

Abstract
Aim
Increasing evidence points to lipoprotein composition rather than reverse cholesterol transport in the cardioprotective properties of high-density lipoproteins (HDLs). HDL binding to receptors at the surface of cardiomyocytes activates signalling pathways promoting survival, but downstream targets are largely unknown. Here, we investigate the pathways by which the sphingosine-1-phosphate (S1P) constituent of HDL limits cell death induced by cardiac ischaemia–reperfusion (I/R).
Methods and results
Apolipoprotein M (ApoM) transgenic (Apom-Tg) mice, in which plasma S1P is increased by 296%, and wild-type (WT) mice were subjected to in vivo I/R. Infarct size, neutrophil infiltration into the infarcted area, and serum Troponin I were less pronounced in Apom-Tg mice. In vitro experiments suggest that this cardioprotection depends on direct effects of S1P on cardiomyocytes, whereas leucocyte recruitment seems only indirectly affected. Importantly, short-term S1P treatment at the onset of reperfusion was sufficient to reduce I/R injury in isolated perfused hearts. Mechanistic in vitro and ex vivo studies revealed that 5 min of S1P treatment induced phosphorylation of the gap junction protein Connexin43 (Cx43) on Serine368 (S368), which was mediated by S1P2 and S1P3, but not by S1P1, receptors in cardiomyocytes. Finally, S1P-induced reduction of infarct size after ex vivo I/R was lost in hearts of mice with a truncated C-terminus of Cx43 (Cx43K258/KO) or in which the S368 is mutated to a non-phosphorylatable alanine (Cx43S368A/S368A).
Conclusion
Our study reveals an important molecular pathway by which modulating the apoM/S1P axis has a therapeutic potential in the fight against I/R injury in the heart.
Keywords: Sphingosine-1-phosphate, Ischaemia–reperfusion injury, Connexin43
1. Introduction
Ischaemic heart disease remains a main cause of morbidity and mortality worldwide. Treatment of acute myocardial infarction consists of procedures that allow the rapid return of blood flow to the ischaemic zone of the myocardium to rescue heart muscle. Although it is essential to re-open the occluded artery as soon as possible, reperfusion may, however, paradoxically lead to further complications involving acceleration of cell death, diminished contractile function (stunning), and arrhythmias. No effective therapy is currently available to protect the heart from this ischaemia–reperfusion (I/R) injury.1
More than three decades ago, the cholesterol carried in high-density lipoprotein (HDL) was shown to be strongly and inversely related to coronary heart disease. Recent studies ascribe to HDL properties that go well beyond its ability to promote cholesterol efflux. HDL is a major transporter of S1P through binding to the HDL-associated apolipoprotein M (apoM), and HDL-associated S1P is bioactive in several ways through activation of its receptors on various types of cells.2,3 On cardiac cells, S1P binds to three G protein-coupled receptors: S1P1, S1P2, and S1P3, leading to the activation of several ‘intracellular’ signalling pathways potentially implicated in cardioprotection.3,4 Two well-known intracellular pathways limiting I/R injury involve RISK (reperfusion injury salvage kinase) and the SAFE (survivor activating factor enhancement) signalling.5 Recent data suggest however that cardioprotection by HDL may also implicate an ‘intercellular’ signalling pathway, namely Connexin43 (Cx43)-mediated gap junctional communication.6
Gap junction channels, made of connexin proteins, form low-resistance pathways between cardiomyocytes ensuring the cell-to-cell flow of electrical current and small molecules. They are essential for the coordination and synchronization of cardiomyocyte contraction and, in addition, allow the intercellular spread of small metabolites guiding towards cellular survival or death. Opening and closure of Cx43 channels is regulated by several factors that are altered during I/R such as intracellular pH and the activity of intracellular kinases.7 Cx43 channels play an important role in the limitation of myocardial infarction.8,9 Although their role in pre-conditioning of the heart is well established,10 there are only a few studies pointing to a role for Cx43 in post-conditioning and the outcomes are controversial.11–13
Here, we investigated the role of Cx43 phosphorylation in the cardioprotection conferred by the apoM/S1P axis. In vitro and ex vivo we demonstrate that short-term S1P treatment induces phosphorylation of Cx43 on Serine368 (S368), via S1P2 and S1P3 receptors on cardiomyocytes. By using ex vivo and in vivo models, we prove for the first time that chronic but also short-term elevation of S1P limits development of I/R injury. Finally, we link the cardioprotective properties of S1P against I/R-induced cell death to the phosphorylation of Cx43 on S368.
2. Methods
Detailed methods are available as Supplementary material online.
2.1. Animals
Apom-Tg and Apom−/−,14 Cx43K258stop/KO,15 and Cx43S368A/S368A 16 mice have been generated as previously described and have been subjected to in vivo and ex vivo protocols of I/R as previously described.6,17 Briefly, in vivo I/R was induced by occlusion of the left anterior descending (LAD) coronary artery for 30 min followed by 24 h of reperfusion and ex vivo I/R was performed with the Langendorff perfusion system in which the heart was submitted to 30 min of no-flow global ischaemia and 60 min of reperfusion. Infarct area was determined by TTC staining. In some experiments, S1P, S1P2, and S1P3 receptor antagonists were added to the perfusion during the first 5 min of reperfusion. Electrophysiological and mechanical measurements were performed as previously described18 on tissue strips of the right ventricle. All animal experimentation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication Eighth Edition, 2011) and was approved by veterinary authorities.
2.2. HPLC-based quantification of S1P
Blood from wild-type (WT), Apom-Tg, and Apom−/− mice was taken from the facial vein, plasma was isolated, and quantification of S1P was performed by HPLC as previously described.19
2.3. In vitro neutrophil transmigration assay
Neutrophils from WT mice were isolated as described previously.20 Neutrophil transmigration was stimulated by the addition of the chemoattractant molecule CXCL2. S1P was added to the neutrophils just before the beginning of the assay. After 1 h of transmigration, the number of migrated neutrophils was counted by flow cytometry.
