

Abstract
The central role of the microbiome in critical illness is supported by a half century of experimental and clinical study. The physiological effects of critical illness and the clinical interventions of intensive care substantially alter the microbiome. In turn, the microbiome predicts patients’ susceptibility to disease, and manipulation of the microbiome has prevented or modulated critical illness in animal models and clinical trials. This Review surveys the microbial ecology of critically ill patients, presents the facts and unanswered questions surrounding gut-derived sepsis, and explores the radically altered ecosystem of the injured alveolus. The revolution in culture-independent microbiology has provided the tools needed to target the microbiome rationally for the prevention and treatment of critical illness, holding great promise to improve the acute and chronic outcomes of the critically ill.
The forgotten organ in multiorgan failure
The common conditions of critical illness (including sepsis, acute respiratory distress syndrome [ARDS], and multiorgan failure) cause tremendous global mortality and an enormous and growing economic burden.1 Although specialties such as oncology and rheumatology have been revolutionised by the breakthroughs of molecular medicine, decades of research into the diseases of critical illness have yielded no targeted therapies. In practice, critical care remains synonymous with supportive care.
There are several possible reasons why no molecular therapies have been developed for these common and fatal diseases. One credible explanation is that the primary focuses of investigation, host inflammation and cellular injury, are downstream consequences of an overlooked upstream source: the diverse ecosystems of microbes on and in the human body. Interest in the microbiome has exploded in the past decade due to the advent of culture-independent methods of identifying microbes.2,3 Although a wealth of clinical and experimental evidence suggests that the microbiome is central to the pathogenesis of critical illness, the common diseases of critical illness have been included in surprisingly few modern microbiome studies. In turn, review articles and clinical guidelines on critical illness largely ignore the microbiome, neglecting what is, effectively, a 1·5 kg organ containing more DNA than every host organ combined.
Critical illness and the interventions of intensive care substantially alter the microbiome. In turn, the microbiome predicts patients’ susceptibility to disease, and manipulation of the microbiome has prevented or modulated critical illness in animal models and clinical trials. In this Review, I describe the altered ecosystem of the microbiome in critically ill patients, focusing on the gut and lungs. I discuss the microbiome’s role in sepsis, ARDS, pneumonia, and exacerbations of chronic lung disease, and identify important unanswered questions that may now be resolved with the techniques of modern microbiology.
The ecological effects of critical illness
The observation at the heart of this Review—that critical illness alters the ecosystem of the body’s microbiota—was first made in a seminal study by Johanson and colleagues4 in 1969, decades before the dawn of high-throughput sequencing. Exposure to the hospital setting has minimal effect on the bacterial communities of the upper respiratory tract: the oropharynges of healthy hospital workers and minimally ill patients staying in hospital are no more frequently colonised by Gram-negative rods than are those in people with no hospital exposure (figure 1). Rather, the change in microbiota seen in patients staying in hospital depends on the severity of their illness rather than their physical location. Critical illness substantially alters the physiology of the host, which in turn alters the environmental conditions and community structures of resident microbes. This clinical observation illustrates an oft-cited tenet in microbial ecology, “Everything is everywhere, but the environment selects”.5 Decades later, we have an incomplete but growing understanding of how the internal environment of critically ill patients creates selective pressure on the relative growth of its microbiota.
Figure 1. The altered ecosystem of the critically ill patient.
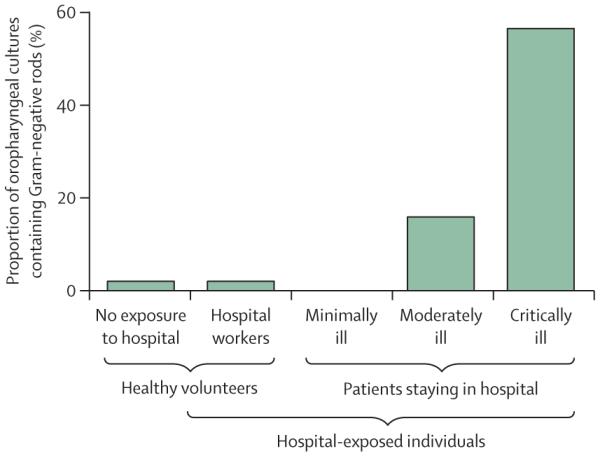
Changes in microbiota depend upon severity of illness rather than physical location and bacterial exposure.4
The composition of every community, microbial or otherwise, is determined by the balance of three ecological factors: immigration into the community, elimination of members from the community, and the relative reproduction rates of the community’s members. Any change in the microbiome, whether it be chronic or acute, must be attributable to some combination of these three forces. All three are greatly altered in the gut and lung ecosystems of critically ill patients by the pathophysiological effects of critical illness and interventions of intensive care (tables 1, 2).
