

Abstract
Background
Preeclampsia (PE) is a prevalent hypertensive disorder of pregnancy and a leading cause of maternal and neonatal morbidity and mortality worldwide. This pathogenic condition is speculated to be due to placental abnormalities that contribute to the maternal syndrome. However, the specific factors and signaling pathways leading to impaired placentas and maternal disease development remain elusive.
Methods and Results
By using two independent animal models of PE—1) genetically-engineered pregnant mice with elevated adenosine exclusively in placentas, and 2) a pathogenic autoantibody-induced PE mouse model—we demonstrated here that chronically elevated placental adenosine was sufficient to induce hallmark features of PE including hypertension, proteinuria, small fetuses, and impaired placental vasculature. Genetic and pharmacologic approaches revealed that elevated placental adenosine coupled with excessive A2B adenosine receptor (ADORA2B) signaling contributed to the development of these features of PE. Mechanistically, we provided both human and mouse evidence that elevated placental CD73 is a key enzyme causing increased placental adenosine, thereby contributing to PE.
Conclusions
We determined that elevated placental adenosine signaling is a previously unrecognized pathogenic factor for PE. Moreover, our findings revealed the molecular basis underlying the elevation of placental adenosine and the detrimental role of excess placental adenosine in the pathophysiology of PE, and, thereby highlight novel therapeutic targets.
Keywords: adenosine, pregnancy, hypertension, preeclampsia
Introduction
Preeclampsia (PE) is a gestation-specific syndrome with a high incidence of mother and infant morbidity and mortality worldwide1. For years, the diagnosis has been made solely by the detection of sudden onset hypertension and proteinuria. Despite intensive research efforts, current strategies for managing PE are inadequate and limited to symptomatic therapy or the termination of pregnancy because the pathogenesis of the disease remains elusive2.
The placenta is a newly formed organ that links mother and fetus throughout pregnancy3. It plays an important role to support intrauterine fetal growth by facilitating the transfer of nutrients and oxygen from the mother to the fetus and by removing fetal waste products4, 5. Additionally, the placenta is an endocrine organ that synthesizes and secretes multiple hormones, neurotransmitters and vasoactive factors5, 6. Impairment in placental development and function is considered to contribute to the pathogenesis of PE1, 7. Considerable evidence indicates that dysregulation of cytoprotective pathways8–10 and increase in antiangiogenic growth factors11, 12, complement activation13, and autoantibodies14 contribute to placental damage and the progression of the disease. However, the placenta-specific molecular basis responsible for placental impairment leading to PE has not been fully understood. Here, we sought to identify a novel pathogenic factor linking placental pathology to the development of PE.
Adenosine is a key signaling molecule that orchestrates the cellular response to hypoxia, energy depletion and tissue damage by activation of G-protein coupled receptors on multiple cell types15, 16. Extracellular adenosine levels are tightly regulated by multiple factors involved in the synthesis from ATP by the sequential action of two ectonucleotidases (CD39 and CD73), degradation by adenosine deaminase (ADA), and cellular uptake by equilibrative nucleoside transporters (ENTs)15, 17. Extracellular adenosine is known to exert its function through the activation of four G-protein coupled cell surface receptors, ADORA1, ADORA2A, ADORA2B, and ADORA315. Acutely elevated adenosine signaling is intended to be brief and beneficial. The response is normally time-limited because of the short half-life of adenosine. In contrast, chronically elevated adenosine is detrimental and is associated with multiple pathologic conditions including chronic kidney disease, pulmonary fibrosis, priapism and sickle cell disease18–20. Intriguingly, previous studies have reported that levels of adenosine are elevated in the maternal or fetal circulation of PE patients compared with normal pregnant women and are correlated to disease severity21, 22. Another earlier study showed that elevated adenosine in PE patients is correlated to Th1/Th2 imbalance23. In vitro studies indicate that elevated adenosine is related to increased platelet aggregation and P-selectin expression24. More recent reports demonstrate that adenosine is capable of inducing sFlt-1 production in rat villous explants25. However, the role of elevated adenosine in the pathophysiology of PE remained unknown and cannot be fully understood using in vitro cell and organ culture systems. Thus, an in vivo animal studies are desperately needed to accurately and fully understand whether elevated adenosine signaling contributes to the pathogenesis of PE. To fully address this question, we sought to i) generate pregnant animals specifically with elevated placental adenosine, ii) determine the pathophysiologic roles of elevated placental adenosine in PE; and iii) delineate the molecular basis for its elevation in PE in mice and humans. Here, we provide both mouse and human evidence that excess placental adenosine coupled with the enhanced ADORA2B signaling contributes to the pathogenesis of PE. Mechanistically, we discovered that elevated placental CD73 is a key enzyme responsible for increased placental adenosine production, and thereby contributes to the development of PE.
Methods
For an expanded Methods sections, please refer to the online-only Data Supplement.
Animals
Fetal liver rescued ADA-deficient mice (Ada−/−/fLi-Tg+ mice) were generated by introducing an ADA minigene (fLi-Tg) that is only expressed in the fetal liver under the control of alpha-fetoprotein (Supplemental Figure 1). Placental rescued ADA-deficient mice (Ada−/−/Pl-Tg+ mice) equipped with an Ada minigene (PL-Tg) that is expressed exclusively in the trophoblast cell lineage were generated and genotyped as previously described26, 27. Details are described in online-only Data Supplement.
Patients
Patients admitted to Memorial Hermann Hospital were identified by the obstetrical faculty of the University of Texas Medical School at Houston. Preeclampsia patients diagnosed based on the definition set by the National High Blood Pressure Education Program Working Group Report28 were included in study. Human subject data were summarized and included in Supplemental Table S1. The research protocol was approved by the Institutional Committee for the Protection of Human Subjects and informed consent was obtained from all the patients.
Statistical analysis
All data were expressed as the mean ± SEM. Mann-Whitney’s U test was applied in two-group analysis. Differences among multiple groups were compared by the Kruskal Wallis test, followed by a Dunn’s post hoc test. Comparison of the data obtained at different time points as repeated measurements in Figures 1D, 1F, 3D, 4D, and 7E were analyzed by two-way repeated measures analysis of variance, followed by the Bonferroni post hoc test. Categorical variables in Supplemental Table S2 were analyzed by the Fisher’s exact test. Statistical significance was set as P<0.05 and analyzed by GraphPad Prism 5 (GraphPad).
Figure 1.