2.4. Immunostaining and western blotting
Neonatal rat ventricular cardiomyocytes (NRVCs) were isolated and cultured as described previously.6 After treatment with S1P, S1P1 agonist, or S1P1-3 receptor antagonists, proteins were extracted in modified radioimmunoprecipitation assay buffer.6 Mitochondrial proteins were obtained with the MITO-ISO2 kit (Sigma-Aldrich). Western blotting was performed using antibodies against Phospho-Cx43 (Ser368), total Cx43, Phospho-ERK1/2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), voltage-dependent anion-selective channel (VDAC), and Na/K/ATPase.
Immunostaining against Phospho-Cx43 (Ser368) or Ly-6G (for neutrophils) were performed on NRVC or heart cryosections. Western blot and immunofluorescence quantifications were performed using NIH Image software (NIH AutoExtractor 1.51; National Institute of Health). Neutrophil content was quantified by dividing the number of positive cells for Ly-6G by the total number of cells in the infarct area.17
2.5. Statistical analysis
All analyses were done with GraphPad Prism 5.01 software, and results are expressed as mean ± SEM. For two group's comparison, unpaired t-test was performed, and for multiple group comparison, one-way ANOVA with Bonferroni's post-test was used. Differences were considered statistically significant at P < 0.05.
3. Results
3.1. Constitutive increase of plasma S1P in Apom-Tg mice protects the myocardium against in vivo I/R-induced cell death
To investigate the involvement of plasma S1P in myocardial I/R pathophysiology, we used Apom-Tg mice with 10-fold increased plasma apoM concentration19 leading to an increase of plasma S1P by 296% (P < 0.01) (Figure 1A). Mice were subjected to 30 min of LAD coronary artery occlusion followed by 24 h of reperfusion (Figure 1B). Whereas the area at risk was not different between Apom-Tg and WT mice (Figure 1C), the infarct area was reduced (P < 0.05) in Apom-Tg mice compared with WT mice (Figure 1D). In apoM-deficient mice (Apom−/−) in which plasma S1P was reduced by 57% (plasma S1P = 0.28 μM, P < 0.01 vs. WT), the infarct area was not affected (area at risk = 30.9 ± 4.1%; infarct area = 32.7 ± 6.4%, n = 9) in comparison with WT mice. In agreement with the measurements of infarct area, serum Troponin I levels were lower (P < 0.01) in Apom-Tg mice compared with WT mice (Figure 1E). Finally, the number of Ly-6G positive cells in the infarct area were significantly lower in Apom-Tg mice than that in WT mice (Figure 1F and G). As S1P is a potent chemoattractant that can regulate endothelial cell and lymphocyte migration,21,22 we performed in vitro neutrophil transmigration assays in the presence of S1P at concentrations (0.5 and 1.8 μM) close to the ones measured in WT and Apom-Tg mice.19 Neutrophil migration was induced by the classical chemoattractant CXCL2. Addition of 0.5 or 1.8 μM S1P to the chemoattraction medium did not change in vitro neutrophil migration (Figure 1H). Taken together, these results show that elevation of plasma S1P limits development of cardiac I/R injury.
Figure 1.

Constitutive increase of plasma S1P in Apom-Tg mice protects the myocardium against in vivo I/R-induced cell death. (A) Plasma S1P concentration is increased in Apom-Tg mice in comparison to WT mice. n = 17/group. **P < 0.01 vs. WT. (B) Representative photographs of heart slices of WT and Apom-Tg mice stained with Evans blue and TTC after I/R. Scale bar = 1 mm. Areas at risk (C) are not different between WT and Apom-Tg mice. Infarct area (D) is decreased in Apom-Tg mice in comparison with WT mice. n = 8–9/group. **P < 0.01 vs. WT. (E) Serum Troponin I level is lower in Apom-Tg mice in comparison with WT mice. n = 3/group. **P < 0.01 vs. WT. (F) Representative images of WT and Apom-Tg heart immunolabelled for Ly6G (neutrophils in green) after I/R. Scale bar = 20 μm. (G) Neutrophil infiltration is reduced in Apom-Tg mice compared with WT mice. n = 3/group. *P < 0.05 vs. WT. (H) In vitro neutrophil migration was induced by the neutrophil chemoattractant CXCL2. Addition of S1P (0.5 or 1.8 μM) to the neutrophils did not modify their transmigratory properties. n = 5/group. #P < 0.05 vs. no CXCL2.
3.2. Myocardial function and sensitivity to I/R is not different in Apom-Tg hearts
To study whether the constitutive increase in plasma apoM/S1P concentration affects heart function, we performed ex vivo functional assays to characterize myocardial left ventricular function and ventricular conduction velocity (CV) of Apom-Tg mice. Left ventricular function was assessed on Langendorff-perfused hearts by introducing a balloon connected to a pressure transducer into the left ventricle of Apom-Tg and WT hearts. CV was measured on strips of right ventricular myocardium of Apom-Tg and WT mice. We did not observe differences in left ventricular developed pressure (LVDP), heart rate, +dP/dt(C), −dP/dt (Figure 2A–D) and CV (Figure 2E and F) between Apom-Tg and WT mice.
Figure 2.

Ex vivo myocardial function and sensitivity to I/R of WT and Apom-Tg mice. LVDP (A), heart rate (B), +dP/dt(C), −dP/dt (D) are not different between WT and Apom-Tg mice. n = 5–6/group. (E) Representative traces of CV measurement on a strip from the right ventricle; bar indicates a 1.376 ms delay between activation under electrode 1 (E1) and 2 (E2). (F) CV is not different between WT and Apom-Tg mice. n = 4/group. (G) Myocardial sensitivity to ischaemia, measured as TTOC, TTMC, and Ischemic Rigor during the ischaemia, is not different between WT and Apom-Tg mice. (H) Representative photographs of heart slices from WT and Apom-Tg mice stained with TTC after I/R. Scale bar = 1 mm. (I) Ex vivo infarct areas are not different between WT and Apom-Tg mice. (J) The recovery of the LVDP after 60 min of reperfusion is not different between WT and Apom-Tg mice. n = 5–6/group.