Table 1.
Ecological effects of critical illness on the gastrointestinal microbiome
| Microbial immigration | Microbial elimination | Environmental growth conditions | |
|---|---|---|---|
| Pathophysiological processes | |||
| Decreased oral intake | Decreased immigration of food-associated microbiota6 |
No direct effect | Shift to stress conditions of nutrient scarcity and altered nutritional substrate6 |
| Altered oropharyngeal microbiota |
Increased immigration of Proteobacteria and potential pathogens4,7 |
No direct effect | No direct effect |
| Intestinal dysmotility | No direct effect | Decreased elimination, increased upper- gastrointestinal community burden |
No direct effect |
| Systemic hyperglycaemia and electrolyte disturbances |
No direct effect | Decreased elimination (intestinal dysmotility)8,9 | No direct effect |
| Gut hypoperfusion, reperfusion injury, impaired mucosal integrity |
No direct effect | Increased elimination via translocation to mesenteric lymphatics10–12 |
Increased mucosal inflammation, increased free radical concentrations and nitrate availability;13 shift from commensal anaerobes to Proteobacteria and select Firmicutes14–16 |
| Decreased bile salt concentration17 |
No direct effect | Decreased elimination of bile-sensitive species (eg, Enterococcus spp)18 |
Selective overgrowth of bile-sensitive species (eg, Enterococcus spp)18 |
| Endogenous opioid production |
No direct effect | Decreased elimination (intestinal dysmotility) | Selective increase in virulence of opioid-responsive species (eg, Pseudomonas aeruginosa),19 disruption of stabilising commensal relationships19,20 |
| Endogenous catecholamine and inflammatory cytokine production |
No direct effect | Decreased elimination (intestinal dysmotility)21 | Selective promotion of growth and virulence of potential pathogens (eg, Pseudomona. aeruginosa),22–24 increased mucosal inflammation (via splanchnic hypoperfusion), decreased oxygen tension and pH |
| Disruption of intestinal mucus layer25,26 |
No direct effect | Increased elimination via translocation to mesenteric lymphatics27,28 |
Altered nutrient supply, altered oxygen gradients,29 loss of mucus reservoir of antibacterial peptides30 |
| Impaired mucosal immunity: decreased IgA and defensin production31,32 |
No direct effect | Decreased elimination of potential pathogens, increased elimination via translocation to mesenteric lymphatics33 |
Loss of growth inhibition for potential pathogens, decreased abundance of commensal Bacteroidetes34,35 |
| Clinical interventions | |||
| Supine positioning | No direct effect | Decreased elimination from upper gastrointestinal tract (intestinal dysmotility)36,37 |
No direct effect |
| Gastric-acid suppression | No direct effect | Decreased elimination from upper gastrointestinal tract (neutralised pH)38,39 |
Selective growth promotion of acid-intolerant bacteria38,39 |
| Enteral feeding | No direct effect | Increased elimination due to antimicrobial actions of luminal bile salts,17 decreased elimination via translocation to mesenteric lymphatics40 |
Altered nutritional substrate,6,41 shift away from stress conditions of nutrient scarcity |
| Parenteral feeding | No direct effect | Increased elimination via translocation to mesenteric lymphatics11,42 |
Loss of growth inhibition for potential pathogens via impaired mucosal immunity (eg, decreased IgA secretion)43 |
| Sedatives, opiates and neuromuscular blockade |
No direct effect | Decreased elimination (intestinal dysmotility) | Selective increase in virulence of opioid-responsive species (eg, Pseudomonas aeruginosa),19 disruption of stabilising commensal relationships19,20 |
| Systemic catecholamines | No direct effect | Decreased elimination (intestinal dysmotility)21 | Selective promotion of growth and virulence of potential pathogens (eg, Pseudomonas aeruginosa),22,23 increased mucosal inflammation (via splanchnic hypoperfusion), decreased oxygen tension and pH |
| Oral decontamination (eg, topical chlorhexadine) |
Decreased immigration of oropharyngeal microbiota |
No direct effect | No direct effect |
| Selective decontamination of the digestive tract |
Decreased immigration of oropharyngeal microbiota |
Increased elimination of select bacteria (eg, Enterobacteriaceae spp)44 |
Selective growth suppression of select bacteria (eg, Enterobacteriaceae spp)44 |
| Systemic antibiotics | No direct effect | Increased elimination of select bacteria (depending on antibiotic regimen) |
Selective growth suppression of bacteria (depending on antibiotic regimen) |
Table 2.
