

Abstract
Ribonucleotide reductase (RR) catalyzes the rate-limiting step of dNTP synthesis and is an established cancer target. Drugs targeting RR are mainly nucleoside in nature. In this study, we sought to identify non-nucleoside small-molecule inhibitors of RR. Using virtual screening, binding affinity, inhibition, and cell toxicity, we have discovered a class of small molecules that alter the equilibrium of inactive hexamers of RR, leading to its inhibition. Several unique chemical categories, including a phthalimide derivative, show micromolar IC50s and KDs while demonstrating cytotoxicity. A crystal structure of an active phthalimide binding at the targeted interface supports the noncompetitive mode of inhibition determined by kinetic studies. Furthermore, the phthalimide shifts the equilibrium from dimer to hexamer. Together, these data identify several novel non-nucleoside inhibitors of human RR which act by stabilizing the inactive form of the enzyme.
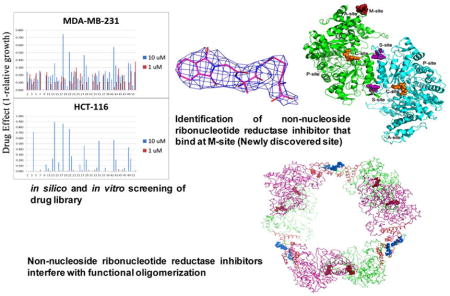
Graphical Abstract
INTRODUCTION
Ribonucleotide reductase (RR) is crucial for rapidly proliferating cells, and inhibition of this enzyme has proven to be an effective strategy for anticancer therapy.1–3 Competitive nucleoside analogue inhibitors such as gemcitabine are some of the few drugs used to treat devastating cancers such as pancreatic cancer.4 Other FDA approved drugs including gemcitabine, fludarabine, clofarabine, and cladarabine are nucleoside analogues that target allosteric sites of RR and irreversibly inhibit DNA replication by incorporation and chain termination.5–10 So far, all of the clinically used drugs that target the large subunit (hRRM1) of hRR are nucleoside analogues.11 Nucleoside analogues lack on-target specificity for hRR.11 For example, gemcitabine is also known to cross-react with numerous other enzymes in addition to hRR including DNA polymerase,12,13 deoxycytidine deaminase (dCMP deaminase), thymidylate synthase, CTP-synthase,14 and topoisomerase 1.15 We wish to discover a novel class of non-nucleoside inhibitors of hRR.16
The development of RR inhibitors has necessarily advanced along with our understanding of its structure and its enzymology, in particular its allosteric regulation. Ribonucleotide reductase is a multiprotein enzyme consisting of a large subunit called hRRM1 (α) containing the catalytic site and allosteric sites and a small subunit called hRRM2 (β) that houses the free radical required for initiating radical-based chemistry.17 The hRRM1 subunit catalyzes the conversion of four ribonucleoside diphosphates (UDP, CDP, GDP, and ADP) to their respective deoxy forms. During the S-phase of the cell cycle, these reduction reactions are allosterically controlled by binding of nucleotide triphosphates to two different sites on RR.18 The S-site is located at the dimer interface of hRRM1 and is involved in allosterically regulating substrate binding specificity (Figure 1A).18–23 ATP is an allosteric activator, while dATP is an allosteric inhibitor, where both bind to the A-site (Figure 1A).18,24
Figure 1.

hRRM1 drug binding sites and fluorescence quenching identifies RR inhibitors. (A) Structure of the hRRM1 dimer with drug-target sites mapped. The M-site is the binding site for the new class of modulators that is the subject of this study. The A-site controls activity. The S-site controls specificity. The C-site is the catalytic site. The P-site binds the smaller R2 subunit derived peptide. (B and C) Tryptophan fluorescence quenching of hRRM1 in the presence of compounds 4 and 10, respectively. (D and E) No tryptophan fluorescence quenching of hRRM1 by compounds 100207 and 184612.
Recent studies with RR have revealed the importance of oligomerization and its regulation as well as its inhibition.25–31 Although the multimerization of RR is still a subject of investigation, the prevailing model is that RR minimally functions as an α2β2 complex. At physiological concentrations of ATP (3 mM), hRRM1 exists predominantly as an active hexamer with a small population of dimer present.25,27–29 When dATP is bound, the large subunit has also been shown to exist as a dimer and hexamer,25,27,29 although these forms of the enzyme are inactive, while baculovirus expressed mouse RR1 was observed to exist as a tetramer.25,32
Stabilization of the inactive dATP bound form might be an effective strategy for RR inhibition. In a recent report, the dATP hexamer was proposed to be stabilized by the protein IRBIT in cells.33 Recently, hexamer formation has been shown to be important for inhibitors such as gemcitabine and clofarabine binding to RR.28,30 For example, gemcitabine was shown to inactivate hRRM1 by inducing α6β6 oligomers, while clofarabine was shown to bind hRRM1 hexamers with nanomolar affinity.28,30 Indeed, the importance of hexamer formation was highlighted in a recent paper characterizing the non-nucleoside drug 5-NITP which is a moderate RR inhibitor.34 While this drug was shown to induce hRRM1 dimers, its low inhibitory potency is proposed to be due to its inability to form inactive hexamers.34
Because of the importance of hexamerization in drug-mediated inactivation, targeting the hexamer interface to develop specific small molecules that bind preferentially to the dATP-induced hexamer can potentially shift the equilibrium toward the inactive conformation. Similar strategies have been used against porphobilinogen synthase (PBGS), phenylalanine hydroxylase, and HIV integrase to discover small molecules that bind at the oligomeric interfaces.35,36 These proteins conform to themorpheein model of allostery,35,37 where the change of oligomeric state is a prerequisite for allosteric regulation.
Here, we describe a method that combines virtual screening with hit validation by biophysical methods, RR activity assays, and growth inhibition using cell culture. In this method, the hexamer interface was chosen as the docking site for virtual screening (M-site on Figure 1A and Figure 2A), and the top 51 of the top 76 hits were subjected to growth inhibition assays to assess cellular uptake and anticancer properties. The top 76 hits obtained from the virtual screen were also subjected to fluorescence quenching assays to verify binding to hRRM1. Collectively, these techniques yielded a broad group of compounds possessing redundant functional group architecture. From this subset, we identified 10 structurally unique chemical scaffolds and subjected themto in vitro enzymatic inhibition assays. We report compounds that exhibit micromolar affinity against RR where PB piperazine (compound 1) demonstrated cytotoxicity against HCT-116 cell line with an IC50 of 2.0 μM. Moreover, we were able to derive the crystal structure for OxoIsoIndoLys (compound 4) which binds at the proposed hexamer interface at the N-terminus of hRRM1, suggesting that we have discovered a new RR modulator that binds at a previously unidentified site (Figure 1A). As these hits are nonnucleoside in nature and are unique chemical entities, they enable us to use a chemical biology platform guided by structure to develop new highly potent anticancer agents.