Myocardial sensitivity to ex vivo ischaemia was evaluated by measuring three parameters characterizing the ischaemic contracture: the time to onset of contracture (TTOC), the time to maximal contracture (TTMC), and the Ischemic Rigor. All three parameters were not different between hearts from Apom-Tg and WT mice (Figure 2G). The infarcted area (Figure 2H and I) and the recovery of LVDP (Figure 2J) measured after 30 min of no-flow ischaemia and 60 min of reperfusion were also similar in hearts from Apom-Tg and WT mice. Thus, heart function per se and the sensitivity of the myocardium to I/R are not affected by chronic elevation in plasma apoM and S1P. Altogether, these results demonstrate that the lower infarct area observed in Apom-Tg mice after in vivo I/R is due to their elevated plasma apoM/S1P and not to intrinsic modifications of their cardiac myocytes.
3.3. Exogenous S1P protects the heart against ex vivo I/R-induced cell death
Next, we investigated whether short-term treatment with S1P at the onset of reperfusion would protect the heart from I/R-induced cell death. WT hearts were submitted to 30 min of no-flow global ischaemia followed by 60 min reperfusion, and S1P was added or not to the perfusion buffer during the first 5 min of reperfusion. As expected, neither the left ventricular function measured during stabilization (Table 1, WT hearts and S1P-treated WT hearts) nor the myocardial sensitivity to ischaemia measured before S1P treatment (Table 2) were different between both groups. Interestingly, the infarct area in S1P-treated hearts was significantly reduced compared with control hearts (Figure 3A and B). After 60 min of reperfusion, the recovery of the left ventricular function was not different between WT and S1P-treated WT hearts (Table 1).
Table 1.
Ex vivo myocardial function of Cx43+/+ (WT), Cx43+/KO, Cx43K258stop/KO, and Cx43S368A/S368A hearts treated or not with S1P
| Untreated WT hearts | S1P-treated WT hearts | Untreated Cx43+/KO hearts | S1P-treated Cx43+/KO hearts | Untreated Cx43K258stop/KO hearts | S1P-treated Cx43K258stop/KO hearts | Untreated Cx43S368A/S368A hearts | S1P-treated Cx43S368A/S368A hearts | |
|---|---|---|---|---|---|---|---|---|
| End of stabilization | ||||||||
| LVEDP (mmHg) | 10 ± 1 | 9 ± 2 | 10 ± 1 | 8 ± 1 | 13 ± 1 | 8 ± 2 | 12 ± 1 | 11 ± 1 |
| LVDP (mmHg) | 85 ± 15 | 86 ± 11 | 125 ± 18 | 98 ± 10 | 83 ± 10 | 86 ± 17 | 91 ± 10 | 74 ± 6 |
| Heart rate (beats/min) | 264 ± 26 | 286 ± 17 | 246 ± 49 | 288 ± 43 | 201 ± 28 | 264 ± 48 | 327 ± 27 | 345 ± 41 |
| +dP/dt (mmHg/s) | 2027 ± 330 | 2246 ± 342 | 2820 ± 480 | 2434 ± 257 | 1704 ± 265 | 2141 ± 479 | 2422 ± 256 | 1936 ± 93 |
| −dP/dt (mmHg/s) | 1428 ± 251 | 1531 ± 235 | 1781 ± 178 | 1735 ± 157 | 1102 ± 172 | 1480 ± 346 | 1672 ± 181 | 1380 ± 95 |
| End of reperfusion | ||||||||
| LVEDP (mmHg) | 47 ± 12 | 37 ± 10 | 65 ± 5 | 42 ± 8 | 55 ± 6 | 47 ± 4 | 52 ± 7 | 43 ± 5 |
| LVDP (% end of stabilization) | 31 ± 6 | 19 ± 4 | 16 ± 5 | 38 ± 14 | 24 ± 6 | 25 ± 7 | 33 ± 5 | 51 ± 8 |
| Heart rate (% end of stabilization) | 90 ± 6 | 71 ± 5 | 116 ± 20 | 78 ± 7 | 101 ± 10 | 84 ± 11 | 72 ± 5 | 62 ± 9 |
| +dP/dt (% end of stabilization) | 33 ± 6 | 21 ± 3 | 17 ± 7 | 51 ± 15 | 28 ± 6 | 29 ± 6 | 32 ± 6 | 52 ± 8 |
| −dP/dt (% end of stabilization) | 33 ± 5 | 22 ± 3 | 15 ± 5 | 48 ± 15 | 30 ± 5 | 28 ± 7 | 32 ± 6 | 50 ± 9 |
After 20 min of stabilization, the left ventricular function characterized by the measurement of left ventricular end-diastolic pressure (LVEDP), LVDP, heart rate, +dP/dt(C), −dP/dt is similar for all four groups. After 60 min of reperfusion, the LVEDP and the recovery of left ventricular function is not affected by S1P treatment in comparison with control hearts. n = 5/group.
Table 2.
Ischaemic sensitivity of mice hearts during 30 min of no-flow global ischaemia
| Untreated WT hearts | S1P-treated WT hearts | Untreated Cx43+/KO hearts | S1P-treated Cx43+/KO hearts | Untreated Cx43K258stop/KO hearts | S1P-treated Cx43K258stop/KO hearts | Untreated Cx43S368A/S368A hearts | S1P-treated Cx43S368A/S368A hearts | |
|---|---|---|---|---|---|---|---|---|
| TTOC (min) | 15 ± 1 | 12 ± 1 | 16 ± 1 | 12 ± 1 | 15 ± 1 | 17 ± 1 | 14 ± 1 | 13 ± 1 |
| TTMC (min) | 21 ± 1 | 20 ± 1 | 25 ± 2 | 18 ± 1 | 21 ± 2 | 26 ± 2 | 21 ± 2 | 20 ± 1 |
| Ischaemic Rigor (mmHg) | 41 ± 5 | 37 ± 6 | 50 ± 1 | 46 ± 8 | 43 ± 4 | 39 ± 4 | 43 ± 3 | 43 ± 2 |
TTOC, TTMC, and Ischaemic Rigor are not different between the different hearts before treatment with S1P. n = 5/group.