Ecological effects of critical illness on the respiratory microbiome
| Microbial immigration | Microbial elimination | Environmental growth conditions | |
|---|---|---|---|
| Pathophysiological processes | |||
| Altered oropharyngeal microbiota |
Increased immigration of Proteobacteria and potential pathogens4,7 |
No direct effect | No direct effect |
| Depressed level of consciousness |
Increased immigration via aspiration of oropharyngeal and gastric contents45 |
Decreased elimination (impaired cough reflex)46 |
No direct effect |
| Aspiration of gastric contents45 |
Increased immigration of gastric microbiota45 |
No direct effect | No direct effect |
| Impaired mucociliary clearance47 |
No direct effect | Decreased elimination (impaired mucociliary escalator)47 |
No direct effect |
| Increased bronchial mucus production |
No direct effect | No direct effect | Increased nutrient substrate, altered gradients of oxygen48 and temperature49 |
| Endogenous catecholamine and inflammatory cytokine production |
No direct effect | Increased elimination via innate and adaptive immune response |
Selective promotion of growth and virulence of potential pathogens (eg, Pseudomonas aeruginosa)23,24,50,51 |
| Recruitment and activation of neutrophils |
No direct effect | Increased elimination of select community members52 |
Selective suppression of bacterial growth,52 increased free radical concentrations and nitrate availability,13,53 altered temperature gradients49,54 |
| Alveolar oedema | No direct effect | No direct effect | Increased and altered nutrient substrate,55,56 altered oxygen gradient |
| Inactivation of alveolar surfactant |
No direct effect | Decreased elimination of surfactant- sensitive bacteria55,57 |
Loss of growth inhibition for selective potential pathogens57 |
| Clinical interventions | |||
| Supine positioning | Increased immigration via aspiration of oropharyngeal and gastric microbiota58 |
No direct effect | No direct effect |
| Head of bed raised | Decreased immigration via aspiration of oropharyngeal and gastric microbiota58 |
Decreased elimination (gravitationally limited mucus clearance59) |
No direct effect |
| Endotracheal intubation | Increased immigration via aspiration of oropharyngeal microbiota |
Decreased elimination (impaired cough and mucociliary escalator) |
Altered airway temperature and humidity |
| Mechanical ventilation | No direct effect | No direct effect | Increased alveolar oedema;60 increased neutrophil, cytokine, and catecholamine concentrations60 |
| Subglottic suctioning | Decreased immigration of oropharyngeal microbiota61 |
No direct effect | No direct effect |
| Gastric-acid suppression | Increased immigration of gastric microbiota38,39 |
No direct effect | No direct effect |
| Sedatives, opiates, and neuromuscular blockade |
No direct effect | Decreased elimination via impaired cough reflex and mucociliary clearance |
No direct effect |
| Systemic catecholamines | No direct effect | No direct effect | Selective promotion of growth and virulence of potential pathogens (eg, Pseudomonas aeruginosa)50,51 |
| Oral decontamination (eg, topical chlorhexadine) |
Decreased immigration of oropharyngeal microbiota |
No direct effect | No direct effect |
| Selective decontamination of the digestive tract |
Decreased immigration of oropharyngeal microbiota |
Increased elimination of select bacteria (eg, Enterobacteriaceae spp)44 |
No direct effect |
| Systemic antibiotics | No direct effect | Increased elimination of select bacteria (depending on antibiotic regimen) |
Selective growth suppression of bacteria (depending on antibiotic regimen)62 |
The primary route of immigration of microbes into the gut microbiome is via the oropharynx, which itself changes strikingly in critical illness. Johanson and colleagues4,7 noted that in critically ill patients, healthy oral microbiota are displaced by gram-negative aerobes (figure 1), including prominent members of the Proteobacteria phylum. The catabolic starvation state of critical illness results in decreased immigration of food-associated bacteria and decreased nutritional supply for commensal microbes.6 Well-studied interventions, such as topical oral decontamination, decrease the bacterial burden of the oropharynx and decrease immigration from the source community.44