Figure 2.

Compound 4 interactions with hRRM1. (A) Model of the hRRM1 hexamer based on the S. cerevisiae RR1 hexamer structure. Ribbon diagram of the hRRM1 hexamer packing arrangement. hRRM1 monomers are green and magenta. All of the four helix ATP-binding cones are red. The 16 N-terminal residues at the hexamer interface are in cyan. Effectors (TTP) bound at the S-site are drawn in brick red spheres, and compound 4 at the hexamer interface is drawn in blue spheres. (B) The |F0| − |Fc| electron density for the phthalimide compound (blue) in complex with hRRM1 orthorhombic crystals. Density contoured at 3σ defines the phthalimide binding to hRRM1. (C) 2|F0| − |Fc| electron density (blue) of the phthalimide compound contoured at 1σ after refinement in the phthalimide-hRRM1 orthorhombic complex. (D) Illustration of the A-site and M-site binding by dATP and compound 4, respectively, Compound 4 is shown in magenta, and dATP is in yellow. (E) Lig plot analysis of compound 4 interactions with hRRM1. The phthalimide compound is shown in purple, carbon atoms in phthalimide are shown in black, oxygen atoms are in red, and nitrogen atoms are in blue. An amino acid residue from hRRM1 interacting with the phthalimide is shown in red.
RESULTS
In Silico Screening Targeting the RR Hexamer Interface
The Cincinnati library consisting of 350,000 compounds was screened in silico using the Schrödinger software suite. The homologous model of the hRRM1 hexamer was constructed using the S. cerevisiae dATP-induced hexamer structure (PDB ID: 3PAW). The docking site was defined as the inactive dATP hexamer interface that consists of the N-terminal 16 residues from adjacent dimers (Figure 2A). When determining hits, we carefully examined the docking poses (Figure S4) where common interactions were a good indication of a consensus binding site. For example, residues Ile 44, Gln 45, Met 1, His 2, Val 51, and Val 43 interact with all 10 compounds in Table 1, which is a good indication that they are binding at the same site. The top 90 hits were subjected to a PAINS filter (http://cbligand.org/PAINS/), which identified 14 hits as violators that were then removed leaving 76 hits.38 A summary of the results from Schrödinger are provided in Table S1. All top ranking hits are referred to by their corresponding Cincinnati library GRI numbers as described in Supporting Information. Compounds discussed in the main text are identified in Table 1.
Table 1.
Identification of 10 Novel hRRM1 Inhibitors Using in Silico Docking, Fluorescence Quenching, RR Inhibition Assays, and Growth Inhibition
| Structure | Docking scores (Schrödinger) | % Fluorescence quenching KD (μM) | Drug Effect (avg)a | RR IC50 (μM) | |
|---|---|---|---|---|---|
| (10 μM) | (1 μM) | ||||
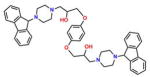 Compound 1 (PB-Piperazine) |
−7.68 | 15; 35.55 ± 3.57 | 0.880 | −0.396 | 23.9 ± 1.1 |
 Compound 2 (tetraHThioDIM) |
−5.28 | 31; ND | 0.531 | −0.199 | 61.74 ± 1.5 |
 Compound 3 (S-DiTDB) |
−4.04 | 34; ND | 0.690 | 0.072 | 47.2 ± 2.1 |
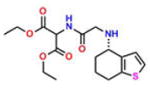
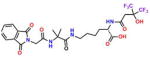 Compound 4 (OxoIsoIndoLys) |
−4.19 | 33; 9.68 ± 2.1 | 0.608 | 0.057 | 32.2 ± 1.3 |
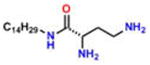
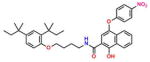 Compound 5 (ButHyNitNap) |
−4.67 | 30; ND | 0.398 | 0.038 | 21.8 ± 1.1 |
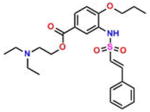 Compound 6 (DPSP) |
−5.28 | 50; 55.29 ± 8.25 | 0.043 | −0.033 | 44.0 ± 5.0 |
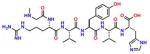 Compound 7 (NmetGAVTVH) |
−7.28 | 43; ND | 0.158 | 0.108 | 45.2 ± 1.2 |
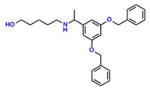 Compound 8 (BoPEAP) |
−4.67 | 35; 10.82 ± 1.86 | 0.036 | −0.076 | 23.6 ± 1.4 |
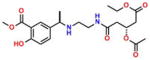 Compound 9 (AEOHydBen) |
−5.84 | 29; ND | 0.096 | 0.098 | 27.2 ± 1.2 |
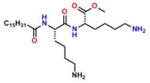 Compound 10 (MePAMLL) |
−5.69 | 32; ND | −0.089 | −0.219 | 35.7 ± 1.9 |
| Hydroxyurea (control) | NA | NA | NA | NA | 997 ± 0.11 |
Drug effect values are averaged from MDA-MB-231 and HCT116 cell lines. ND: KD’s not determined by fluorescence quenching due to the lack of sufficient quantities of the compound
Analysis of compound binding using intrinsic protein fluorescence
The top 76 hits from the in silico screen were subjected to fluorescence quenching assays for binding to hRRM1 (Table S1). Ligands that exhibited 25% or more quenching were considered to have sufficient affinity for hRRM1. On the basis of this criterion, 51% of the ligands tested were considered as binding to hRRM1. As shown in Figure 1B–C and Table S1, compound 4 shows 35% quenching, and compound 10 shows 40% quenching. The compounds that did not show any quenching (Figure 1D and E) were not selected for further screening or studies. To test whether the observed quenching of tryptophan fluorescence of hRRM1 was due to binding controls, the ability of the compounds to quench the fluorescence of the free tryptophan analogue NATA was measured, and the binding of selected compounds was confirmed by thermal denaturation of hRRM1 in the presence of compounds (data not shown).39 Furthermore, nonspecific and artificial inhibition was eliminated by testing two unrelated compounds to the 76 hits using fluorescence quenching (Figure S1) which demonstrated no binding. Four of the 10 compounds reported in Table 1 were subject to KD determination using fluorescence quenching. The KD’s ranged from 10–55 μM (Table 1 and Figure S5).