Figure 3.
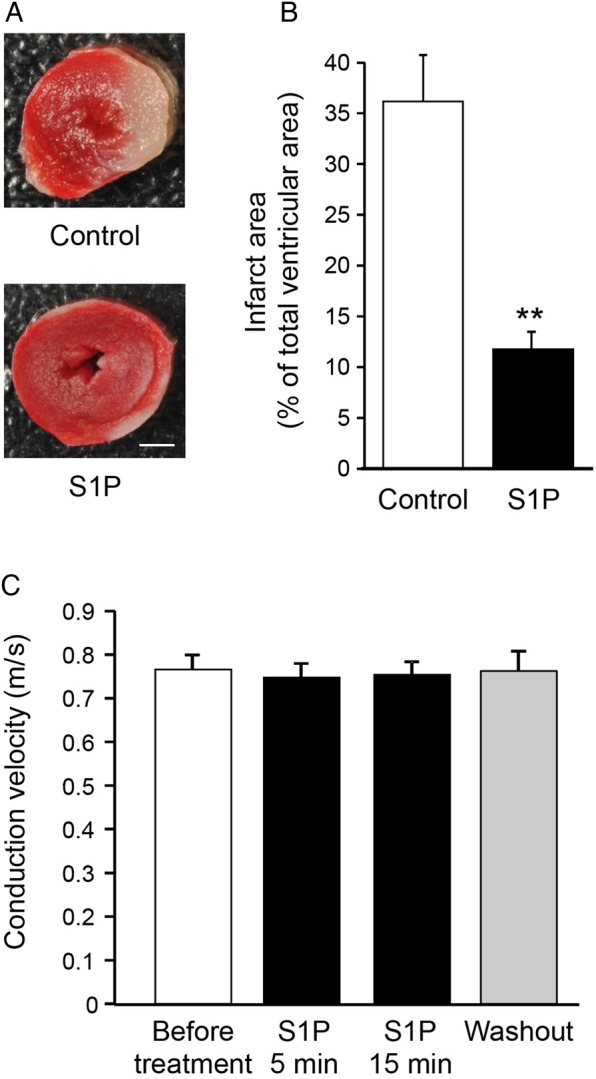
Exogenous S1P protects the heart against ex vivo I/R-induced cell death. (A) Representative photographs of S1P-treated and untreated (control) heart slices stained with TTC after I/R. Scale bar = 1 mm. (B) Infarct area is lower in S1P-treated hearts in comparison with control hearts. n = 6–7/group. **P < 0.01 vs. control. (C) CV measured on ventricular strips is not modified after 5 or 15 min of treatment with S1P or after washout as compared before treatment. n = 4/group.
Next, we studied whether short-term S1P treatment would affect CV in ventricular strips of WT hearts. Myocardial CV was not affected by 5 or 15 min treatment with S1P nor after washout of the molecule (Figure 3C). Thus, short-term S1P treatment at the onset of the reperfusion significantly limits I/R-induced cell death without negatively affecting myocardial CV or recovery of the heart.
3.4. Exogenous S1P maintains phosphorylation of Cx43 shortly after ex vivo I/R
We have recently shown in vitro that short-term treatment with HDL induces phosphorylation of cardiac Cx43 at the serine in Position 368 in the C-terminal tail of the gap junction protein.6 This specific phosphorylation of Cx43 reduced the diffusion of large tracer molecules between cardiomyocytes, whereas action potential conduction was maintained.6 To identify Cx43-S368 phosphorylation in hearts submitted to ex vivo I/R, we quantified immunofluorescent staining of Cx43 with specific antibodies recognizing phosphorylated S368. As shown in Figure 4A, B and G, 30 min of global ischaemia increased Cx43-S368 phosphorylation at the intercalated disc in comparison to hearts after stabilization (before ischaemia). After reperfusion, S368 phosphorylation of Cx43 returned immediately to basal levels in untreated hearts (Figure 4C, D and G). However, short-term treatment with S1P at the onset of reperfusion maintained myocardial Cx43-S368 phosphorylation 5 min after reperfusion was initiated (Figure 4E and G), whereas the level of Cx43-S368 phosphorylation only returned to basal after 60 min of reperfusion (Figure 4F and G).
Figure 4.

Phospho-Cx43 (Ser368) immunofluorescent staining (in green) in Langendorff-perfused hearts submitted to ex vivo I/R. Thirty minutes of no-flow global ischaemia (B, G) favoured Cx43-S368 phosphorylation in comparison to stabilization (A, G). Five minute S1P treatment increased this phosphorylation (E, G) in comparison with 5 min reperfusion without treatment (C, G). After 60 min of reperfusion, there is no difference between hearts treated with (F, G) or without (D, G) S1P. Scale bar = 20 μm. n = 3/group. *P < 0.05 vs. stabilization.
Next, we studied whether S1P solely induced Cx43 phosphorylation in the plasma membrane at cell–cell contacts between cardiomyocytes or whether mitochondrial Cx43 might be involved as well. As shown in Figure 5A, we first confirmed in vitro that S1P induced Cx43-S368 phosphorylation at cell–cell contacts between NRVCs. Western blots of mitochondrial protein extracted from NRVCs were immunoreactive for Cx43, Cx43-S368, and the mitochondrial protein VDAC (Figure 5B and C) and showed no immunosignal for GAPDH and Na/K-ATPase, as expected (not shown). Five minute treatment with S1P led to an increased Cx43-S368 phosphorylation (Figure 5B) without affecting the total level of Cx43 present in the mitochondrial fraction (Figure 5C). Altogether, these results demonstrate that short-term S1P treatment increased Cx43-S368 phosphorylation at cell–cell contacts and in mitochondria of cardiomyocytes.
