12.5 min pre-treatment with 100 μM 2APB prior to recording. (A) Recordings from a representative population of GBA1wt/wt55, GBA1mut/mut55GD and GBA1wt/mut55PD cells. (B) Summary data from 2 independent passages analysing 72–88 cells. (C) Recordings from a representative population of control GBA1wt/wt fibroblasts treated with 10 μM CBE for 7–9 days. (D) Summary data from 2 independent treatments analysing 43–72 cells. ***p < 0.001. ns, not significant.
4. Discussion
Ca2+ stores represent a major source of Ca2+ signals but their role in PD is largely unknown. In the present study, we identify age-dependent changes in ER Ca2+ release in both type I GD and GBA1-linked PD fibroblasts. Additionally, we report disturbances in lysosomal morphology and lysosomal Ca2+ content in these cells.
Patient fibroblasts represent a robust, tractable system for disease study. They harbour cumulative damage for a given subject, perhaps particularly relevant to late onset neurodegenerative disease. Nevertheless, a limitation of fibroblasts is their non-neuronal nature. Our data demonstrating exaggerated ER Ca2+ release in GBA1-PD fibroblasts however is consistent with a recent report using induced pluripotent stem cell-derived dopaminergic neurons which showed enhanced Ca2+ release to the ryanodine receptor agonist, caffeine [29]. In that study, lines were derived from patients with an L444P mutation in GBA1 and asymptomatic carriers were unavailable. Defects reported here were not manifest in asymptomatic carriers and presented only in the younger patients. We interpret the defect as being “non-additive” with ageing which we report is also associated with similar perturbations in ER Ca2+ signalling. Notably, strategies that increase ER Ca2+ content improve mutant β-glucocerebrosidase folding [30]. Enhanced ER Ca2+ content, although beneficial with respect to protein folding, may render cells more sensitive to apoptotic stimuli and thus link Ca2+ disturbances to cell death.
Reduced lysosomal Ca2+ content in GBA1-PD fibroblasts is similar to that reported in Niemann–Pick type C1 diseased fibroblasts [16] and Presenilin-1 knockout mouse embryonic fibroblasts [31], [32]. Functionally, reduced lysosomal Ca2+ content might affect Ca2+-dependent membrane trafficking events within the endo-lysosomal system [33], thereby accounting for altered lysosome morphology. Similar lysosome morphology alterations have been reported in fibroblasts from patients with mutations in ATP13A2 (PARK9) [34], a lysosomal ATPase and LRRK2 (PARK8) for which evidence of an endolysosomal locus of action continues to accrue [24], [35], [36], [37].
How mutant GBA1 disposes to PD is unclear. Both loss- and gain-of function models have been proposed [27]. For-example, knock-down of β-glucocerebrosidase in mouse models is associated with increases in the substrate glucocerebroside which stabilises α-synuclein, a component of Lewy bodies characteristic of the disease [38]. Concomitantly, α-synuclein also reduces trafficking of β-glucocerebrosidase to the lysosome pointing to a positive feedback loop triggered by a reduction in β-glucocerebrosidase activity that might precipitate disease [38]. However, increases in α-synuclein levels do not always correlate with β-glucocerebrosidase activity [27]. Notably, the E326K mutation in β-glucocerebrosidase, which is linked to early onset PD, has a more modest effect on β-glucocerebrosidase activity than other mutations and does not cause Gaucher disease [39]. Furthermore, we have shown that substrate does not accumulate in the brains of GBA1 carrier-PD patients [40] despite demonstrable reduction in β-glucocerebrosidase activity [41]. That many mutant forms of β-glucocerebrosidase accumulate in the ER supports the alternative gain-of function mechanism for toxicity [27]. Our findings reported here, showing that neither ER nor lysosomal defects were recapitulated upon inhibiting/depleting β-glucocerebrosidase, support such a gain-of-function mechanism for pathogenic GBA1. However, we cannot rule out that residual β-glucocerebrosidase activity (albeit modest) is sufficient to maintain homeostasis.