Chemical Classification and Inhibitory Potency of Non-nucleoside Ligands Inhibiting RR Reveals Broader Pharmacophore Diversity
On the basis of their structure, the collection of hits can be broadly classified into 10 groups (Table S2) with distinct chemical scaffolds (see Table S7 and Figure S2 for HRMS and 1H NMR data). A group of fluorenyl piperazines represented by PB-piperazine (compound 1), which consists of a C2-symmetric p-cresol core tethered to two glycerol units that are derivatized as their corresponding fluorenylpiperazine units on either ends. The next class represented by TetraHThioDIM (compound 2) featured varying substituent groups on a tetrahydrobenzothiophene heterocycle. The next class contained a diamino butanamide moiety connected to aliphatic chains of varying lengths. A representative member of this class is S-DiTDB (compound 3), generally referred to as the DiaminoTDBamide class of inhibitors. The class represented by compound 4 (OxoIsoIndoLys) is uniquely defined through the presence of the phthalimide ring system at one end of the molecule and a hexafluoromonoketide group at the other terminus, tethered with an L-lysine-α,α-dimethylglycine dipeptide scaffold. ButHyNitNap (compound 5), generally referred to as the Hydroxy Naphthamide class, is representative of a class consisting of an o-naphthamide functionality featuring a polar p-nitrophenol substituent and tethered to a hydrophobic tert-pentyl phenoxy group through an amide linkage. An additional class of molecules is styrenyl sulfonamides, represented by DPSP (compound 6, with a general name, DPS-benzoate) possessing a trans-configured sulfonamide moiety. Nmet GAVTVH (compound 7) is peptidyl-like containing an N-methyl-acetylated guanidine-containing arginine at one of its termini and a series of L-amino acids terminating with L-histidine at its carboxy terminus. A class is defined by a (3,5-bis(benzyloxy)-phenyl-ethyl) amino pentanol group, represented by BoPEAP (compound 8). A linear 3-hydroxy-keto amide represented the next class we identified, which also featured a peptidyl core and an acetoxy-ethoxy-oxopentamide scaffold, represented by AEOHydBen (compound 9) (generally referred to as the AcetoxyHydBenzoate class). MePAMLL (compound 10) was defined by L-lysinyl L-lysinate groups containing an aliphatic chain of varying length (C15H31 for instance). In addition to the chemical classification names, the pharmacologic properties (including AlogP, polar surface area, and the corresponding binding efficiency) for each representative member are listed in Table S2.
RR Inhibition
A representative of each class was tested for enzyme inhibition and revealed approximate IC50 values in the micromolar range (Table 1 and Table S3). As a positive control, the well-known RR drug hydroxyurea’s IC50 was determined using our two-point IC50 derivation method, which was in extremely good agreement with published results29 (Table 1). Surprisingly, among the top four of the most potent inhibitors, compounds 5 and 8 and 1 and 9 (21.8, 23.6, 23.9, and 27.2 μM RR IC50, respectively), there is little structural similarity. They all possess multiple flat aromatic rings with 5 and 8 having distinct structural features that impart high hydrophobicity at one terminus and a polar functionality at the other. Compounds 1 and 9 like-wise possess distinct structural features yet have similar potency against the catalytic RR. The lack of apparent correlations between structural parameters such as polar surface area, solubility property (AlogP), and RR inhibition suggests that identifying pharmacophore a priori would be difficult.
Growth Inhibition of Established Cancer Cell Lines Including MDA-MB-231, HCT116, A549, and Panc1
Approximately 51 compounds were screened for their ability to inhibit growth and/or induce cell death of well-characterized cell lines representing common cancer types that are generally difficult to treat (triple negative breast cancer and colon cancer). Pancreatic cancer was also considered informative because gemcitabine is a core component of the current standard of care chemotherapy for this disease. From these screens, compounds were identified that showed significant (>50%) growth inhibitory activity against both MDA-MB-231 (triple negative breast cancer) and HCT-116 (DNA mismatch repair deficient colon cancer) cell lines at 1 μM, 10 μM, or 50 μM after a three day continuous exposure (Figure 3). As shown in Table S4, approximately 64% of the compounds did not show any significant growth inhibitory activity at 50 μM, and an additional 36% did not show significant activity at 10 μM. Although none of the compounds showed significant activity at 1 μM, 8.5% of the compounds did show significant activity at 10 μM. Figure 4 shows a more detailed growth inhibition study in the initially screened cell lines (MDA-MB-231 and HCT116) as well as two additional cell lines (A549 and Panc1) for compound 1. The median effect doses (Dm) calculated using Calcusyn 2.0 are shown in Table S5.
Figure 3.

Growth inhibition of MDA-MB-231 and HCT-116 cancer cells in a moderate throughput screen of candidate RR inhibitors. Cells were treated with 3 doses of candidate drugs (1 μM, 10 μM, and 50 μM) for 3 days in a standard growth inhibition assay in 96 well plates. Growth inhibition in duplicate wells of each drug/dose/cell line was assessed by measuring relative DNA content per well compared with that of untreated cells. Drug effect (1-relative growth) is plotted for the 10 μM and 1 μM dose groups (50 μM groups were not included for clarity).
Figure 4.

Growth inhibition of cancer cell lines by compound 1. Cells were seeded into 96 well plates (2000 cells/well) and the following day incubated with the indicated concentrations of gemcitabine alone or in combination with compound 1 for 24 h. At this time, drugs were removed and replaced with control media in the gemcitabine alone groups, or compound 1 at the indicated concentrations in the compound 1 alone or combination groups for an additional 48 h. Relative growth was assessed by measuring DNA content in each well. Each drug concentration was assayed utilizing five replicates for each cell line. Results are representative of at least 2 experiments.
Compound 1 demonstrated the greatest efficacy in this set of cell lines. Interestingly, compound 1 showed a very dramatic dose–response in three of the cell lines, with little growth inhibitory activity below 5 μM and nearly 100% growth inhibition at the next highest dose (10 μM). This is in contrast to the more gradual growth inhibitory activity of the other compounds tested in these experiments.
Combination studies were undertaken to determine if sublethal amounts of compound 1 could enhance the cytotoxicity of other agents including gemcitabine. MDA-MB-231 and HCT-116 cells were treated with sublethal doses of compound 1 (2.5 μM for MDA-MB-231 and 1.0 μM for HCT-116) in addition to a standard range of gemcitabine doses. Gemcitabine containing media were removed after 24 h, and media containing compound 1 was replaced and remained on the cells for the duration of the experiment (72 h). As shown in Figure 4 and Table S5, the addition of sublethal amounts of compound 1 enhanced the cytotoxicity of gemcitabine, decreasing the relative Dms by 90% (MDA-MB-231, gemcitabine alone 0.92 μM; to 0.1 μM for gemcitabine plus compound 1).
X-ray Structure Determination of the Phthalimide-Based Compound 4 in Complex with hRRM1
Of our 10 potential hits (Table 1), compound 4 (a phthalimide) was then subjected to X-ray crystallography studies in an effort to obtain - a crystal structure of hRRM1 in complex with these M-site modulators.