Figure 5.

Membrane and mitochondrial Cx43 phosphorylation after S1P treatment. (A) Five minute S1P treatment induces an increase in the signal for phosphorylated Cx43 on S368 (in red) at the plasma membrane of NRVCs. Scale bar = 50 μm. Representative western blots of mitochondrial Phospho-Cx43 (Ser368) (B) and total Cx43 (C) and VDAC in the absence of S1P and after 5 min S1P treatment. S1P treatment increases mitochondrial phosphorylation of Cx43 on S368 (B) but does not affect the total amount of Cx43 (C). n = 4/group. *P < 0.05 vs. no treatment.
3.5. S1P induces phosphorylation of Cx43 by S1P2 and S1P3, but not S1P1, receptors
The biological effects of S1P are mediated by activation of G-protein-coupled S1P receptors. S1P binding to S1P1-3 receptors in the heart activates downstream signalling pathways that promote myocyte survival. To identify the receptors implicated in Cx43 phosphorylation, NRVCs were treated with specific antagonists of S1P1, S1P2, or S1P3 receptors before addition of S1P. As shown in Figure 6A and B, 5 min treatment with S1P induced an increase in Cx43 phosphorylation at S368, which was abolished by pre-treating the cells with S1P2 or S1P3 receptor antagonists but not by an S1P1 receptor antagonist. Moreover, treatment of cardiomyocytes with S1P1 agonist did also not alter Cx43 phosphorylation at S368 (Figure 6B). However, the phosphorylation of ERK1/2 induced by S1P was inhibited by pre-incubation with the S1P1 antagonist (see Supplementary material online, Figure S1A), and the S1P1 agonist induced phosphorylation of ERK1/2 to a level comparable with that induced by S1P (see Supplementary material online, Figure S1B), thus confirming the presence and activity of S1P1 receptors on the cells. Interestingly, co-treatment with S1P and S1P2/S1P3 receptor antagonists abolished the maintenance of Cx43-S368 phosphorylation that was observed ex vivo after short-term treatment at the onset of reperfusion with S1P alone (Figures 4E and G, 6C). Moreover, infarct size was significantly larger after 60 min of reperfusion in hearts co-treated with S1P and S1P2/S1P3 receptor antagonists in comparison with hearts receiving S1P alone (Figure 6D). Collectively, these results suggest that S1P treatment at the onset of reperfusion activates S1P2 and S1P3 receptors on cardiomyocytes leading to maintenance of Cx43-S368 phosphorylation, which possibly confers protection against I/R-induced cell death.
Figure 6.

S1P induces phosphorylation of Cx43 through S1P2 and S1P3, but not S1P1 receptors. (A) Representative western blot of Phospho-Cx43 (Ser368) and GAPDH in the absence of S1P, after 5 min of treatment with S1P or after 30 min of pre-incubation with S1P1, S1P2, or S1P3 receptor antagonist followed by 5 min of S1P treatment. (B) Phosphorylation of Cx43 on Ser368 induced by S1P is inhibited by pre-incubation with S1P2 and S1P3 receptor antagonists, but not with an S1P1 receptor antagonist. S1P1 agonist does not affect the S368 phosphorylation status of Cx43. (C) After 5 min of reperfusion, Cx43 phosphorylation on Ser368 is increased in the presence of S1P in comparison of reperfusion without treatment. Co-treatment with S1P2 and S1P3 receptor antagonists inhibits Cx43 phosphorylation induced by S1P. (D) Infarct size reduction induced by S1P treatment is abolished in the presence of S1P2 and S1P3 receptor antagonists. n = 3–14/group. *P < 0.05 vs. no treatment, #P < 0.05 vs. S1P, ##P < 0.01 vs. S1P.
3.6. Limitation of myocardial infarction by S1P implicates Cx43 phosphorylation
To study the implication of phosphorylation of Cx43 at S368 in S1P-induced cardioprotection, we used Cx43K258stop/KO mice in which the C-terminal tail of the protein has been deleted15 and Cx43S368A/S368A mice in which the S368 has been mutated to alanine, a non-phosphorylatable amino acid.16 Hearts of these mice were submitted to 30 min of no-flow global ischaemia followed by 60 min reperfusion, and S1P was added or not to the perfusion buffer during the first 5 min of reperfusion. At the end of the stabilization, the myocardial function was not different between all groups (Table 1). Moreover, the myocardial sensitivity to ischaemia (TTOC, TTMC, and Ischemic Rigor) was not different between all hearts (Table 2). Interestingly, addition of S1P to the perfusion buffer at the onset of the reperfusion did not affect infarct size in Cx43K258stop/KO mice or in Cx43S368A/S368A mice (Figure 7). Infarct size measured in Cx43+/KO was also not significantly different from the infarct size measured in Cx43K258stop/KO mice both under control conditions and after S1P (32.0 ± 4.0% and 28.0 ± 5.0%, n = 5, respectively). Finally, the recovery of the myocardial function after 60 min of reperfusion was similar between control and S1P-treated groups (Table 1). Thus, phosphorylation of Cx43 at S368 mediates S1P-induced cardioprotection against I/R-induced cell death.
Figure 7.

The absence of S1P cardioprotection against ex vivo I/R-induced cell death in Cx43K258stop/KO hearts (A) and Cx43S368A/S368A hearts (B). n = 5/group.