PD has a complex aetiopathogenesis, which likely results from interplay between genetic and environmental cues. Although it is established that mutations in GBA1 substantially increase risk of developing PD not all carriers succumb. These data strongly suggest that pathology is not a sole consequence of the mutant GBA1 allele. We show here that ER Ca2+ and lysosomal morphology defects identified in PD cells are not present in asymptomatic GBA1 carriers. Thus, defects correlate with pathology despite similar GBA1 status. It remains to be established whether these phenotypes are contributing causal factors for the disease or a consequence. Nevertheless, disruptions in fibroblast Ca2+ store homeostasis and lysosomal morphology described here might serve as biomarkers for GBA1-linked PD given incomplete penetrance in GBA1 carriers. Further work, however, is required using additional patient cell lines to validate our findings.
In summary, we identify age-dependent disturbances in both ER and lysosomal Ca2+ stores of potential relevance to the pathology of PD and GD.
Author contributions
BSK performed the Ca2+ and Lysotracker imaging, immunocytochemistry and Western blotting. MEG and BSK performed enzyme activity measurements, JM and MJWC provided the SH-SY5Y cells. MSB and AM obtained the fibroblasts. DB and GCC synthesised the cADPR-AM. MRD, AHS and SP conceived the study. BSK and SP wrote the paper with input from all authors.
Funding
This work was supported by an IMPACT studentship from UCL (to BSK), Parkinson's UK grants K-1107, K-1412 and H-1202 (to SP, AHS and MRD), Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698 to AHS and MRD), an MRC CoEN award (to AHS). AHS is a NIHR Senior Investigator and is supported by the NIHR UCLH BRC.
Acknowledgements
We thank Laura D. Osellame and Zhi Yao for help with pilot experiments, and Leanne N. Hockey for help with analyses.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ceca.2015.11.002.
Contributor Information
Bethan S. Kilpatrick, Email: bethan.kilpatrick.10@ucl.ac.uk.
Sandip Patel, Email: patel.s@ucl.ac.uk.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Berridge M.J., Lipp P., Bootman M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Clapham D.E. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 3.Lee H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012;287:31633–31640. doi: 10.1074/jbc.R112.349464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel S., Docampo R. Acidic calcium stores open for business: expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010;20:277–286. doi: 10.1016/j.tcb.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel S., Muallem S. Acidic Ca(2+) stores come to the fore. Cell Calcium. 2011;50:109–112. doi: 10.1016/j.ceca.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Kilpatrick B.S., Eden E.R., Schapira A.H., Futter C.E., Patel S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013;126:60–66. doi: 10.1242/jcs.118836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitner E.B., Platt F.M., Futerman A.H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 2010;285:20423–20427. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platt F.M. Sphingolipid lysosomal storage disorders. Nature. 2014;510:68–75. doi: 10.1038/nature13476. [DOI] [PubMed] [Google Scholar]
- 9.Sidransky E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dehay B., Martinez-Vicente M., Caldwell G.A., Caldwell K.A., Yue Z., Cookson M.R. Lysosomal impairment in Parkinson's disease. Mov. Disord. 2013;28:725–732. doi: 10.1002/mds.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beavan M.S., Schapira A.H.V. Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann. Med. 2013;45:511–521. doi: 10.3109/07853890.2013.849003. [DOI] [PubMed] [Google Scholar]
- 12.Osellame L.D., Rahim A.A., Hargreaves I.P., Gegg M.E., Richard-Londt A., Brandner S. Mitochondria and quality control defects in a mouse model of Gaucher disease-links to Parkinson's disease. Cell Metab. 2013;17:941–953. doi: 10.1016/j.cmet.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schapira A.H.V., Olanow C.W., Greenamyre J.T., Bezard E. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014;384:545–555. doi: 10.1016/S0140-6736(14)61010-2. [DOI] [PubMed] [Google Scholar]
- 14.Surmeier D.J., Guzman J.N., Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium. 2010;47:175–182. doi: 10.1016/j.ceca.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korkotian E., Schwarz A., Pelled D., Schwarzmann G., Segal M., Futerman A.H. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 1999;274:21673–21678. doi: 10.1074/jbc.274.31.21673. [DOI] [PubMed] [Google Scholar]