The X-ray structure of compound 4 was determined to 3.7 Å resolution in complex with hRRM1 (Table 2). The binding of the compound was modeled to a homologous model derived from the hexameric S. cerevisiae structure that we had determined by X-ray crystallography, reported in 2011. Since S. cerevisiae and human enzymes share a sequence identity of 68% and structural homology greater than 80%, where the RMSD between the Cα atoms is less than 2 Å, it is easy to superimpose each RR1 dimer from one species to the other; hence, the same orientation matrix relating the dimeric structures from each species can be used to transpose the inhibitor to its correct location in the model. As the hRRM1 crystal form belongs to the orthorhombic class of crystals, the structure was easily transformed into the hexameric form of the homologous model using superposition (Figure 2A). This can be easily done by superimposing the orthorhombic dimer onto each of the three hexameric dimers in the coordinates PDB ID 3PAW. Usually, when protein molecules are superimposed, the bound ligands are automatically placed in the correct orientation, providing a model complex structure. This transformation is required as the direct solution of the complex in the hexameric crystal form would lack the resolution (approximately 6 Å) to provide useful molecular details. Upon several rounds of refinement, the 2FO − FC and FO − FC Fourier omit maps revealed that there was ligand density at the proposed N-terminal hexamer interface (Figure 2B and C). Almost the entire compound 4 was visible in the 2FO − FC Fourier difference electron density map with the exception of the two terminal fluorinated methyl groups. Compound 4 makes mostly nonpolar interactions with the N-terminus of one dimer at the hexamer interface (Figure 2D,E). Figure 2E depicts a ligand plot of the protein–ligand interactions. A comprehensive list of interactions is given in Table S6, while the crucial interactions are summarized below. The oxygen atom which is bound to the C12 atom of the phthalimide ring forms a polar contact with the main chain nitrogen atom of Ala 49. Additionally, hydrophobic interactions are observed among C9, C10, C11, and C12 of compound 4 and Ala 48, Ala 49, and Ala 53. The Cβ atom of Ala 48 forms hydrophobic interactions with C10 of compound 4 at a distance of 3.5 Å. The nitrogen atom of Ala 49 forms hydrophobic interactions with C9, C10, C11, and C12 of the phthalimide ring at a distance of 3.5, 3.6, 3.7, and 3.6 Å, respectively (Figure 2D,E). Additionally, Cβ of Ala 49 interacts with C11 and C12 atoms of the phthalimide ring at 3.6 and 3.7 Å, respectively. The Cβ atom of Ala 53 forms a van der Waals interaction with O7 of compound 4. Also, residues that span the loop region formed by residues 45–52 are believed to be an important part of the dimer–dimer interface in the hRRM1 hexamer.29 The surface accessible area of the phthalimide ligand when bound to the protein was 83 Å2 derived with the AREAIMOL program using the solvent/probe radius to 1.4 Å.40
Table 2.
Data Collection and Refinement Statistics for X-ray Crystal Structures of hRRM1 Bound with Compound 4
| Cell Dimensions | |
|---|---|
| space group | P212121 |
| a, b, c, (Å) | 72.21, 112.11, 218.27 |
| wavelength (Å) | 0.98 |
| resolution (Å) | 200.00–3.70 |
| monomer per asymmetric unit | 2 |
| unique reflections | 19896 |
| Rsyma | 21.8 (89.7)c |
| I/σ(I) | 28.8 (4.4) |
| (%) completeness | 95.5 (95.7) |
| redundancy | 3.2 (3.0) |
|
| |
| refinement | |
|
| |
| number of reflections | 18327 |
| Rwork/Rfreeb | 23.2/28.9 |
| number of atoms | 11384 |
| protein | 11296 |
|
| |
| RMS deviation from ideal | |
|
| |
| bond length (Å) | 0.009 |
| bond angle (deg) | 1.78 |
|
| |
| Ramachandran plot, residues in (%) | |
|
| |
| core regions | 80.6 |
| allowed regions | 17.5 |
| generously allowed regions | 1.5 |
Rsym = ΣhklΣi/Ii(hkl) − 〈I(hkl)〉/ΣhklΣiIi(hkl), where Ii(hkl) is the ith observation of reflection hkl, and 〈I(hkl)〉i is the weighted average intensity for all observations i of reflection hkl.
Rwork and Rfree = Σ||Fo| − |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. For the calculation of Rfree, 5% of the reflection data were selected and omitted from refinement.
Values in parentheses are used for the highest-resolution shell.
Mechanism of hRRM1 Inhibition by Compound 4
The mechanism of inhibition of hRRM1 by compound 4 was analyzed using steady-state kinetics (Figure S3). A plot of reaction velocity versus substrate concentration shows that the vmax is reached at NDP concentrations greater than 1 mM. Likewise, the plots for assays in the presence of compound 4 also showed substrate saturation at NDP concentrations greater than 1 mM; however, the kcat in the presence of compound 4 decreases in response to increasing inhibitor concentration (0.67:1 at 32 μM; 0.30:1 at 64 μM) consistent with noncompetitive inhibition. A double reciprocal plot further demonstrates that compound 4 results in a decrease in kcat while Km is minimally affected. To quantitatively assess the mode of inhibition, the alpha value for the data set was calculated. A value of α = 1 denotes noncompetitive inhibition, α ≫ 1 denotes competitive inhibition, and α ≪ 1 denotes noncompetitive inhibition. An alpha value of 1.047 was obtained for compound 4 further supporting the interpretation of a noncompetitive mode of inhibition.
Next, gel filtration experiments were conducted to study the impact of compound 4 on the oligomeric state of hRRM1 (see Figure 5). hRRM1 mainly exists as a monomer (greater than 90%) with a small fraction of dimer (less than 10%) (Figure 5A). The addition of 1mMphthalimide results in an increased population of the dimer compared to hRRM1 alone (approximately 40%) (Figure 5C and D). As previously observed,29 the addition of 50 μM dATP results in hexamer formation (Figure 5C). Upon addition of 1 mM phthalimide, the dimer peak diminishes significantly, while the hexamer appears to be enhanced considerably (Figure 5D). This is observed when the area under the dATP hexamer peak in the presence of phthalimide was integrated and compared to the area under the native dATP hexamer peak (Figure 5C and D). In the presence of dATP, the area under the hexamer peak is three times that of the dimer peak. Upon addition of phthalimide, the area under the hexamer peak increases to seven times that of the dimer (Figure 5C and D) indicating that the presence of compound 4 can affect hRRM1 multimerization equilibrium.
Figure 5.