In healthy individuals, the primary means of microbial elimination from the gut microbiome is transit through and from the gastrointestinal tract, which is normally rapid. Via defecation, a healthy adult expels about 1014 bacterial cells per day.63 In critically ill patients, transit time is substantially slowed by various pathophysiological (glucose and electrolyte disturbances8,9 and endogenous opioid production) and therapeutic (sedatives, opiates, and systemic catecholamines21) factors. In the stomach, which is normally fast to empty and extremely acidic, transit time slows36 and pH is neutralised by the use of agents to suppress the production of gastric acid.38,39 Other mechanisms of microbial elimination are impaired in critical illness: bile salt production drops,17 IgA production is impaired,31 and the dense mucosal barrier of secreted antimicrobial peptides is lost.25,26,32 The net effect is reduced elimination of bacteria, especially in the upper gastrointestinal tract, which is transformed into a pH-neutral reservoir that quickly becomes overgrown by Gram-negative bacteria.64
Environmental growth conditions of the gut are transformed in critical illness, which affects the relative reproduction rates of community members. Hypoperfusion and reperfusion of the intestinal wall results in intense mucosal inflammation, leading to a cascade of environmental changes. Increased nitrate concentrations13 and an altered mucosal oxygen gradient29 favour the growth of microbes in the Proteobacteria phylum, which contains many clinically familiar gram-negative rods, such as Pseudomonas aeruginosa and Escherichia coli, and some members of the Firmicutes phylum, such as Staphylococcus aureus and Enterococcus spp.14–16 Importantly, in many critically ill patients, the dense intestinal mucus layer is thinned, disrupted, or absent.25,26 This crucial anatomical component of gut anatomy harbours its own protective microbiota and provides a physical barrier between the intestinal ecosystem and the host. Almost every common clinical intervention used in intensive care (eg, enteral feeding,43 proton-pump inhibitors,38,39 systemic catecholamines,22,23 and systemic antibiotics65,66) changes environmental growth conditions for intestinal bacteria (table 1).
The net effect of these alterations in ecology is an unstable and often collapsed community with catastrophically low diversity. The stomach and proximal small intestine, which are usually sparsely populated, become overgrown by a small number of species, such as E coli, P aeruginosa, and Enterococcus spp.67,68 The upper gastrointestinal tract becomes a stagnant reservoir of potential pathogens, the presence of which is predictive of extra-abdominal infections and multiorgan failure.64,67 The lower gastrointestinal tract, which in healthy people contains hundreds of distinct bacterial species, loses diversity, and the community is overrun by a few (in some cases only one) bacterial species.20,69,70 Dominant species include S aureus, Enterococcus spp, and members of the Enterobacteriaceae family (including E coli and Klebsiella spp). P aeruginosa, which is normally low in abundance, grows in prominence.20,69,71 Additionally, normally rare fungi, such as Candida spp, bloom and thrive;20 culture-based detection of candidaemia is a marker of disease severity and predictive of a poor outcome.72 Viruses, archaea, and eukaryotes comprise less than 10% of the gut community in healthy individuals,73 and the effects of critical illness on abundance and behaviour of these organisms are unknown. This catastrophic drop in bacterial diversity, compared with the relatively subtle differences seen across chronic disease states, is astounding. In critical illness the gut microbiome resembles an infection rather than a community.
The absence of specific bacteria in the gut is just as important as the presence of others. The resident microbes of the lower gastrointestinal tract normally serve essential metabolic and immunomodulatory functions. Even slight differences in the abundance of healthy gut bacteria have been implicated in diverse systemic diseases.74 The lower gastrointestinal tract in critically ill patients becomes an inhospitable desert for these stabilising resident microbes. For example, butyrate is the primary energy source for the epithelial cells that line the colon. Without butyrate these cells are starved and shrivel and degrade.75 Butyrate also dampens the intestinal and systemic immune response by stimulating the development of regulatory T cells.76 In studies of the gut microbiome in critically ill patients, butyrate-producing bacteria are uncommon or absent,20,69–71 and butyrate production is at a minimum.71 The pathophysiological consequences of this condition are predictable (epithelial cell death and dysregulated inflammation), but the clinical consequences are unknown.