- 16.Lloyd-Evans E., Morgan A.J., He X., Smith D.A., Elliot-Smith E., Sillence D.J. Niemann–Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 17.Shachar T., Lo Bianco C., Recchia A., Wiessner C., Raas-Rothschild A., Futerman A.H. Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov. Disord. 2011;26:1593–1604. doi: 10.1002/mds.23774. [DOI] [PubMed] [Google Scholar]
- 18.Guzman J.N., Sánchez-Padilla J., Chan C.S., Surmeier D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan C.S., Guzman J.N., Ilijic E., Mercer J.N., Rick C., Tkatch T. “Rejuvenation” protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 20.Guzman J.N., Sanchez-Padilla J., Wokosin D., Kondapalli J., Ilijic E., Schumacker P.T. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNeill A., Magalhaes J., Shen C., Chau K.-Y., Hughes D., Mehta A. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137:1481–1495. doi: 10.1093/brain/awu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleeter M.W.J., Chau K.-Y., Gluck C., Mehta A., a Hughes D., Duchen M. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013;62:1–7. doi: 10.1016/j.neuint.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosen D., Bloor-Young D., Squires J., Parkesh R., Waters G., Vasudevan S.R. Synthesis and use of cell-permeant cyclic ADP-ribose. Biochem. Biophys. Res. Commun. 2012;418:353–358. doi: 10.1016/j.bbrc.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 24.Hockey L.N., Kilpatrick B.S., Eden E.R., Lin-Moshier Y., Brailoiu G.C., Brailoiu E. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 2015;128:232–238. doi: 10.1242/jcs.164152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kilpatrick B.S., Eden E.R., Hockey L.N., Futter C.E., Patel S. Methods for monitoring lysosomal morphology. Methods Cell Biol. 2015;126:1–19. doi: 10.1016/bs.mcb.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Collier T.J., Kanaan N.M., Kordower J.H. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011;12:359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sidransky E., Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penny C.J., Kilpatrick B.S., Han J.M., Sneyd J., Patel S. A computational model of lysosome-ER Ca2+ microdomains. J. Cell Sci. 2014;127:2934–2943. doi: 10.1242/jcs.149047. [DOI] [PubMed] [Google Scholar]
- 29.Schöndorf D.C., Aureli M., McAllister F.E., Hindley C.J., Mayer F., Schmid B. iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014;5:4028. doi: 10.1038/ncomms5028. [DOI] [PubMed] [Google Scholar]
- 30.Ong D.S.T., Mu T.-W., Palmer A.E., Kelly J.W. Endoplasmic reticulum Ca2+ increases enhance mutant glucocerebrosidase proteostasis. Nat. Chem. Biol. 2010;6:424–432. doi: 10.1038/nchembio.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coen K., Flannagan R.S., Baron S., Carraro-Lacroix L.R., Wang D., Vermeire W. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J.-H., McBrayer M.K., Wolfe D.M., Haslett L.J., Kumar A., Sato Y. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luzio J.P., Bright N.A., Pryor P.R. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem. Soc. Trans. 2007;35:1088–1091. doi: 10.1042/BST0351088. [DOI] [PubMed] [Google Scholar]
- 34.Dehay B., Ramirez A., Martinez-Vicente M., Perier C., Canron M.-H., Doudnikoff E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gómez-Suaga P., Luzón-Toro B., Churamani D., Zhang L., Bloor-Young D., Patel S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012;21:511–525. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dodson M.W., Zhang T., Jiang C., Chen S., Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012;21:1350–1363. doi: 10.1093/hmg/ddr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schreij A.M.A., Chaineau M., Ruan W., Lin S., Barker P.A., Fon E.A. LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep. 2015;16:79–86. doi: 10.15252/embr.201438714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazzulli J.R., Xu Y.-H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duran R., Mencacci N.E., Angeli A.V., Shoai M., Deas E., Houlden H. The glucocerobrosidase E326K variant predisposes to Parkinson's disease, but does not cause Gaucher's disease. Mov. Disord. 2013;28:232–236. doi: 10.1002/mds.25248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gegg M.E., Sweet L., Wang B.H., Shihabuddin L.S., Sardi S.P., Schapira A.H.V. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015;30:1085–1089. doi: 10.1002/mds.26278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gegg M.E., Burke D., Heales S.J.R., Cooper J.M., Hardy J., Wood N.W. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012;72:455–463. doi: 10.1002/ana.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.