4. Discussion
S1P is a bioactive lysophospholipid that regulates many important cellular processes. Plasma S1P is principally produced by endothelial cells, erythrocytes, platelets, and hepatocytes, and ∼60% of plasma S1P is transported by HDL. Whereas the anti-inflammatory properties and vascular effects of S1P are well-known, recent evidence points to the functional importance of S1P for cardiac myocytes, and in particular to its cardioprotective properties against I/R injury.4
We took advantage of Apom-Tg mice in which plasma S1P is increased by 296% to study in vivo the effects of a chronic alteration in plasma S1P on I/R injury. We observed a reduction in myocardial infarct size and serum Troponin I in Apom-Tg mice when compared with WT mice, demonstrating that chronic elevation of plasma apoM/S1P is cardioprotective. Exposing Apom−/−- mice, in which plasma S1P is reduced by 57%, to I/R did not worsen the outcome. Thus, even in the absence of apoM, plasma S1P bound to, for example, albumin protects the heart from damaging conditions.
The atheroprotective properties of HDL have been attributed to the ability of S1P to preserve endothelial function and to inhibit pro-inflammatory signalling in leucocytes.23 In the process of myocardial infarction, the infiltration of inflammatory cells occurs quickly after the onset of reperfusion and a peak of neutrophil recruitment is usually observed 24 h later.24 We observed smaller myocardial infarcts and reduced neutrophil infiltration into the infarcted area of Apom-Tg mice when compared with WT mice. Chemoattraction of neutrophils in vitro was, however, not affected by S1P at the concentrations found in Apom-Tg or WT mice. Thus, our results suggest that cardioprotection conferred by S1P against in vivo I/R is due to a direct effect of S1P on cardiac myocytes and not to inhibition of pro-inflammatory signalling in leucocytes. These results are in accordance with previous studies, showing that S1P directly mediates survival of cardiomyocytes subjected to stress.25,26 However, as reperfusion injury depends in part on microvascular endothelial integrity, we cannot exclude that a direct effect of the chronic rise in plasma S1P on endothelial cells contributes as well to the reduction of myocardial infarction in Apom-Tg mice.
The actions of S1P on cardiac cells are mediated via various receptors. S1P1 is the main S1P receptor expressed in cardiomyocytes and it typically signals through the RISK and SAFE pathways for cardioprotection.4,5 Various intracellular signalling pathways, including ERK, Akt, or PKC, have been proposed to mediate the cardioprotective effects of S1P2 and S1P3 receptors.5,27,28 Importantly, the combined deletion of S1P2 and S1P3 receptors in mice increased infarct size in vivo after I/R, whereas no differences in infarct size were observed in S1P2−/− and S1P3−/− mice,29 suggesting that only the joint action of S1P2 and S1P3 receptors protects the heart from I/R-induced cell death. Here, we show in vitro and ex vivo that activation of S1P2 and S1P3 receptors by S1P led to the phosphorylation of Cx43 at S368, whereas activation of S1P1 was not implicated in this effect. S368 is a well-known phosphorylation site for PKC in the C-terminal tail of Cx43, and PKC-induced phosphorylation by the phorbol ester TPA has been shown to increase gap junctional conductance (Gj) between pairs of NRVCs, while at the same time the transfer of the large fluorescent molecular tracer Lucifer Yellow (MW 457) was reduced.30 These opposite effects of PKC-induced modulation of electrical and metabolic coupling were explained by parallel effects on the channels open probability (supposedly increasing Gj) and unitary conductance (supposedly decreasing dye transfer).30,31 The latter hypothesis was proved in studies using cells expressing Cx43 in which the S368 was changed for the non-phosphorylable residue alanine in which a reduced incidence of the larger-sized unitary conductances32,33 and a severely reduced (by 10-fold) selective permeability of the junctions for a small cationic dye32 were observed. In addition, phosphorylation of S368 by PKC induces a conformational change of Cx43 that results in a decrease in channel permeability.34 We have recently demonstrated that PKC-dependent phosphorylation of Cx43-S368 by S1P also limits the exchange of the molecular tracer Lucifer Yellow between cardiomyocytes.6 A continuously increasing number of large molecular death signals have been described to pass through gap junctions since the seminal observation by Mesnil et al.35 that bystander killing of cancer cells by herpes simplex virus thymidine kinase gene was mediated by Cx43. As shown in Figure 4, the level of Cx43-S368 phosphorylation is rapidly reduced by reperfusion in control hearts, whereas S1P treatment retains Cx43-S368 phosphorylation after the onset of reperfusion. In addition, we demonstrate that treatment with S1P2/S1P3 receptor antagonists abolished S1P-induced cardioprotection against I/R injury (Figure 6). Together, these observations suggest that S1P2 and S1P3 receptors mediate S1P-induced Cx43-S368 phosphorylation that, in turn, limits the spread of death signals through gap junctions to neighbouring cells and consequently reduces I/R-induced cell death.
In a pre-conditioning setting, mitochondrial Cx43 is involved in the resistance to myocardial I/R injury.36,37 Indeed, mitochondrial Cx43 may impact respiratory function,38,39 mitochondrial permeability,40 on reactive oxygen species production,41 and calcium-induced permeability transition pore opening.42,43 In our study, we showed that 5 min of treatment with S1P did not change the total amount of mitochondrial Cx43 but significantly increased its phosphorylation in S368 (Figure 5). Interestingly, dephosphorylation of Cx43 at S368 lead to the opening of the permeability transition pore of rat brain mitochondria resulting in cell death.42 Together, these observations warrant detailed future studies onto the effect of S1P on mitochondrial Cx43 in post-conditioning protection against cardiac I/R injury.