Effect of phthalimide binding on oligomerization in hRRM1 using gel filtration chromatography. (A) Chromatogram of hRRM1 with 1 mM concentration of compound 4 is shown in red, where the native hRRM1 in the absence of compound 4 is shown in black. (B) Standard curve for the determination of molecular masses (Mr) of RR. Kav = (Ve − V0)/(Vt − V0), where Ve = elution volume, V0 = void volume, and Vt = total volume. (C) hRRM1 hexamerization in the presence of dATP; at 50 μMdATP, the hexamers species are predominant with a small amount of dimers. The hexamer to dimer ratio based on integration of the peaks is approximately 3-fold. (D) The chromatogram of hRRM1 in the presence of 1 mM compound 4 and 50 μM dATP. The hexamer to dimer ratio based on integration of the peaks is approximately 7-fold.
DISCUSSION AND CONCLUSIONS
Ribonucleotide reductase is a major cancer target. Drugs such as hydroxyurea, fludarabine, clofarabine, gemcitabine, and cladarabine are FDA-approved drugs for the treatment of cancer that target RR.5,7,41,42 Hydroxyurea targets the small subunit of RR, while the others are nucleoside analogues that target the large subunit (Figure 1A). Our aim was to discover a class of small-molecule inhibitors that targeted hRRM1.
In this study, we screened for small molecules that target RR using virtual screening where the hits were confirmed by fluorescence quenching and RR inhibition assays. We targeted the hexamer interface of RR in our in silico screen (Figure 1A and Figure 2A). Recent reports have revealed the advantage of targeting protein–protein interfaces in drug discovery.43–45 Out of the 76 compounds that were screened for binding to hRRM1 using fluorescence quenching, 51% of them produced at least 25% quenching indicating that they were binders (Table S1). We were able to obtain sufficient amounts for compounds 1, 4, 6, and 8 to determine the dissociation constant using fluorescence quenching. These compounds’ KD’s range from 10–55 μM (Figure S5).
As most of the results were considered chemically redundant, the compounds were condensed into groups of unique chemical classes (Table 1). From a medicinal chemistry perspective, a few of the scaffolds represented in this group of 10 inhibitors have prior non-RR medicinal activities. The ethyl ester version of compound 1 (with PubChem ID: NSC-282192 and CAS 7325-88-4) was identified through NCI’s anticancer screen against mec2-1, rad50, and rad14 assays. Related compounds have also been reported as vectors for cell transformation and as antimicrobial agents.46,47 Compound 3 has a C14 alkyl chain; however, its C12 analogue has been reported in the literature as antibacterial. The peptidyl scaffold presented in compound 7 is a commonly observed substructure in multiple angiotensin-related peptides. Compound 6, the styrenyl sulfonate DPS Benzoate is an analogue of proparacaine, that has activity in ion channels and aryl hydrocarbon receptor and sigma-1-receptor modulation. No definitive medicinal activity reports are available, in the context of inhibition against RR, for compounds 4 (OxoIsoIndoLys), 5 (BHNaphthamide, CAS: 18643-47-5), 8 (BoPEAP), and 9 (AEOHydBen), and this study therefore is the first to report them.
Of these, compound 5 (ButHyNitNap) was the most potent inhibitor of RR with an IC50 of 21 ± 1.1 μM. In general, the IC50’s of these compounds ranged from 21–60 μM, closely shadowing their KD’s. This suggests that the potency could be enhanced using a medicinal chemistry and/or structure-based drug design approach.48 Although cocrystallization attempts with the phthalimide (compound 4) derivative were conducted, they did not produce cocrystals possibly due to the high percentage of DMSO, which was essential for solubilizing the ligand.
Soaking experiments with the phthalimide derivative successfully yielded its X-ray structure defining its binding site on the hRRM1 hexamer interface (Figure 1A and Figure 2A–C). The structure reveals that the phthalimide binds at a surface pocket interacting with the β-cap at the ATP-binding cone. The surface accessibility of the free ligand is 606 Å2 where upon binding the protein, the surface accessibility is reduced to 461 Å2 indicating that 24% of the ligand is buried upon binding hRRM1, suggesting that the ligand binds in a surface pocket (Figure 2A). The loop including residues 47–49 in the β cap has been identified as an important region crucial for dATP and ATP binding, required for inactivation and activation of the enzyme, respectively.49 The pocket defined by phthalimide binding has not been previously observed as a ligand binding site for hRRM1. However, this binding pocket lies within approximately 8 Å of the activity site. Figure 2D shows the proximity and relationship between the M-site and A-site. Portions of the N-terminal β-cap (1–14, 48–51) and helix H3 (residues 53–70) are shared between the two sites (Figure 2D). Interestingly, a surface pocket has been identified in a similar study for nuclear receptors where modulators bind.50
Our structural findings were further confirmed by analyzing the mechanism of inhibition of hRRM1 by compound 4 using steady-state kinetics (Figure S3). The velocity over substrate plots at multiple inhibition concentrations and the Lineweaver–Burk plots have profiles reminiscent of noncompetitive inhibitors where Km remains the same, while kcat changes. On the basis of this mode of inhibition, compound 4 will bind both the free enzyme and the enzyme–substrate complex. This model is consistent with our X-ray crystal structure where compound 4 binds at a site independent of the catalytic site.
Targeting protein–protein interfaces such as the hexamer interface is gaining momentum in the postgenome era due to the abundance of such targets in the human genome.43,44 Although in the past protein–protein interface drug targets have been considered to be extremely challenging, new breakthroughs in drug discovery have produced several promising examples that are progressing through the late stages of clinical trials. The flat shapeless surfaces of the protein–protein interface were considered to be a major hurdle for drug design.43,45 However, there are hotspots within the protein–protein interface of a few residues that can be targeted by small-molecule fragments to obtain high affinity inhibitors.44,51 The structure of compound 4 suggests that the phthalimide fragment binds in such a hotspot at the hexamer interface, allowing us to design highly potent RR modulators (Figure 2B–E). Fragment-based drug design is considered a prudent strategy for targeting protein–protein interfaces.43 The phthalimide structure provides a good starting point for fragment-based drug design. Although the resolution of our structure is limited to 3.7 Å, similar resolution structures in combination with isothermal titration calorimetry (ITC) have been successfully used with the acetylcholine binding protein in fragment-based drug design.52
The impact of the phthalimide compound on the oligomeric states of hRRM1 was investigated using gel filtration chromatography (Figure 5). Our studies show that the addition of the phthalimide into nucleoside-free hRRM1 shifts the equilibrium from monomer to dimer (Figure 5A). This is an interesting finding, as our structure (Figure 2) reveals that the phthalimide binds at the hexamer interface, which is far from the dimerization domain near loop 1 (Figure 1A). These results indicate that phthalimide binding at the hexamer interface possibly stabilizes and promotes enhancement of the dimer population. Our studies on how the phthalimide affects the dATP hexamer is shown in Figure 5C and D. We observed that the dimer to hexamer equilibrium shifts in favor of the hexamer. These results support the notion that the phthalimide fragment binding at the hexamer interface strengthens the inactive hexamer formation. Proteins such as PBGS, phenylalanine hydroxylase, HIV-integrase, and pyruvate kinase, which follow the morpheein model of allostery, also are regulated by oligomerization, and small-molecule modulators have been shown to shift the equilibrium from lower order to higher order oligomers.35–37
Independently, 51 of the 76 hits from the in silico screen were tested for growth inhibition and cytotoxicity using cultured cancer cell lines (Figure 3). Compound 1, a fluoronyl piperazine, was observed to enhance the cytotoxicity of gemcitabine when administered together (Figure 4). This observation suggests that RR modulators may be able to enhance the toxicity of existing drugs, when administered in combination. By varying the structural scaffold of these compounds systematically, more optimal structures may be found. Such studies are currently being pursued for two of most promising hits identified from this study.