The ecological effects of critical illness are similarly extreme in the respiratory tract (table 2). Although even healthy lungs are subject to constant immigration from oropharyngeal microbes via microaspiration,77–79 this immigration is accelerated due to depressed consciousness and endotracheal intubation. The dynamics of the aerodigestive tract become inverted during critical illness: whereas in health, the oropharynx is the primary source community for the lungs and the stomach,80 the overgrown microbial reservoir of the stomach and small intestine becomes the primary source community for the mouth and lungs.64,67 The oropharynx is usually populated by benign Prevotella spp and Veillonella spp,2,77,78 but becomes overrun by potentially pathogenic bacteria, including prominent Proteobacteria, such as P aeruginosa and K pneumoniae.4,7,81
Although elimination of microbes from the respiratory tract is accelerated in critical illness partly by the activation of immune defences, most pathophysiological and clinical factors decrease the rate of microbial elimination. Depressed consciousness and sedation blunt the cough reflex,46 and endotracheal intubation and acute illness impair the mucociliary escalator.47 Elevation of the head of the bed decreases the immigration rate of gastric microbiota,58 but it also impedes microbial elimination, which is predominantly gravity-dependent when cough and mucociliary clearance are impaired.59 The inactivation of alveolar surfactant decreases the elimination of surfactant-sensitive bacteria.55,57
Finally, as discussed in detail below, acute illness substantially changes the environmental growth conditions of the lungs. The normally nutrient-poor environment of the alveolus is flooded with nutrient-rich oedema,55 pockets of oxygen and heterogeneous temperature gradients are established,48,49 and the signalling molecules of the host stress response selectively promote the growth of potential pathogens.23,50,51 The ubiquitous use of systemic antibiotics further alters the relative reproduction rates of community members. The predicted effect of these ecological forces in the lungs, therefore, is a state of increased immigration, decreased elimination, and favourable growth conditions for potential pathogens.61,82–84 Understanding of these ecological forces will be informed by longitudinal, culture-independent surveys of microbial com munities in the upper and lower respiratory tracts in critically ill patients.
Gut-derived sepsis: the inarguable and the unknown
The suspicion that the intestinal microbiome can be turned against the host is as old as germ theory. In 1868, contemporaneous with Pasteur, Herman Senator speculated that “self-infection” within the gastrointestinal tract could release systemic factors that cause fever, tachycardia, and obtundation.85 In 1952, a decade after the introduction of penicillin,86 Fine and colleagues87 reported that pretreating the gut with enteric antibiotics significantly lessened the risk of death in an animal model of haemorrhagic shock. In 1972, 5 years after the first description of ARDS,88 Cuevas and colleagues89 showed that the disease could be prevented in animal models of shock by pretreatment with enteric antibiotics.
During severe systemic illness, such as sepsis or haemorrhagic shock, the bacterial content of the gut determines the severity of systemic injury (figure 2). When the bacterial burden of the gut is minimised, either by pretreatment with enteric antibiotics or by use of germ-free animals, the inflammation and injury sustained by distal organs in shock is lessened. This relation has been reported consistently across species (mice,90,93 rats,94 rabbits,89 and dogs87), types of shock (haemorrhage,87 sepsis,89 and ischaemia–reperfusion90), and decades of rigorous inquiry. The microbiome, therefore, is of clear relevance to any discussion of precision medicine in critical care: the treatment groups in these studies differed not in genetics or exposure history but rather only in their microbiota (figure 2).
Figure 2. Manipulation of the microbiome and the prevention of critical illness.