To further define the role of Cx43 in S1P-induced cardioprotection, we used mouse lines with genetically modified Cx43: Cx43K258stop/KO mice, in which the C-terminal tail of the protein is no longer present15 and Cx43S368A/S368A mice in which the S368 is mutated to the non-phosphorylatable amino acid alanine.16 Hearts from adult Cx43K258stop/KO mice displayed a normal morphology and, as shown using laser scanning microscopy and transmission electron microscopy, the spatial distribution of the Cx43-K258stop gap junction channels is altered, i.e. these mutant channels localize to the periphery of the intercalated disc.44 Cx43-K258stop gap junction plaque size is increased while the number of plaques is reduced, and Cx43-K258stop cardiomyocytes are functionally coupled.44 Exposing these hearts to ex vivo I/R, we observed that the cardioprotective effect of S1P against I/R-induced cell death was lost, illustrating the importance of the C-terminal tail of Cx43 in this pharmacological post-conditioning. As in vitro experiments revealed that S1P specifically induced phosphorylation of Cx43 on S368, we next used Cx43S368A/S368A mice.16 Cx43 density and localization in heart ventricle is similar in Cx43S368A/S368A and WT hearts.16 As expected, the ex vivo myocardial function of Cx43S368A/S368A hearts was normal. We found that S1P-induced cardioprotection against I/R injury is abolished in Cx43S368A/S368A mice underlining the crucial role of the phosphorylation of the S368 residue of Cx43 in this process.
Ischaemic heart disease is associated with high morbidity and mortality. Rapid re-establishment of blood flow to the ischaemic zone is mandatory to allow for survival of cardiac myocytes, but the effectiveness of this therapy is often hampered by I/R injury. This study reveals an important molecular pathway by which modulating the apoM/S1P axis has a therapeutic potential in the fight against I/R injury. It shows for the first time that S1P-induced cardioprotection involves intercellular signalling via the gap junction protein Cx43. S1P treatment at the onset of reperfusion activates S1P2 and S1P3 receptors on cardiomyocytes leading to phosphorylation of Cx43-S368 and limitation of I/R-induced cell death without negatively affecting recovery of the heart.
Supplementary material
Supplementary Material is available at Cardiovascular Research online.
Funding
This work was supported by the Swiss National Science Foundation (310030_143343/1 and 310030_162579/1 to B.R.K. and 310030_152639/1 to Fa.M.), Swiss Life Foundation (to S.M.), the Schmidheiny Foundation (to S.M.), the Fondation Prévot (to S.M.), the Swiss Heart Foundation (to B.R.K.), the US National Institutes of Health (GM55632 to P.D.L.), the Danish National Research Foundation (to L.N.A. and M.S.N.), the Danish Research Council (1331-00337B to L.B.N. and 09-073571 to C.C.), the Novo Nordisk Foundation (to C.C.), and the Lundbeck Foundation (to C.C.).
Acknowledgements
We thank Esther Sutter, Bernard Foglia, Graziano Pelli, and Marc Bachetta for excellent technical assistance.
Conflict of interest: none declared.
References
- 1.Sluijter JP, Condorelli G, Davidson SM, Engel FB, Ferdinandy P, Hausenloy DJ, Lecour S, Madonna R, Ovize M, Ruiz-Meana M, Schulz R, Van Laake LW. Novel therapeutic strategies for cardioprotection. Pharmacol Ther 2014;144:60–70. [DOI] [PubMed] [Google Scholar]
- 2.James RW, Frias MA. High density lipoproteins and ischemia reperfusion injury: the therapeutic potential of HDL to modulate cell survival pathways. Adv Exp Med Biol 2014;824:19–26. [DOI] [PubMed] [Google Scholar]
- 3.Waeber C, Walther T. Sphingosine-1-phosphate as a potential target for the treatment of myocardial infarction. Circ J 2014;78:795–802. [DOI] [PubMed] [Google Scholar]
- 4.Karliner JS. Sphingosine kinase and sphingosine 1-phosphate in the heart: a decade of progress. Biochim Biophys Acta 2013;1831:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Somers SJ, Frias M, Lacerda L, Opie LH, Lecour S. Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc Drugs Ther 2012;26:227–237. [DOI] [PubMed] [Google Scholar]
- 6.Morel S, Frias MA, Rosker C, James RW, Rohr S, Kwak BR. The natural cardioprotective particle HDL modulates connexin43 gap junction channels. Cardiovasc Res 2012;93:41–49. [DOI] [PubMed] [Google Scholar]
- 7.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol 2004;36:1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanno S, Kovacs A, Yamada KA, Saffitz JE. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J Am Coll Cardiol 2003;41:681–686. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc Res 2004;61:386–401. [DOI] [PubMed] [Google Scholar]
- 10.Schulz R, Heusch G. Connexin43 and ischemic preconditioning. Adv Cardiol 2006;42:213–227. [DOI] [PubMed] [Google Scholar]
- 11.Heusch G, Buchert A, Feldhaus S, Schulz R. No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res Cardiol 2006;101:354–356. [DOI] [PubMed] [Google Scholar]
- 12.Skyschally A, Walter B, Schultz Hansen R, Heusch G. The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn Schmiedebergs Arch Pharmacol 2013;386:383–391. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y, Gu EW, Zhu Y, Zhang L, Liu XQ, Fang WP. Sufentanil limits the myocardial infarct size by preservation of the phosphorylated connexin 43. Int Immunopharmacol 2012;13:341–346. [DOI] [PubMed] [Google Scholar]
- 14.Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, Dahlback B, Nielsen LB. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem 2008;283:1839–1847. [DOI] [PubMed] [Google Scholar]