In summary, we have conducted an in silico screen of potential inhibitors of hRRM1. Our studies have identified 10 potential hits that target hRRM1 with low to medium micromolar IC50’s. Remarkably, no 2 of these 10 compounds resemble each other closely. The structure of the phthalimide compound bound to hRRM1 confirms that it binds at the hexamer interface. In general, most of the compounds tested inhibited RR with IC50 values in the micromolar range. This bears implications for exploration of these chemical spaces as novel non-nucleoside inhibitors of RR. Future work will involve the study of the structure–activity relationship (SAR) and the mechanism of inhibition of the remaining nine potential hits (Table 1) using enzyme kinetics and X-ray crystallography. These studies will definitively identify their respective binding sites. The strategy used in this study can be adapted to obtain novel lead compounds directed against hRRM1 using additional chemical libraries.
EXPERIMENTAL SECTION
Virtual Screening of the Cincinnati Library against hRRM1
In order to conduct virtual screening against hRRM1, we used a homologous model of the dATP-induced hexamer that was based on the S. cerevisiae structure (Figure 2A29). S. cerevisiae shares a 68% sequence identity and a greater than 80% structural homology with the human enzyme. The model was made by substituting the hRRM1 sequence onto the S. cerevisiae structure followed by energy minimization.
In silico docking of the Cincinnati chemical library (formerly the Proctor & Gamble chemical library) was performed independently using the Glide docking module of the Schrödinger 9.3 modeling software suite.53–55 The hits were scored using a docking function and a glide scoring function (glide score). The docking process is described in further detail in the Supporting Information. When considering the best hits, more weight was given to careful examination of the docking poses; especially, consensus interactions with the same residues defining the docking site were high in our rankings. The top 90 hits were subjected to the PAINS filter using the Web site http://cbligand.org/PAINS/.38 This filter removed 14 hits.
Protein Expression and Purification
The hRRM1 protein was expressed in E. coli BL21-codon plus (DE3)-RIL cells and purified using peptide affinity chromatography to homogeneity as previously described by Fairman et al.29 The hRRM2 protein was also expressed in E. coli BL21-codon plus (DE3) cells and purified to homogeneity using Ni-NTA affinity chromatography, and the protein concentrations were measured as described.56
Fluorescence Quenching
Tryptophan fluorescence spectra of hRRM1 at 0.1 mg/mL in 50 mM Tris, pH 8.0, 5% glycerol, 5 mM MgCl2, and 10 mM DTT (Assay buffer) were recorded using a Jobin Yvon-Spex fluorescence spectrophotometer by exciting the sample at 295 nm. The protein samples were treated with ligand at a fixed concentration of 50 μM with the 74 compounds obtained from the virtual screen. The spectra were corrected for the inherent fluorescence contributions made by the ligand. Compounds exhibiting quenching greater than 25% were kept for further evaluation. We have used 0.1 mg/mL bovine serum albumin (BSA) and 1 μM N-acetyltryptophanamide (NATA) as controls. Controls for nonspecific or artificial inhibition were tested by subjecting two independent phthalimide derivatives derived from an unrelated chemical library to the Cincinnati library using the fluorescence quenching assay shown in Figure S1.
KD Determination Using Fluorescence Quenching
We attempted to measure the dissociation constant (KD) for four compounds in Table 1 (compounds 1, 4, 6, and 8) using the following procedure. Unfortunately, the six remaining compounds were not available in sufficient quantities to conduct the same experiments. Tryptophan fluorescence spectra of hRRM1 at 0.3 mg/mL in assay buffer were recorded using a Jobin Yvon-Spex fluorescence spectrophotometer by exciting the sample with 295 nm light. The hRRM1 sample was titrated with increasing concentrations (1.25 μM–400 μM) of the individual compounds at room temperature. The data were fitted by nonlinear regression using the one-site binding (hyperbola) equation Y = Bmax·X/(Kd(app) + X), where Bmax is the maximum extent of quenching, and Kd(app) is the apparent dissociation constant, using GraphPad Prism 6.0 software (Figure S5). Measurements were recorded in duplicate in order to estimate error.
Ribonucleotide Reductase Inhibition Assays and Mechanism
The specific activity of hRR was determined in vitro using 14C-ADP reduction assays as previously described.28,29 The full assay protocol, including purification of hRRM2, is described in detail in the Supporting Information. Briefly, we adopted a two-point method for IC50 determination using the procedure described by Krippendorff et al.57 as only limited amounts of the hits were available. On the basis of this method, we used 5 and 100 μM concentrations of the ligand for measuring the IC50. As a control we used hydroxyurea, a common RR drug used against cancer, to validate our two-point method for deriving IC50. For instance, the two-point method gave an IC50 of 0.997 μM, while a traditional IC50 measurement using multiple points gave us an IC50 of 1.07 μM, confirming that both methods give almost identical numbers. The product 14C-dADP that formed during the reaction was separated from substrate 14C-ADP using boronate affinity chromatography. 28 14C-dADP was quantified by liquid scintillation counting using a Beckman LS6500 liquid scintillation counter. The IC50 was defined as the concentration of any compound that reduced the specific activity of hRRM1 to 50% of the control activity. The inhibition mechanism was analyzed by generating double-reciprocal plots and fitting with the mixed-model equations as described in the Supporting Information.
Growth Inhibition Screening Assays for Determining Cellular Toxicity
To assess cellular toxicity, growth inhibition assays were conducted using the standard MTT assay with the cancer cell lines MDA-MB-231, HCT-116, A549, and Panc1. Median effect doses (Dm) were calculated using Calcusyn, version 2.0. A detailed description of the procedures is described in the Supporting Information.
Crystallographic Studies of Compound 4 Bound to hRRM1
A full description of the crystallization and structure solution are provided in the Supporting Information. Briefly, hRRM1 was crystallized as described.29 As compound 4 could not be cocrystallized with hRRM1, the TTP-bound orthorhombic crystals were soaked with 100–500 μM of compound 4 for 2 h, and the crystals were cryogenized and data collected at the NE-CAT beamline at APS. A full description of the refinement and model building is provided in the Supporting Information.