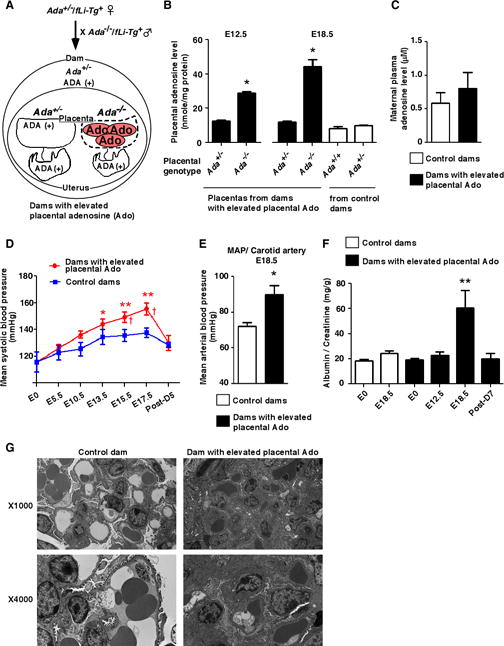
Pregnant mice with elevated placental adenosine display spontaneously developed hallmark features of preeclampsia. (A) Schema of mating strategy to generate pregnant mice with elevated placental adenosine (Ado). (B) Levels of adenosine in mouse placentas obtained on E12.5 or E18.5 detected by HPLC. (n=4 on E12.5, n=8 on E18.5 each), (*P<0.05 vs Ada+/− on E12.5, *P<0.05 vs other groups on E18.5). (C) No significance of aternal circulating adenosine levels on E18.5. (n=4 each). (D) Blood pressure measured by the tail-cuff method from non-pregnant state (E0) until postpartum day 5 (Post-D5). (Control dams: n=10, Dams with elevated placental Ado: n=9), (*P<0.05, **P<0.01 vs E0, †P<0.05 vs control dams at the same time point). (E) Intra carotid mean arterial blood pressure (MAP) measured on E18.5. (Control dams: n=3, Dams with elevated Ado: n=4), (*P<0.05 vs Control dams). (F) Urine protein (ratio of albumin/creatinine in urine) was measured by ELISA. (Control dams: n=9, Dams with elevated Ado: n=7), (**P<0.01 vs other time points). (G) Electron microscopy of glomeruli. Glomeruli from dams with elevated placental adenosine possessed capillaries with markedly decreased lumenal spaces, with endothelial cytoplasmic swelling and a moderate expansion of the mesangium by mesangial cells and matrix. The visceral epithelial cells showed focal foot process effacement.
Figure 3.
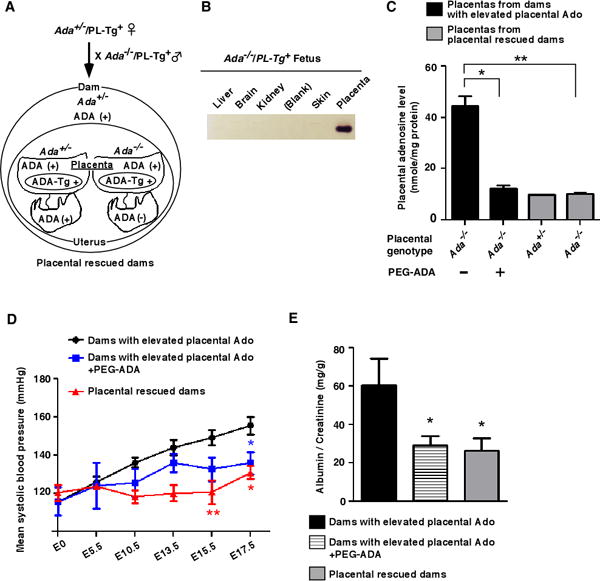
Preeclamptic features caused by elevated placental adenosine are prevented by genetically restoring ADA to placentas or by ADA-enzyme therapy. (A) Schema of mating strategy of placental rescued ADA-deficient mice (Ada−/−/Pl-Tg+). (B) ADA enzymatic activity in tissues determined by ADA zymogram analysis. (C) Adenosine levels in placentas from dams with elevated placental adenosine with or without PEG-ADA treatment (5 units on E5.5 and E12.5 by retro-orbital sinus injection) and placental ADA-rescued dams were determined on E18.5 by HPLC. (n=8 each group), (*P<0.05, **P<0.01). (D and E) Blood pressure measured by the tail-cuff method (D) and proteinuria (E) measured by ELISA. (Dams with elevated placental Ado: n=9, PEG-ADA treatment: n=5, Placental rescued dams: n=8), (D: *P<0.05, **P<0.01 vs Dams with elevated placental Ado at the same time point, E: *P<0.05 vs Dams with elevated placental Ado).
Figure 4.
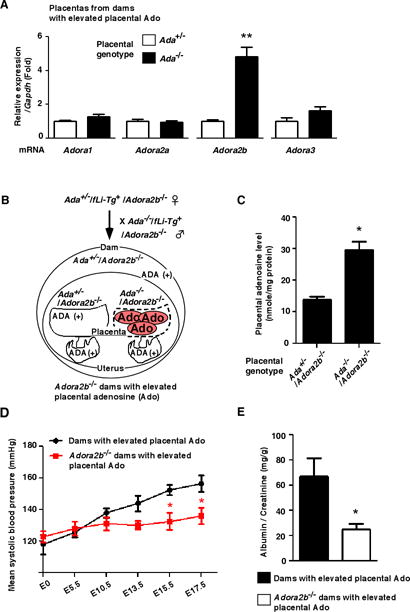
Genetic deletion of ADORA2B prevents placental elevated adenosine-induced preeclamptic features. (A) mRNA expression levels of adenosine receptors in placentas from dams with elevated placental adenosine were detected by real-time RT-PCR. Each value was expressed as a fold induction relative to Ada+/− placentas. (n=6 each), (**P<0.01 vs Ada+/−). (B) Schema of mating strategy to generate Adora2b−/− dams with elevated placental adenosine. (C) Levels of adenosine in placentas from Adora2b−/− dams with elevated placental adenosine were determined on E18.5 by HPLC. (n=4 each group), (*P<0.05 vs Ada+/−/Adora2b−/−). (D and E) Blood pressure measured by the tail-cuff method (D) and proteinuria (E) measured by ELISA. (Dams with elevated placental Ado: n=9, Adora2b−/− dams: n=8), (D: *P<0.05 vs Dams with elevated placental Ado at the same time point, E: *P<0.05 vs Dams with elevated placental Ado).
Figure 7.