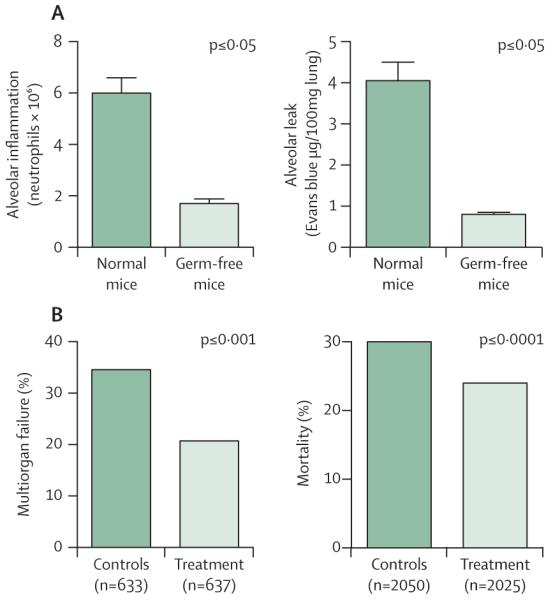
(A) In diverse models of shock, germ-free mice are protected from the alveolar inflammation and injury seen in acute respiratory distress syndrome.90 (B) In clinical trials, manipulation of gut microbiota with antibiotics (selective decontamination of the digestive tract) protects against extra-abdominal infections, multiorgan failure, and death.91,92 Part A was adapted from reference 90 by permission of the American Association of Immunologists.
In the 1980s, these experimental observations prompted clinical investigation of the suppression of gut bacteria in patients at risk of critical illness. Selective decontamin ation of the digestive tract (SDD) is achieved by prophylactic administration of antibiotics tailored to keep overgrowth of potential pathogens in the gut to a minimum. Since the first (which was also the first positive) randomised controlled trial in 1987,95 SDD has been tested in more than 65 randomised controlled trials studying more than 15000 patients.96 The findings are unambiguous: patients who receive SDD are less likely to develop multiorgan failure91 or die96 than patients who do not (figure 2). Nevertheless, clinical use of SDD remains uncommon, especially in North America, due to perceived risk of antimicrobial resistance, although this concern is not supported by large clinical trials and meta-analyses.97 Although the ecological effects of SDD on antibiotic-resistant pathogens at the intensive-care-unit level remain controversial,98 the reality of the patient-level benefits are beyond debate.
This connection between patients’ microbiota and their susceptibility to critical illness has been reinforced by an even broader scope of study. When more than 10000 hospital inpatients were stratified according to estimated degrees of intestinal dysbiosis, a strong and consistent dose–response relation was uncovered between disorder of the microbiome and subsequent development of severe sepsis.99 This association between the microbiome and susceptibility to critical illness has, therefore, been shown at every level of inquiry: the laboratory bench, clinical trials, meta-analyses, and population studies. Yet, despite the clarity of this biological signal, the mechanisms behind it remain controversial and incompletely understood.
The oldest, most intuitive explanation for so-called gut-derived sepsis is that in states of critical illness, bacteria and bacterial products escape from the gut and translocate via the bloodstream to distal organs, where they provoke inflammation and injury. The intestinal wall of critically ill patients is permeable, and the degree of permeability correlates with subsequent risks of organ injury and death.100 However, in a study of trauma patients at high risk of multiorgan failure,101 serial blood cultures drawn from indwelling portal vein catheters have shown minimum evidence of bacterial translocation and no association between portal vein bacteraemia and subsequent illness. The explanation of bacterial translocation, at least via a blood-borne route, therefore, waned in popularity. The explanation was subsequently refined after consideration of intestinal anatomy.12 The lower gastrointestinal tract drains not only into the portal circulation but also into mesenteric lymph nodes. These nodes drain to the thoracic duct, which in turn empties into the left subclavian vein. Therefore, the lungs are the first capillary bed in the body to filter the 1–4 L chyle per day that is emptied into the blood via the thoracic duct. These anatomical considerations gave rise to the so-called gut-lymph hypothesis.12
Substantial clinical and experimental evidence supports the gut-lymph hypothesis. In clinical studies of critically ill high-risk surgical patients and in animal studies of shock, bacteria have been cultured from the mesenteric lymph nodes.10,12,102 Furthermore, detection of bacteria in mesenteric lymph is predictive of subsequent sepsis and infectious complications.10,103 In animal studies of shock, ligation of the mesenteric duct protected against lung injury,102 and the harvested mesenteric lymph of critically ill animals can provoke lung injury in otherwise healthy animals.104 Of note, the toxicity of this lymph does not depend on the presence of endotoxin or of detectable bacteria,104 which suggests that other bacterial or tissue injury factors are important mediators of injury.