- 15.Maass K, Ghanem A, Kim JS, Saathoff M, Urschel S, Kirfel G, Grummer R, Kretz M, Lewalter T, Tiemann K, Winterhager E, Herzog V, Willecke K. Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol Biol Cell 2004;15:4597–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang GY, Xie LJ, Linask KL, Zhang C, Zhao XQ, Yang Y, Zhou GM, Wu YJ, Marquez-Rosado L, McElhinney DB, Goldmuntz E, Liu C, Lampe PD, Chatterjee B, Lo CW. Evaluating the role of connexin43 in congenital heart disease: screening for mutations in patients with outflow tract anomalies and the analysis of knock-in mouse models. J Cardiovasc Dis Res 2011;2:206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morel S, Braunersreuther V, Chanson M, Bouis D, Rochemont V, Foglia B, Pelli G, Sutter E, Pinsky DJ, Mach F, Kwak BR. Endothelial Cx40 limits myocardial ischaemia/reperfusion injury in mice. Cardiovasc Res 2014;102:329–337. [DOI] [PubMed] [Google Scholar]
- 18.Olsen KB, Axelsen LN, Braunstein TH, Sorensen CM, Andersen CB, Ploug T, Holstein-Rathlou NH, Nielsen MS. Myocardial impulse propagation is impaired in right ventricular tissue of Zucker diabetic fatty (ZDF) rats. Cardiovasc Diabetol 2013;12:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, Dahlback B. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA 2011;108:9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morel S, Chanson M, Nguyen TD, Glass AM, Richani Sarieddine MZ, Meens MJ, Burnier L, Kwak BR, Taffet SM. Titration of the gap junction protein Connexin43 reduces atherogenesis. Thromb Haemost 2014;112:390–401. [DOI] [PubMed] [Google Scholar]
- 21.English D, Welch Z, Kovala AT, Harvey K, Volpert OV, Brindley DN, Garcia JG. Sphingosine 1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemostasis and angiogenesis. FASEB J 2000;14:2255–2265. [DOI] [PubMed] [Google Scholar]
- 22.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007;316:295–298. [DOI] [PubMed] [Google Scholar]
- 23.Poti F, Simoni M, Nofer JR. Atheroprotective role of high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P). Cardiovasc Res 2014;103:395–404. [DOI] [PubMed] [Google Scholar]
- 24.Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 2013;62:24–35. [DOI] [PubMed] [Google Scholar]
- 25.Tao R, Hoover HE, Honbo N, Kalinowski M, Alano CC, Karliner JS, Raffai R. High-density lipoprotein determines adult mouse cardiomyocyte fate after hypoxia-reoxygenation through lipoprotein-associated sphingosine 1-phosphate. Am J Physiol Heart Circ Physiol 2010;298:H1022–H1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frias MA, Lang U, Gerber-Wicht C, James RW. Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc Res 2010;85:118–126. [DOI] [PubMed] [Google Scholar]
- 27.Kawabori M, Kacimi R, Karliner JS, Yenari MA. Sphingolipids in cardiovascular and cerebrovascular systems: Pathological implications and potential therapeutic targets. World J Cardiol 2013;5:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, Schober O, Hildebrand R, Schulz R, Heusch G, Haude M, von Wnuck Lipinski K, Herzog C, Schmitz M, Erbel R, Chun J, Levkau B. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 2006;114:1403–1409. [DOI] [PubMed] [Google Scholar]
- 29.Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, Chun J, Brown JH. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2007;292:H2944–H2951. [DOI] [PubMed] [Google Scholar]
- 30.Kwak BR, van Veen TA, Analbers LJ, Jongsma HJ. TPA increases conductance but decreases permeability in neonatal rat cardiomyocyte gap junction channels. Exp Cell Res 1995;220:456–463. [DOI] [PubMed] [Google Scholar]
- 31.Kwak BR, Hermans MM, De Jonge HR, Lohmann SM, Jongsma HJ, Chanson M. Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol Biol Cell 1995;6:1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res 2006;98:1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol 2000;149:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bao X, Reuss L, Altenberg GA. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of Serine 368. J Biol Chem 2004;279:20058–20066. [DOI] [PubMed] [Google Scholar]
- 35.Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci USA 1996;93:1831–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Meana M, Rodriguez-Sinovas A, Cabestrero A, Boengler K, Heusch G, Garcia-Dorado D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc Res 2008;77:325–333. [DOI] [PubMed] [Google Scholar]
- 37.Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C, Garcia-Dorado D, Di Lisa F, Heusch G, Schulz R. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res 2005;67:234–244. [DOI] [PubMed] [Google Scholar]
- 38.Boengler K, Ruiz-Meana M, Gent S, Ungefug E, Soetkamp D, Miro-Casas E, Cabestrero A, Fernandez-Sanz C, Semenzato M, Di Lisa F, Rohrbach S, Garcia-Dorado D, Heusch G, Schulz R. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med 2012;16:1649–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruiz-Meana M, Nunez E, Miro-Casas E, Martinez-Acedo P, Barba I, Rodriguez-Sinovas A, Inserte J, Fernandez-Sanz C, Hernando V, Vazquez J, Garcia-Dorado D. Ischemic preconditioning protects cardiomyocyte mitochondria through mechanisms independent of cytosol. J Mol Cell Cardiol 2014;68:79–88. [DOI] [PubMed] [Google Scholar]
- 40.Miro-Casas E, Ruiz-Meana M, Agullo E, Stahlhofen S, Rodriguez-Sinovas A, Cabestrero A, Jorge I, Torre I, Vazquez J, Boengler K, Schulz R, Heusch G, Garcia-Dorado D. Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res 2009;83:747–756. [DOI] [PubMed] [Google Scholar]
- 41.Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia-Dorado D, Di Lisa F, Schulz R, Heusch G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 2005;97:583–586. [DOI] [PubMed] [Google Scholar]
- 42.Azarashvili T, Baburina Y, Grachev D, Krestinina O, Evtodienko Y, Stricker R, Reiser G. Calcium-induced permeability transition in rat brain mitochondria is promoted by carbenoxolone through targeting connexin43. Am J Physiol Cell Physiol 2011;300:C707–C720. [DOI] [PubMed] [Google Scholar]
- 43.Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KB, Kardami E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc Res 2014;103:72–80. [DOI] [PubMed] [Google Scholar]
- 44.Maass K, Shibayama J, Chase SE, Willecke K, Delmar M. C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circulation Research 2007;101:1283–1291. [DOI] [PubMed] [Google Scholar]