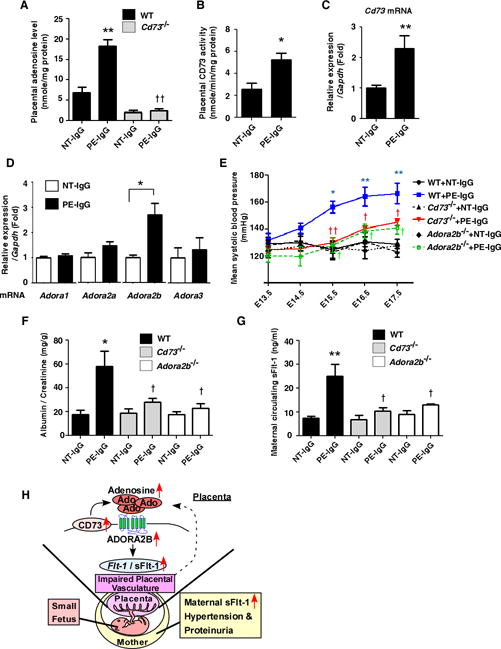
Elevated CD73 underlies increased placental adenosine and subsequent disease development via ADORA2B activation in an autoantibody-induced mouse model of PE. (A) Adenosine levels in mouse placentas on E18.5 were detected by HPLC. (n=5–8 per group), (**P<0.01 vs WT+NT-IgG, ††P<0.01 vs WT+PE-IgG). (B) CD73 activities in IgG-injected mouse placentas were determined by enzyme-based assay. (n=4 each), (*P<0.05 vs NT-IgG). (C) Increased expression of Cd73 mRNA in placentas from PE-IgG-injected dams. (NT-IgG; n=6, PE-IgG; n=7), (**P<0.01 vs NT-IgG). (D) mRNA expression levels of adenosine receptors in mouse placentas were determined by real-time RT-PCR (n=6 each), (*P<0.05). (E and F) Blood pressure measured by the tail-cuff method (E) and proteinuria (F) measured by ELISA. (n=7–11 per group), (E: *P<0.05, **P<0.01 vs WT+NT-IgG at the same time point, †P<0.05, ††P<0.01 vs WT+PE-IgG at the same time point, F: *P<0.05 vs WT+NT-IgG, †P<0.05 vs WT+PE-IgG). (G) Maternal circulating sFlt-1 levels on E18.5 det. (n=5–7 per group), (**P<0.01 vs WT+NT-IgG, †P<0.05 vs WT+PE-IgG). (H) Working model: Elevated placental adenosine coupled with enhanced ADORA2B signaling as a novel pathogenic factor for PE.
Results
Pregnant mice with elevated placental adenosine display spontaneously-developed hallmark features of PE
To examine the pathologic consequences of excessive placental adenosine we needed an experimental approach to generate pregnant mice with placentas with elevated adenosine. One approach is to generate placentas that lack adenosine deaminase (ADA), the enzyme that irreversibly degrades adenosine to inosine. However, global ADA deficient mice die late in gestation as a result of impaired liver function29, 30. To circumvent this complication and to produce viable embryos with excessive placental adenosine, we genetically rescued ADA-deficient embryos from prenatal lethality by introducing an ADA minigene that is only expressed in the fetal liver (for details see Methods in online-only Data Supplement, Supplemental Figure 1A and 1B). Next, we took advantage of fetal liver rescued ADA-deficient mice (Ada−/−/fLi-Tg+) to assess the impact of elevated placental adenosine throughout pregnancy in vivo. Specifically, we designed a mating strategy by crossing Ada−/−/fLi-Tg+ males with Ada+/−/fLi-Tg+ females to generate pregnant mice in which half of the placentas are expected to be ADA deficient and have elevated adenosine (dams with elevated placental adenosine) (Figure 1A). Dams were positive for ADA and fetuses expressed ADA activity only in the fetal liver (Figure 1A, Supplemental Figure 1C and 1D). In control crosses of Ada+/−/fLi-Tg+ females with Ada+/+ males, all placentas were ADA-positive (control dams). We found that placental adenosine was significantly elevated in the ADA-negative placentas compared to the ADA-positive placentas on embryonic day 12.5 (E12.5) and remained elevated through E18.5 (Figure 1B). In contrast, the placentas in control dams with either Ada+/− or Ada+/+ genotype contained similar levels of adenosine in the normal range on E18.5 (Figure 1B, empty bar). Correspondingly, we confirmed that ADA protein and enzymatic activity were not detected in Ada−/− placentas whereas ADA-positive placentas with Ada+/− or Ada+/+ genotype displayed ADA enzymatic activity (Supplemental Figure 1C and 1D). Additionally, because dams with elevated placental adenosine and control dams are both positive for ADA expression (maternal genotype ADA+/−), there was no increase in adenosine levels in the maternal circulation of pregnant mice harboring ADA-negative placentas compared to control dams with all ADA-positive placentas (Figure 1C). Thus, fetal liver rescued ADA-deficient mice provided us a genetic investigative tool to assess the impact of elevated placental adenosine throughout pregnancy in vivo.
We first monitored hallmark features of PE in dams with elevated placental adenosine and control dams and found that dams with elevated placental adenosine displayed a significant increase in mean systolic blood pressure late in pregnancy beginning on embryonic day 15.5 (E15.5) and remained significantly elevated through E17.5 compared with the control dams. Elevated systolic blood pressure returned to normal by day 5 postpartum (Figure 1D). To validate tail cuff measurement of blood pressure, we conducted invasive measurement of mean arterial pressure (MAP) of the carotid artery. We found that MAP was significantly elevated on E18.5 of dams with elevated placental adenosine compared with the control dams (Figure 1E). Additionally, we found that urinary protein (ratio of albumin/creatinine) was not significantly increased in the dams with elevated placental adenosine on E12.5 while it was significantly elevated on E18.5 (Figure 1F). By postpartum day 7, proteinuria was reduced to the levels similar to that of the non-pregnant state (Figure 1F). Interestingly, Ada+/−/fLi-Tg+ dams crossed with Ada+/−/fLi-Tg+ males, in which 25% of the placentas are expected to be ADA negative and have elevated adenosine, did not display an increase in blood pressure or proteinuria (data not shown), suggesting that there is a threshold for the number of ADA-deficient placentas with excess placental adenosine to trigger maternal features of PE. Histological studies revealed pathologic changes in the kidneys including swollen glomeruli with narrowed capillary and Bowman’s spaces in the pregnant mice with elevated placental adenosine (Supplemental Figure 2A and 2B). Furthermore, electron microscopic studies revealed that glomeruli in dams with elevated placental adenosine showed the typical pathologic change “glomerular endotheliosis” as seen in PE patients (Figure 1G). Thus, our findings showed that the elevation of placental adenosine occurs prior to the onset of maternal PE features and suggest that elevated placental adenosine contributes to the pathogenesis of PE.