A final explanation for gut-derived sepsis posits that translocation of microbes and microbial products is not necessary for the microbiome to cause systemic inflammation and injury.22,105,106 Just as the community composition of the gut microbiome is altered by the intestinal environment in critically ill patients, the behaviour and virulence of individual community members are also changed.22 A bacterial strain that is normally inert and invisible to the host immune system can be transformed by the conditions of critical illness, gaining virulence that ignites systemic inflammation and sepsis. The virulence of pathogens familiar in intensive care is promoted by conditions of nutrient scarcity, competition from neighbouring community members, disruption of stabilising commensal relationships,20 and exposure to the mediators of the host stress response (eg, catecholamines, inflammatory cytokines, and endogenous opioids39,47,48).
In all likelihood, the pathogenesis of gut-derived sepsis, like most processes in critical illness, is multifactorial, replete with biological redundancy.106,107 All three hypotheses (systemic translocation, gut-lymph translocation, and in-situ virulence) probably explain complementary features of a complex pathogenesis of multiorgan failure, and all three will be informed by the revolution in culture-independent microbiology. The detection and identification of translocated bacteria and characterisation of collapsing gut communities are no longer limited by insensitive culture-based techniques, which cannot detect most gut bacteria.108 Modern techniques will also inform understanding of how clinical interventions contribute to these parallel processes. Many daily therapies and interventions in intensive care increase intestinal permeability (eg, nonsteroidal anti-inflammatory drugs109 and parenteral feeding55,82), bacterial translocation (eg, antibiotics,65 corticosteroids,110 and opiates111), and bacterial virulence (eg, opiates19 and catecholamines22,51). With modern techniques, the mechanisms behind the microbiome’s role in the progression from acute injury to systemic inflammation to multiorgan failure to death can finally be unfolded.
The radically altered ecology of the injured alveolus
Even in healthy individuals the lungs are subject to constant bombardment by bacteria from the upper respiratory tract.77–80 Unlike the gut, however, the alveolar space is an ecologically unfavourable environment for most bacteria and reproduction is minimal.77,112 An important reason for low reproduction is the lack of nutrient substrate for bacterial metabolism. Whereas the gut lumen offers an abundance of protein and carbohydrate energy sources, the alveolus is empty except for the thin bactericidal layer of lipid-rich surfactant that lines the epithelium. From the perspective of bacteria, healthy alveoli are inhospitable. In states of alveolar injury, however, such as in ARDS or pneumonia, the environmental conditions shift abruptly (figure 3). The previously empty alveoli are flooded with protein-rich fluid, providing a newly abundant energy source for reproducing microbes. The bactericidal surfactant layer is inactivated55,57 and microbial elimination is slowed by impairment of mucociliary clearance.47 Ecologically, the injured alveoli begin to resemble the gut more than healthy lungs and, therefore, it is unsurprising that most pathogens that arise in critical illness are of enteric origin. The microbiome and alveolar injury can propel each other in a dysregulated feedback loop that spans the host–microbiome divide (figure 3).55,113
Figure 3. Alteration of bacterial ecology in injured alveoli.

(A) Unlike in the healthy gut, the environment in healthy lungs is nutrient poor for bacteria and the protein content of alveolar lavage fluid is at a minimum. (B) In states of health, bacterial growth in the alveolar space is limited by the local inflammatory response it provokes and by its depletion of available nutrients. In conditions of alveolar injury, such as in ARDS and pneumonia, the alveolar space is flooded with nutrient-rich fluid, which promotes bacterial growth that in turn perpetuates a positive-feedback loop of inflammation, injury, alveolar oedema, and further dysbiosis. BAL=bronchoalveolar lavage. ARDS=acute respiratory distress syndrome. Part B was reproduced from reference 113 by permission of Elsevier.
Important features of the relation between alveolar injury and lung microbiota have been validated by innovative animal studies.56 Sterile direct lung injury in mice leads to increases in the bacterial content of the lungs, indicating increased reproduction. The lung community membership shifts towards overgrowth of specific community members that were present in small numbers before injury. Lavage fluid from injured lungs contains the specific nutrients that are metabolised by the newly enriched species, as predicted by the hypothesis that lung injury alters the microbiome via changes in nutrient availability. Finally, when the bacterial communities from injured lungs are introduced into the lungs of otherwise healthy mice, they provoke more inflammation and injury than do bacteria acquired from uninjured lungs. These novel findings reveal numerous new targets for clinical intervention. Virtually all preventive and therapeutic strategies for ARDS have been aimed at blunting host inflammation and injury. This model suggests that the dynamic interface between the host and its disordered lung communities (figure 3) is a ripe, unexplored target for intervention.