Gel Filtration Chromatography
The effect of compound 4 on the oligomeric state of hRRM1 was assessed using gel filtration chromatography using a Superdex 200 (GE Healthcare) column as described.29 A full description of the procedure is found in the Supporting Information.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health, grants R01GM100887 and R01CA100827. T.M. was supported by training grant 5R25CA148052-05. This research was also supported by the Translational Research & Pharmacology Core Facility of the Case Comprehensive Cancer Center (P30 CA43703) and the Early Clinical Trials of Anti-Cancer Agents with Phase I Emphasis UO1 grant (U01 CA062502). We thank the Case Western Reserve University School of Medicine and the University of Cincinnati for making the chemical library available to us for screening. We thank the members of NE-CAT at the APS and X29 at NSLS for assistance with data collection. The NE-CAT beamlines are supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We acknowledge Mr. Andrew Burr for assisting us with ligand interaction analysis. We appreciate William Seibel for sharing his expert knowledge on the Cincinnati library. We would like to acknowledge Banumathi Sankaran for screening crystals at the ALS beamline. We thank Kathleen Lundberg for her assistance in the mass spectrometry analysis.
ABBREVIATIONS USED
- NATA
N-acetyl-L-tryptophanamide
- hRR
human ribonucleotide reductase
- hRRM1
large subunit of human ribonucleotide reductase
- Kav
partition coefficient
- hRRM2
small subunit of human ribonucleotide reductase
- PAINS
pan assay interference compounds
- dATP
deoxyadenosine 5′-triphosphate
- Dm
median effect dose
Footnotes
Accession Codes
4X3V.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00929.
Experimental methods for virtual screening, RR inhibition assays, inhibition mechanism, growth inhibition screening assays for determining cellular toxicity, crystallization of hRRM1, data collection, and structure determination; docking scores for Schrödinger; fluorescence quenching data; medicinal chemistry data; growth inhibition data for all compounds tested in MDA-MB-231 breast cancer and HCT-116 colon cancer cell lines; median effect doses (Dm) for compound 1; interactions between compound 4 and hRRM1 (within 5 Å); enzyme kinetic mechanism of compound 4 inhibition of hRRM1; docking poses for the 10 compounds in Table 1 (PDF)
References
- 1.Jordheim LP, Seve P, Tredan O, Dumontet C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol. 2011;12:693–702. doi: 10.1016/S1470-2045(10)70244-8. [DOI] [PubMed] [Google Scholar]
- 2.Jordheim LP, Guittet O, Lepoivre M, Galmarini CM, Dumontet C. Increased expression of the large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol Cancer Ther. 2005;4:1268–1276. doi: 10.1158/1535-7163.MCT-05-0121. [DOI] [PubMed] [Google Scholar]
- 3.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets. 2006;6:409–431. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 4.Tang SC, Chen YC. Novel therapeutic targets for pancreatic cancer. World J Gastroenterol. 2014;20:10825–10844. doi: 10.3748/wjg.v20.i31.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang P, Chubb S, Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J Biol Chem. 1990;265:16617–16625. [PubMed] [Google Scholar]
- 6.Avery TL, Rehg JE, Lumm WC, Harwood FC, Santana VM, Blakley RL. Biochemical pharmacology of 2-chlorodeoxyadenosine in malignant human hematopoietic cell lines and therapeutic effects of 2-bromodeoxyadenosine in drug combinations in mice. Cancer Res. 1989;49:4972–4978. [PubMed] [Google Scholar]
- 7.Griffig J, Koob R, Blakley RL. Mechanisms of inhibition of DNA synthesis by 2-chlorodeoxyadenosine in human lymphoblastic cells. Cancer Res. 1989;49:6923–6928. [PubMed] [Google Scholar]
- 8.Parker WB, Shaddix SC, Chang CH, White EL, Rose LM, Brockman RW, Shortnacy AT, Montgomery JA, Secrist JA, 3rd, Bennett LL., Jr Effects of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5′-triphosphate. Cancer Res. 1991;51:2386–2394. [PubMed] [Google Scholar]
- 9.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567–572. [PubMed] [Google Scholar]
- 10.Parker WB, Shaddix SC, Rose LM, Shewach DS, Hertel LW, Secrist JA, 3rd, Montgomery JA, Bennett LL., Jr Comparison of the mechanism of cytotoxicity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)adenine, 2-chloro-9-(2-deoxy-2-fluorobeta-D-ribofuranosyl)adenine, 2-chloro-9-(2-deoxy-2, 2-difluorobeta-D-ribofuranosyl)adenine in CEM cells. Mol Pharmacol. 1999;55:515–520. [PubMed] [Google Scholar]
- 11.Cerqueira NM, Fernandes PA, Ramos MJ. Ribonucleotide reductase: a critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat Anti-Cancer Drug Discovery. 2007;2:11–29. doi: 10.2174/157489207779561408. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Lohman GJ, Stubbe J. Mechanism of inactivation of human ribonucleotide reductase with p53R2 by gemcitabine 5′-diphosphate. Biochemistry. 2009;48:11612–11621. doi: 10.1021/bi901588z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–6117. [PubMed] [Google Scholar]
- 14.Heinemann V, Schulz L, Issels RD, Plunkett W. Gemcitabine: a modulator of intracellular nucleotide and deoxynucleotide metabolism. Semin Oncol. 1995;22:11–18. [PubMed] [Google Scholar]
- 15.Gmeiner WH, Yu S, Pon RT, Pourquier P, Pommier Y. Structural basis for topoisomerase I inhibition by nucleoside analogs. Nucleosides, Nucleotides Nucleic Acids. 2003;22:653–658. doi: 10.1081/NCN-120022604. [DOI] [PubMed] [Google Scholar]
- 16.Zhou B, Su L, Hu S, Hu W, Yip ML, Wu J, Gaur S, Smith DL, Yuan YC, Synold TW, Horne D, Yen Y. A small-molecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res. 2013;73:6484–6493. doi: 10.1158/0008-5472.CAN-13-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown NC, Canellakis ZN, Lundin B, Reichard P, Thelander L. Ribonucleoside diphosphate reductase. Purification of the two subunits, proteins B1 and B2. Eur J Biochem. 1969;9:561–573. doi: 10.1111/j.1432-1033.1969.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 18.Brown NC, Reichard P. Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J Mol Biol. 1969;46:39–55. doi: 10.1016/0022-2836(69)90056-4. [DOI] [PubMed] [Google Scholar]