Mice with elevated placental adenosine present with impaired placental vasculature and fetal growth restriction
In addition to maternal PE features, we found that ADA-deficient placentas with elevated adenosine were smaller and weighed significantly less than ADA-positive placentas with normal levels of adenosine from dams with PE features (Supplemental Table S2). Likewise the fetuses associated with ADA-deficient placentas with elevated placental adenosine were smaller and weighed significantly less than fetuses associated with ADA-positive placentas without morphological abnormalities (Figure 2A and Supplemental Table S2). To further validate our findings, we compared the fetal and placental weights to those observed in control dams. We found that ADA-negative placentas and their associated fetuses from the PE dams with elevated placental adenosine were significantly smaller and weighed less than those from the control dams.
Figure 2.
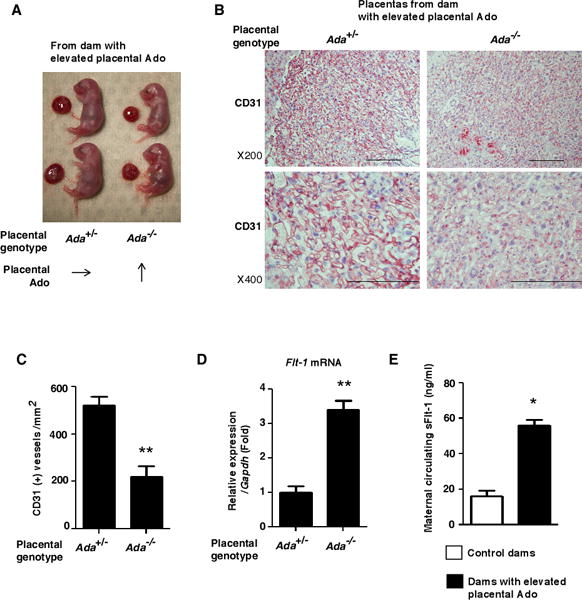
Pregnant mice with placental excess adenosine present with impaired placental vasculature with increased Flt-1 gene expression and elevated sFlt-1 levels in the maternal circulation. (A) Representative image of fetuses and placentas on E18.5 from dams with elevated placental adenosine. (B) Vascular structure of placentas from dams with elevated placental adenosine was assessed by CD31 immunostaining on E18.5. Scale bar; 200μm. (C) CD31-positive vessel density was determined. The data was quantified by counting CD31-positive vessels obtained from every three center areas in the labyrinth zone of three different samples in each group. (**P<0.01 vs Ada+/−) (D) Flt-1 mRNA expression in the placentas on E18.5 determined by real-time RT-PCR. (n=6 each), (**P<0.01 vs Ada+/−). (E) Maternal circulating sFlt-1 levels on E18.5. (Control dams: n=6, Dams with elevated Ado: n=4), (*P<0.05 vs Control dams).
Histological analysis of placentas by using CD31 staining revealed that ADA-negative placentas showed disorganized and impaired vasculature in the labyrinthine zone compared to ADA-positive placentas (Figure 2B). Semi-quantification of CD31 staining demonstrated the CD31-positive vessels were significantly reduced in Ada−/− placentas with elevated adenosine (Figure 2C). Supporting this finding, we found that ADA-deficient placentas with elevated adenosine contained significantly elevated Flt-1 mRNA compared to ADA-positive placentas (Figure 2D). Accordingly, we found that maternal circulating sFlt-1 levels in dams with elevated placental adenosine were significantly higher than those of the control dams (Figure 2E). These studies provide genetic evidence that increased placental adenosine is associated with small fetuses and small placentas featured with impaired vasculature and increased Flt-1 gene expression.
Elevated placental adenosine contributes to the onset of maternal PE features, impaired placentas, and small fetuses in pregnant mice
Next, to determine if elevated placental adenosine causes the placental impairment, small fetuses and maternal PE features, we used a transgenic approach to genetically restore ADA exclusively to the placentas of ADA-deficient mice to lower placental adenosine (genotype Ada−/−/PL-Tg+) (Figure 3A)26, 27. We found that ADA enzyme activity was only observed in the placentas and absent in fetal organs of Ada−/−/PL-Tg+ mice (Figure 3B). More importantly, restoring ADA to placentas completely restored placental adenosine levels to the normal range (Figure 3C, gray bar) compared to the placenta from fetal liver rescued dam with elevated placental adenosine (Figure 3C, black bar) on E18.5. Consequently, placental and fetal weights were significantly increased compared to the dams with elevated placental adenosine (Supplemental Table S2). Moreover, expressing ADA only in the placentas restored the normal placental vasculature and returned placental Flt-1 gene expression to levels found in controls (Supplemental Figure 3 and Supplemental Figure 4A) and abolished elevated maternal circulating sFlt-1 levels (Supplemental Figure 4B), hypertension, proteinuria (Figure 3D and 3E), and kidney pathohistologic changes (Supplemental Figure 5). Altogether, these results provide strong genetic evidence that elevated placental adenosine is associated with impaired placental vasculature, fetal growth restriction, and maternal features of PE.
Next, we performed pharmacological studies using polyethyleneglycol-ADA (PEG-ADA) enzyme therapy to prevent the accumulation of adenosine in Ada−/− placentas. We found that PEG-ADA treatment significantly reduced placental adenosine levels in these mice (Figure 3C). PEG-ADA treatment significantly ameliorated all the features observed in dams with elevated placental adenosine, including impaired placentas (Supplemental Figure 3), maternal PE features (Figure 3D, 3E, and Supplemental Figure 4), and renal pathologic changes (Supplemental Figure 5). Taken together, we provide both genetic and pharmacological evidence that elevated placental adenosine underlies impaired placental vasculature, fetal growth restriction, and maternal PE features in mice.
Genetic deletion of A2B adenosine receptor (ADORA2B) prevents the elevated placental adenosine-induced placental impairment, fetal growth restriction, and maternal features of PE
To determine which adenosine receptor may be responsible for elevated adenosine-induced placental impairment, small fetuses, and maternal PE features, we determined the gene expression profiles of the four adenosine receptors in placentas with normal and elevated adenosine. We found that among adenosine receptors only Adora2b gene expression was significantly increased in ADA-deficient placentas with elevated adenosine compared to ADA-positive placentas (Figure 4A), suggesting that elevated ADORA2B signaling may mediate the development of PE features in dams with elevated placental adenosine. To test this possibility, we generated mice deficient in ADORA2B on the background of fetal liver rescued ADA deficient mice (Ada−/−/fLi-Tg+/Adora2b−/− mice) (Figure 4B and see Methods in online-only Data Supplement). We found that genetic deletion of Adora2b did not affect the levels of elevated placental adenosine compared to the controls (Figure 4C). However, genetic deletion of Adora2b resulted in a significant increase in placental and fetal weight (Supplemental Table S2). CD31 staining showed that the placental vasculature was more organized and uniform in Adora2b−/− placentas with elevated adenosine (Supplemental Figure 3). Additionally, all the preeclamptic features observed in dams with elevated placental adenosine, including hypertension and proteinuria (Figure 4D and 4E), increase in maternal circulating sFlt-1 (Supplemental Figure 4B), and renal pathologic changes (Supplemental Figure 5), were ameliorated by the genetic deletion of Adora2b. Thus, we provided genetic evidence that excess placental adenosine coupled with enhanced ADOAR2B signaling is responsible for maternal PE features, impaired placentas, and subsequent fetal growth restriction.
Adenosine levels and ADORA2B expression are significantly increased in the placentas of preeclampsia patients, and elevated ADORA2B signaling directly induces sFlt-1 production
To examine the translational relevance of our mouse findings to human pregnancy, we first measured adenosine levels in the placentas of normotensive pregnant individuals (NT) and preeclampsia patients (PE). We found that adenosine levels were significantly elevated in the placentas of women with PE compared to placentas from NT pregnant women (Figure 5A). Similarly, ADORA2B gene expression and protein level were found to be significantly elevated in the PE placentas compared to the NT placentas (Figure 5B and 5C).
Figure 5.
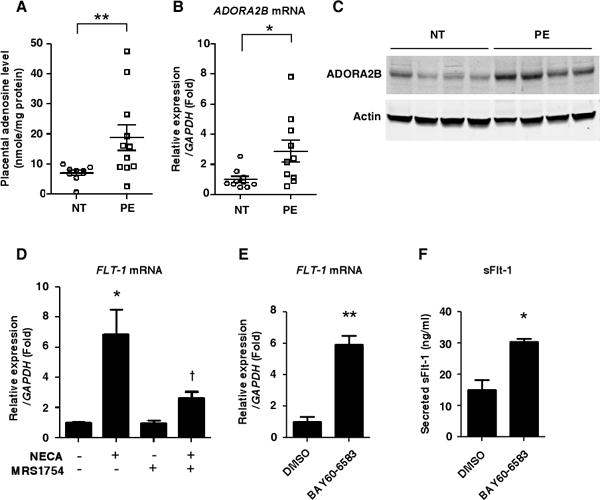
Importance of excess adenosine coupled with enhanced ADORA2B signaling in placentas of preeclampsia patients. (A) Adenosine levels in the placentas of preeclampsia patients (PE) or normotensive pregnant women (NT) were detected by HPLC. (NT: n=9, PE: n=11), (**P<0.01). (B) Levels of ADORA2B mRNA in human placentas determined by real-time RT-PCR. (n=10 per group), (*P<0.05). (C) Levels of ADORA2B protein in human placentas detected by immunoblotting. (D) FLT-1 mRNA expression levels in human villous explants determined by real-time RT-PCR. Human villous explants were pretreated with or without 10 nM MRS1754 for 30 min and then treated with 1μM NECA for 24 hours. (n=3), (*P<0.05 vs non-treatment, †P<0.05 vs NECA treatment only). (E and F) ADORA2B agonist treatment significantly induced FLT-1 mRNA (E) and sFlt-1 secretion (F) in the cultured human villous explants. Human villous explants were treated with 1μM BAY60-6583 for 24 hours. (n=3), (*P<0.05, **P<0.01).
To test whether elevated ADORA2B signaling may contribute to features of PE in humans, we conducted experiments using human placental villous explants cultured from NT placentas. First, we found that NECA, a non-metabolized adenosine analogue, significantly induced FLT-1 gene expression and that the stimulation of FLT-1 gene expression was attenuated by an ADORA2B specific antagonist (MRS1754) (Figure 5D). Moreover, we found that the ADORA2B specific agonist, BAY60-6583, significantly induced FLT-1 gene expression (Figure 5E) and sFlt-1 secretion (Figure 5F) under normoxic conditions in the villous explants. These results translate our mouse finding of the significance of excess placental adenosine signaling in PE to human pregnancy by showing that 1) placental adenosine levels are increased in PE patients, and 2) elevated adenosine signaling via ADORA2B contributes to elevated Flt-1 gene expression and subsequent sFlt-1 secretion from human villous explants.
CD73 is elevated in preeclampsia patient placentas
Next, to examine the molecular mechanisms underlying the elevation of placental adenosine in PE patients, we initially assessed the possible involvement of ADA. In contrast to mouse placentas, ADA enzyme activity was extremely low in the normal human placentas without significant difference in enzyme activity between the placentas of normotensive pregnant women and those of PE patients (Figure 6A). Thus, we concluded that reduction in ADA levels is not a major factor contributing to increased placental adenosine in PE patients.
Figure 6.
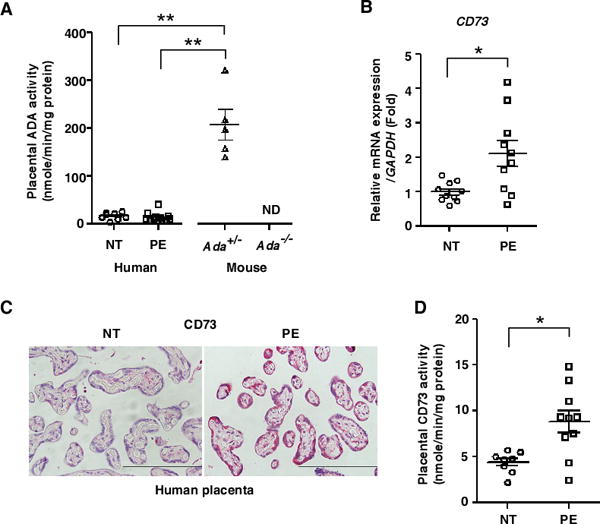
Expression profiling of purinergic molecules in human placentas revealed that CD73 was elevated in preeclampsia patients. (A) ADA activities in human placentas (NT: n=9, PE: n=12) or mouse placentas from the dams with elevated placental adenosine (n=5 per group) were determined using a spectrophotometric assay. (**P<0.01, ND: not detected). (B) mRNA expression levels of CD73 in human placentas. (n=10 per group), (*P<0.05 vs NT). (C) Representative images of CD73 immunostaining in human placentas from NT or PE. Scale bar, 200μm. (D) CD73 activities to convert AMP to adenosine in human placentas were determined by enzyme-based assay. (NT: n=8, PE: n=10), (*P<0.05).