- 19.Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnstrom K, Sjoberg BM, Eklund H. Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure. 1997;5:1077–1092. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 20.Larsson KM, Jordan A, Eliasson R, Reichard P, Logan DT, Nordlund P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat Struct Mol Biol. 2004;11:1142–1149. doi: 10.1038/nsmb838. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Faber C, Uchiki T, Fairman JW, Racca J, Dealwis C. Structures of eukaryotic ribonucleotide reductase I provide insights into dNTP regulation. Proc Natl Acad Sci U S A. 2006;103:4022–4027. doi: 10.1073/pnas.0600443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmad MF, Kaushal PS, Wan Q, Wijerathna SR, An X, Huang M, Dealwis CG. Role of arginine 293 and glutamine 288 in communication between catalytic and allosteric sites in yeast ribonucleotide reductase. J Mol Biol. 2012;419:315–329. doi: 10.1016/j.jmb.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uhlin U, Eklund H. Structure of ribonucleotide reductase protein R1. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 25.Kashlan OB, Scott CP, Lear JD, Cooperman BS. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry. 2002;41:462–474. doi: 10.1021/bi011653a. [DOI] [PubMed] [Google Scholar]
- 26.Cooperman BS, Kashlan OB. A comprehensive model for the allosteric regulation of Class Ia ribonucleotide reductases. Adv Enzyme Regul. 2003;43:167–82. doi: 10.1016/s0065-2571(02)00035-3. [DOI] [PubMed] [Google Scholar]
- 27.Rofougaran R, Vodnala M, Hofer A. Enzymatically active mammalian ribonucleotide reductase exists primarily as an alpha6beta2 octamer. J Biol Chem. 2006;281:27705–27711. doi: 10.1074/jbc.M605573200. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Lohman GJ, Stubbe J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc Natl Acad Sci U S A. 2007;104:14324–14329. doi: 10.1073/pnas.0706803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T, Dealwis CG. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat Struct Mol Biol. 2011;18:316–322. doi: 10.1038/nsmb.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aye Y, Stubbe J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc Natl Acad Sci U S A. 2011;108:9815–9820. doi: 10.1073/pnas.1013274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aye Y, Brignole EJ, Long MJ, Chittuluru J, Drennan CL, Asturias FJ, Stubbe J. Clofarabine targets the large subunit (alpha) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem Biol. 2012;19:799–805. doi: 10.1016/j.chembiol.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kashlan OB, Cooperman BS. Comprehensive model for allosteric regulation of mammalian ribonucleotide reductase: refinements and consequences. Biochemistry. 2003;42:1696–1706. doi: 10.1021/bi020634d. [DOI] [PubMed] [Google Scholar]
- 33.Arnaoutov A, Dasso M. IRBIT is a novel regulator of ribonucleotide reductase in higher eukaryotes. Science. 2014;345:1512–1515. doi: 10.1126/science.1251550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmad MF, Wan Q, Jha S, Motea E, Berdis A, Dealwis C. Evaluating the therapeutic potential of a non-natural nucleotide that inhibits human ribonucleotide reductase. Mol Cancer Ther. 2012;11:2077–2086. doi: 10.1158/1535-7163.MCT-12-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaffe EK, Stith L, Lawrence SH, Andrake M, Dunbrack RL. A new model for allosteric regulation of phenylalanine hydroxylase: implications for disease and therapeutics. Arch Biochem Biophys. 2013;530:73–82. doi: 10.1016/j.abb.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaffe EK. Impact of quaternary structure dynamics on allosteric drug discovery. Curr Top Med Chem. 2013;13:55–63. doi: 10.2174/1568026611313010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaffe EK. Morpheeins - A new pathway for allosteric drug discovery. Open Conf Proc J. 2010;1:1–6. doi: 10.2174/2210289201001010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 39.Wan KF, Wang S, Brown CJ, Yu VC, Entzeroth M, Lane DP, Lee MA. Differential scanning fluorimetry as secondary screening platform for small molecule inhibitors of Bcl-XL. Cell Cycle. 2009;8:3943–3952. doi: 10.4161/cc.8.23.10114. [DOI] [PubMed] [Google Scholar]
- 40.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J Mol Biol. 1973;79:351–371. doi: 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
- 41.Stearns B, Losee KA, Bernstein J. Hydroxyurea. A new type of potential antitumor agent. J Med Chem. 1963;6:201. doi: 10.1021/jm00338a026. [DOI] [PubMed] [Google Scholar]
- 42.Donehower RC. An overview of the clinical experience with hydroxyurea. Semin Oncol. 1992;19:11–19. [PubMed] [Google Scholar]
- 43.Winter A, Higueruelo AP, Marsh M, Sigurdardottir A, Pitt WR, Blundell TL. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: applications in structure-guided drug discovery. Q Rev Biophys. 2012;45:383–426. doi: 10.1017/S0033583512000108. [DOI] [PubMed] [Google Scholar]
- 44.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 45.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 46.Mills CJ, Richardson T, Jasensky RD. Antimicrobial effects of N alpha-palmitoyl-L-lysyl-L-lysine ethyl ester dihydrochloride and its use to extend the shelf life of creamed cottage cheese. J Agric Food Chem. 1980;28:812–817. doi: 10.1021/jf60230a035. [DOI] [PubMed] [Google Scholar]
- 47.Cegielska A, Taylor A. The sfiA11 mutation prevents filamentation in a response to cell wall damage only in a recA+ genetic background. Mol Gen Genet. 1985;201:537–542. doi: 10.1007/BF00331352. [DOI] [PubMed] [Google Scholar]
- 48.Blundell TL. Structure-based drug design. Nature. 1996;384:23–26. doi: 10.1038/384023a0. [DOI] [PubMed] [Google Scholar]
- 49.Hofer A, Crona M, Logan DT, Sjoberg BM. DNA building blocks: keeping control of manufacture. Crit Rev Biochem Mol Biol. 2012;47:50–63. doi: 10.3109/10409238.2011.630372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Estebanez-Perpina E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ciulli A, Williams G, Smith AG, Blundell TL, Abell C. Probing hot spots at protein-ligand binding sites: a fragment-based approach using biophysical methods. J Med Chem. 2006;49:4992–5000. doi: 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- 52.Edink E, Rucktooa P, Retra K, Akdemir A, Nahar T, Zuiderveld O, van Elk R, Janssen E, van Nierop P, van Muijlwijk-Koezen J, Smit AB, Sixma TK, Leurs R, de Esch IJ. Fragment growing induces conformational changes in acetylcholine-binding protein: a structural and thermodynamic analysis. J Am Chem Soc. 2011;133:5363–5371. doi: 10.1021/ja110571r. [DOI] [PubMed] [Google Scholar]
- 53.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 54.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 55.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 56.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krippendorff BF, Lienau P, Reichel A, Huisinga W. Optimizing classification of drug-drug interaction potential for CYP450 isoenzyme inhibition assays in early drug discovery. J Biomol Screening. 2007;12:92–99. doi: 10.1177/1087057106295897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.