In an effort to identify the factors causing elevated adenosine in the placentas of PE patients, we conducted expression profiling of purinergic molecules. Among the genes screened, we found that mRNA encoding CD73, a key ectonucleotidase producing extracellular adenosine from AMP, was significantly elevated in placentas of PE patients (Figure 6B). Additionally, we found that the protein level and enzyme activity of CD73 were significantly increased in placentas of PE patients (Figure 6C and 6D). In contrast, the mRNA levels encoding CD39 (an ectonucleotidase that converts ATP to AMP), ENT1 and ENT2 (adenosine transporters) showed no significant difference between NT and PE placentas (Supplemental Figure 6). Thus, analysis of human placentas indicated that elevated CD73 is responsible for increased placental adenosine and subsequent disease development.
Elevated CD73 underlies increased placental adenosine and contributes to pathophysiology of PE via ADORA2B activation in an experimental model of PE
To test the generality of this intriguing hypothesis that elevated placental adenosine in PE patients is due to elevated CD73-mediated production of adenosine, we took advantage of an established experimental mouse PE model induced by adoptive transfer of PE patient-derived IgG (PE-IgG) known to contain the pathogenic autoantibodies that activate the angiotensin II type 1 receptor agonistic autoantibody (AT1-AA)14. As with PE patients, we found that placental CD73 activity and adenosine levels were significantly induced in PE-IgG-injected dams compared to normotensive pregnant women-derived IgG (NT-IgG)-injected dams (Figure 7A and 7B). In contrast, adenosine levels were not increased in the maternal circulation nor in the maternal kidneys of PE-IgG-injected dams compared to the dams injected with NT-IgG (Supplemental Figure 7A and 7B). Similar to human studies, gene expression analysis revealed that mRNA and protein levels of CD73 were significantly increased in the placentas of PE-IgG-injected dams (Figure 7C and Supplemental Figure 8). Additionally, we found that among adenosine receptors only Adora2b mRNA was significantly elevated in the placentas of PE-IgG-treated mice (Figure 7D). Similarly, the placental expression of Ada, Cd39, and Ent genes, showed no significant difference between NT-IgG- and PE-IgG-treated dams (Supplemental Figure 6).
Next, to determine whether the increased expression of CD73 was responsible for PE-IgG-induced placental adenosine production and the pathogenesis of PE, we injected PE-IgG into CD73-deficient dams (Cd73−/− females mated with Cd73−/− males). Five days after PE-IgG injection, we found that CD73 deficiency resulted in significantly reduced PE-IgG-induced production of adenosine in the placentas (Figure 7A), indicating that elevated CD73 was required for the increase of placental adenosine induced by PE-IgG. Additionally, we found that the key diagnostic features of PE, hypertension and proteinuria, were significantly attenuated in Cd73−/− dams injected with PE-IgG compared with PE-IgG-injected WT dams (Figure 7E, 7F, and Supplemental Figure 9). In addition, we found that levels of maternal circulating sFlt-1 induced by PE-IgG were significantly suppressed in Cd73−/− dams as compared with those in PE-IgG-injected WT dams (Figure 7G). Thus, this study revealed that elevated CD73 is essential for increased placental adenosine and that excess Ado is responsible for disease development in the pathogenic autoantibody-induced animal model of PE.
Finally, to determine whether elevated Adora2b expression in the placentas has pathophysiologic significance, we injected PE-IgG into Adora2b−/− dams (Adora2b−/− females mated with Adora2b−/− males). We found that all the preeclamptic features, including hypertension, proteinuria, and elevation of circulating sFlt-1, which were observed in PE-IgG-injected WT dams were significantly suppressed in Adora2b−/− dams (Figure 7E, 7F, 7G, and Supplemental Fig. 9). These results provide genetic evidence that elevated CD73-mediated chronically elevated adenosine in placentas exerts its detrimental effects in PE-IgG-injected pregnant mice thorough enhanced ADORA2B signaling.
Discussion
The involvement of adenosine signaling in the pathogenesis of PE was unknown prior to our current study. Here we report the use of genetic approaches to successfully generate pregnant mice with elevated adenosine only in placentas. We demonstrated the pathogenic role of elevated placental adenosine signaling via excessive ADOAR2B signaling, which induces placental impairment associated with sFlt-1 induction, small fetuses, and maternal PE features. Moreover, we have provided both mouse and human findings that elevated CD73 is a key enzyme underlying increased placental adenosine and subsequent disease development. Overall, our findings reveal the pathogenic consequences of chronically elevated placental adenosine, the molecular basis for its elevation and the specific signaling pathways leading to clinical features of PE.
PE is commonly considered to result from impaired placental development secondary to hypoxia or other hypoxia-independent mediators including cytokines, complement and autoantibody13, 14, 31. Adenosine is a key signaling molecule that orchestrates a rapid multi-cellular physiological response to hypoxia or tissue damage16. However, certain disease states are associated with chronically elevated adenosine and the resulting persistent adenosine signaling is detrimental18, 19, 32. A potentially detrimental role for adenosine signaling in PE placentas is suggested by a recent study showing that adenosine stimulates increased sFlt-1 production in cultured rat placental villous explants25. To our knowledge, in vivo evidence for a role of elevated placental adenosine signaling in PE has not been previously reported until we observed hallmark features of PE in pregnant mice with excessively accumulated placental adenosine due to absence of placental ADA. It is interesting to note that elevated placental adenosine occurs on E12.5 prior to the maternal symptoms (i.e. hypertension and proteinuria) which develop around E15.5 and disappear postpartum. Thus, our studies provide strong in vivo evidence that elevated placental adenosine is a causative factor to induce maternal PE features. The detrimental role of elevated placental adenosine in PE is also supported by another animal model of PE based on the injection of pathogenic autoantibodies purified from PE patients. Altogether, in two independent animal models of PE, one with placental ADA deficiency and another with injection of pathogenic autoantibodies, we demonstrated the common pathogenic role of chronically elevated placental adenosine in PE in vivo. Our mouse findings were further validated in humans by showing that placental adenosine levels were also significantly elevated in women with PE. Thus, we have provided significant mouse and human evidence that elevated placental adenosine contributes to the pathogenesis of PE.
Extracellular adenosine levels are regulated by multiple factors involved in the synthesis (CD73), degradation (ADA) and cellular uptake (ENTs) of adenosine15, 17. Our initial mouse studies created placentas with elevated adenosine using genetic strategies to create ADA-deficient placentas. Earlier studies demonstrated that placental and circulating ADA activities were quite low and that slightly higher levels of circulating and placental ADA activity were observed in the PE patients33, 34. In these studies the reported increase in ADA activity was relatively minor and not correlated to the disease severity. Because human placental ADA activity is normally low it is not likely that further reduction in placental ADA would have a large impact on placental adenosine levels. Instead, we found that the expression and activity of CD73, a key enzyme responsible for the extracellular synthesis of adenosine from AMP, were significantly elevated in the placentas of women with PE. Similar to these findings in women with PE, we found that an autoantibody-injection model of PE in mice also displays increased levels of placental adenosine and CD73. The pathologic significance of increased placental CD73 and adenosine was demonstrated by experiments showing that genetic deletion of Cd73 prevented PE-IgG-induced placental adenosine production and attenuated hypertension, proteinuria, and sFlt-1 secretion in the pregnant mice. Thus, elevated CD73, not reduced ADA, underlies increased placental adenosine production and subsequent disease development.
In contrast to the work of Espinoza et al. which focuses on elevated fetal plasma adenosine22 and earlier work of others showing elevated adenosine in the maternal circulation21, our studies reported here focus on the detrimental consequences of locally elevated adenosine in the placenta. Additionally, our results with two mouse models of PE (genetically engineered mice with ADA-deficient placentas and PE-IgG injected pregnant mice) show that the elevation of placental adenosine was not accompanied with an elevated level of maternal circulating adenosine. Thus, our findings indicate that a local increase in adenosine in the placenta is sufficient to trigger features of PE in two different mouse models of PE. As a limitation of human evidence for elevated placenta adenosine, the determination of placental adenosine levels during human pregnancy prior to the onset of disease is not feasible. Nevertheless, our finding implicates that elevated circulating adenosine observed in PE patients may be secondary to an initial increase in placental adenosine and in this way may serve as a presymptomatic biomarker for PE.
The metabolic consequences of elevated adenosine include intracellular accumulation of S-adenosylhomocysteine, a potent inhibitor of many celluar methyltransferase enzymes, including those that methylate DNA. For this reason elevated adenosine is associated with DNA hypomethylation35, 36. It is likely that the elevated placental adenosine in women with PE, and in placentas from our mouse models of PE, contributes to DNA hypomethylation and changes in gene expression. However, it is important to point out the pathophysiological consequences of elevated placental adenosine that we report here are mediated through ADORA2B receptor signaling, a process distinct from the metabolic consequences of elevated adenosine on methyltransferase reactions. Adenosine is a signaling nucleoside that activates four receptors, ADORA1, ADORA2A, ADORA2B and ADORA315. In two independent animal models of PE, we demonstrated that excess placental adenosine signaling via elevated ADORA2B underlying pathophysiology of PE. Moreover, the significance of ADORA2B signaling in humans was evident from translational studies showing that ADORA2B levels were elevated in placentas of PE patients relative to controls and that enhanced ADORA2B signaling contributed to elevated sFlt-1 production in human placental villous explants.
Our studies support a novel but compelling concept of pathogenesis of PE: 1) Elevated CD73 underlies increased placental adenosine. 2) Chronic excess placental adenosine preferentially signaling via elevated ADORA2B induces sFlt-1 production, impaired placentas, small fetuses, and maternal PE features. Without interference, placental damage due to elevated sFlt-1-medialted impaired vasculature leads to further elevation of adenosine. As such, elevated CD73-mediated elevated placental adenosine, enhanced ADORA2B activation, and placental vasculature impairment function as a malicious cycle to promote the progression of maternal disease development by continuously inducing local placental accumulation of adenosine (Figure 7H). These findings suggest multiple possible therapeutic options for the treatment of PE including the use of 1) PEG-ADA to reduce adenosine levels, 2) an ADORA2B antagonist to specifically inhibit the detrimental adenosine signaling, or 3) a CD73 inhibitor to prevent the excess production of adenosine. In particular, PEG-ADA is a FDA-approved drug that has been successfully used to treat ADA-deficient patients for several decades. It is our hope that the use of adenosine-based therapies to prevent features of PE resulting from excessive placental adenosine signaling will reduce the morbidity and mortality of PE in humans in the future.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported by National Institute of Health Grants HL119549 (to Y.X.), RC4HD067977 and HD34130 (to Y.X and R.E.K.), HL73430 (to T.S.C.) and by China National Science Foundation 81228004 (to Y.X.) and by John Sealy grant (to T.S.C.).
Footnotes
Journal Subject Codes: [130] Animal models of human disease, [14] Other hypertension
Disclosures: None.
References
- 1.Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376:631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- 2.Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: Preeclampsia and beyond. Circ Res. 2013;113:78–87. doi: 10.1161/CIRCRESAHA.113.300752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23:3–19. doi: 10.1053/plac.2001.0738. [DOI] [PubMed] [Google Scholar]
- 4.Thornburg KL, Louey S. Uteroplacental circulation and fetal vascular function and development. Curr vasc pharmacol. 2013;11:748–757. doi: 10.2174/1570161111311050012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dilworth MR, Sibley CP. Review: Transport across the placenta of mice and women. Placenta. 2013;34(Suppl):S34–39. doi: 10.1016/j.placenta.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Newbern D, Freemark M. Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol Diabetes Obes. 2011;18:409–416. doi: 10.1097/MED.0b013e32834c800d. [DOI] [PubMed] [Google Scholar]
- 7.Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Annu Rev Pathol. 2010;5:173–192. doi: 10.1146/annurev-pathol-121808-102149. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed A, Rahman M, Zhang X, Acevedo CH, Nijjar S, Rushton I, Bussolati B, St John J. Induction of placental heme oxygenase-1 is protective against tnfalpha-induced cytotoxicity and promotes vessel relaxation. Mol Med. 2000;6:391–409. [PMC free article] [PubMed] [Google Scholar]
- 9.Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, Devey LR, Wigmore SJ, Abbas A, Hewett PW, Ahmed A. Negative regulation of soluble flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115:1789–1797. doi: 10.1161/CIRCULATIONAHA.106.660134. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacos E, Buhimschi IA, Buhimschi CS, Ahmed A. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation. 2013;127:2514–2522. doi: 10.1161/CIRCULATIONAHA.113.001631. [DOI] [PubMed] [Google Scholar]
- 11.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sflt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmad S, Ahmed A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ Res. 2004;95:884–891. doi: 10.1161/01.RES.0000147365.86159.f5. [DOI] [PubMed] [Google Scholar]
- 13.Lynch AM, Salmon JE. Dysregulated complement activation as a common pathway of injury in preeclampsia and other pregnancy complications. Placenta. 2010;31:561–567. doi: 10.1016/j.placenta.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. 2008;14:855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karmouty-Quintana H, Xia Y, Blackburn MR. Adenosine signaling during acute and chronic disease states. J Mol Med. 2013;91:173–181. doi: 10.1007/s00109-013-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Xia Y. Adenosine signaling in normal and sickle erythrocytes and beyond. Microbes Infect. 2012;14:863–873. doi: 10.1016/j.micinf.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Schneider DJ, Blackburn MR. Adenosine signaling and the regulation of chronic lung disease. Pharmacol Ther. 2009;123:105–116. doi: 10.1016/j.pharmthera.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H, Tao L, Lu G, Alexander DC, Milburn MV, Carter-Dawson L, Lewis DE, Zhang W, Eltzschig HK, Kellems RE, Blackburn MR, Juneja HS, Xia Y. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17:79–86. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi T, Abbasi S, Zhang H, Uray K, Chunn JL, Xia LW, Molina JG, Weisbrodt NW, Kellems RE, Blackburn MR, Xia Y. Excess adenosine in murine penile erectile tissues contributes to priapism via a2b adenosine receptor signaling. J Clin Invest. 2008;118:1491–1501. doi: 10.1172/JCI33467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoneyama Y, Suzuki S, Sawa R, Yoneyama K, Power GG, Araki T. Increased plasma adenosine concentrations and the severity of preeclampsia. Obstet Gynecol. 2002;100:1266–1270. doi: 10.1016/s0029-7844(02)02247-0. [DOI] [PubMed] [Google Scholar]
- 22.Espinoza J, Espinoza AF, Power GG. High fetal plasma adenosine concentration: A role for the fetus in preeclampsia? Am J Obstet Gynecol. 2011;205:485, e424–487. doi: 10.1016/j.ajog.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 23.Yoneyama Y, Suzuki S, Sawa R, Yoneyama K, Power GG, Araki T. Relation between adenosine and t-helper 1/t-helper 2 imbalance in women with preeclampsia. Obstet Gynecol. 2002;99:641–646. doi: 10.1016/s0029-7844(02)01657-5. [DOI] [PubMed] [Google Scholar]
- 24.Matsubara S, Sato I. Plasma adenosine levels and p-selectin expression on platelets in preeclampsia. Obstet Gynecol. 2001;98:354–355. doi: 10.1016/s0029-7844(01)01467-3. [DOI] [PubMed] [Google Scholar]
- 25.George EM, Cockrell K, Adair TH, Granger JP. Regulation of sflt-1 and vegf secretion by adenosine under hypoxic conditions in rat placental villous explants. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1629–1633. doi: 10.1152/ajpregu.00330.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- 27.Blackburn MR, Wakamiya M, Caskey CT, Kellems RE. Tissue-specific rescue suggests that placental adenosine deaminase is important for fetal development in mice. J Biol Chem. 1995;270:23891–23894. doi: 10.1074/jbc.270.41.23891. [DOI] [PubMed] [Google Scholar]
- 28.Roberts JM, Pearson G, Cutler J, Lindheimer M, Pregnancy NWGoRoHD Summary of the nhlbi working group on research on hypertension during pregnancy. Hypertension. 2003;41:437–445. doi: 10.1161/01.HYP.0000054981.03589.E9. [DOI] [PubMed] [Google Scholar]
- 29.Migchielsen AA, Breuer ML, van Roon MA, te Riele H, Zurcher C, Ossendorp F, Toutain S, Hershfield MS, Berns A, Valerio D. Adenosine-deaminase-deficient mice die perinatally and exhibit liver-cell degeneration, atelectasis and small intestinal cell death. Nat Genet. 1995;10:279–287. doi: 10.1038/ng0795-279. [DOI] [PubMed] [Google Scholar]
- 30.Wakamiya M, Blackburn MR, Jurecic R, McArthur MJ, Geske RS, Cartwright J, Jr, Mitani K, Vaishnav S, Belmont JW, Kellems RE, et al. Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc Natl Acad Sci U S A. 1995;92:3673–3677. doi: 10.1073/pnas.92.9.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep. 2007;9:480–485. doi: 10.1007/s11906-007-0088-1. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, Sun K, Glover L, Grenz A, Sun H, Tao L, Zhang W, Colgan SP, Blackburn MR, Eltzschig HK, Kellems RE, Xia Y. Elevated ecto-5′-nucleotidase-mediated increased renal adenosine signaling via a2b adenosine receptor contributes to chronic hypertension. Circ Res. 2013;112:1466–1478. doi: 10.1161/CIRCRESAHA.111.300166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneyama Y, Sawa R, Suzuki S, Doi D, Yoneyama K, Otsubo Y, Araki T. Relationship between plasma malondialdehyde levels and adenosine deaminase activities in preeclampsia. Clin Chim Acta. 2002;322:169–173. doi: 10.1016/s0009-8981(02)00175-4. [DOI] [PubMed] [Google Scholar]
- 34.Kafkasli A, Karabulut AB, Atmaca R, Laurini R. Clinical correlation between adenosine deaminase activity and pre-eclampsia severity. J Int Med Res. 2006;34:247–255. doi: 10.1177/147323000603400303. [DOI] [PubMed] [Google Scholar]
- 35.Johnston JM, Kredich NM. Inhibition of methylation by adenosine in adenosine deaminase-inhibited, phytohemagglutinin-stimulated human lymphocytes. J Immunol. 1979;123:97–103. [PubMed] [Google Scholar]
- 36.Kredich NM, Hershfield MS. S-adenosylhomocysteine toxicity in normal and adenosine kinase-deficient lymphoblasts of human origin. Proc Natl Acad Sci U S A. 1979;76:2450–2454. doi: 10.1073/pnas.76.5.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.