This model of pathogenesis can apply to ARDS and to pneumonia, and might explain why such extensive clinical overlap exists between the two disorders. Pneumonia is the most common cause of ARDS,114 and roughly half of patients with established ARDS develop pneumonia during intensive care.114,115 In the most convincing study so far to test the preventive value of lung-protective ventilation in patients without ARDS, the intraoperative use of larger tidal volumes (which induce alveolar injury and leak,60 figure 3) increased the rate of postoperative pneumonia by a factor of five (from 1·5% to 8·0%).116
Nutrient supply is not the only way the ecology of the alveolus changes in critically ill patients. The influx of oedema creates steep oxygen gradients, which shape bacterial community structure.29,48 Surfactant is inactivated, which disinhibits the growth of sensitive bacteria,55,57 and mucociliary clearance is impaired.47 The cells of innate immunity (macrophages and neutrophils) increase in number and activation, which causes the alveolar concentration of molecules related to the host stress response to increase.117
These molecular stress signals—increased concentrations of catecholamines and inflammatory cytokines—affect lung bacteria.118,119 In vitro, the growth of P aeruginosa is increased by the presence of catecholamines (figure 4).51 In human bronchoalveolar lavage samples, increased alveolar catecholamine concentrations correlate strongly with collapse of the lung microbiome around one dominant species (most frequently P aeruginosa, figure 4).50 Thus any source of alveolar injury and inflammation, whether direct (eg, aspiration or ventilator-induced lung injury60) or indirect (eg, sepsis or shock) can trigger a cascade of inflammation leading to increased concentrations of intra-alveolar catechol amines,120 which in turn promote the growth and virulence of select bacterial community members and a disordered bacterial community that perpetuates alveolar inflammation (figure 4). Bacterial growth promotion by host stress molecules is not unique to P aeruginosa, and is also seen with Streptococcus pneumoniae,121 S aureus,122 and Klebsiella pneumoniae.123 Additionally, as well as catecholamines, growth promotion is seen with TNFα, interleukins 1, 6, and 8, and glucocorticoids.23,24,124,125 The web of interactions between the lung microbiome and alveolar inflammation is complex, dynamic, and bidirectional.
Figure 4. Catecholamines and disorder in the alveolar bacterial ecosystem.

(A) The growth of bacteria, such as Pseudomonas aeruginosa, is promoted in vitro by catecholamines, such as norepinephrine and dopamine.51 (B) In the human lung microbiome, increased catecholamine concentrations are strongly associated with community collapse and the emergence of one dominant species.50 (C) In states of critical illness, direct and indirect lung injury provoke alveolar inflammation, which promotes catecholamine production and creates a positive-feedback loop of dysbiosis and inflammation.50 CFU=colony forming unit. VILI=ventilator-induced lung injury. Part A adapted from reference 51 by permission of American College of Chest Physicians. Part B adapted from reference 50 by permission of American Thoracic Society.
Exacerbations of chronic lung disease are not acute bacterial infections
Not all respiratory failure in intensive care is attributable to alveolar injury. A common presentation is the clinical exacerbation of chronic airway diseases, such as asthma, chronic obstructive pulmonary disease (COPD), bronchiectasis, and cystic fibrosis. These exacerbations are associated with increased and persistent airway inflammation, and result in severe morbidity and death and high expense related to intensive care.126
Although viral infections have an unambiguous role as a common precipitant of exacerbations, the role of bacteria in the pathogenesis of exacerbations has been controversial for decades.126 The theory that exacerbations represent acute bacterial infections ranges from universally assumed (cystic fibrosis127 and bronchiectasis128) to highly controversial (COPD53) to widely dismissed (asthma129). Confusion and debate on this issue stems from the poor sensitivity of culture-based approaches in the characterisation of lung communities.2,126 Culture-independent techniques have helped to clarify this long-debated relation between bacteria, infections, and exacerbations.
Ecologically, infections are characterised by an increase in microbial burden and a decrease in community diversity, coupled with increased host inflammation and tissue injury. Bacterial pneumonia, a true lung infection, exemplifies these features: it is characterised by increased bacterial burden and low community diversity (generally one dominant pathogen).62,83,130 These features correlate tightly with multiple indices of host inflammation, including alveolar neutrophilia93 and high alveolar concentrations of catecholamines50 and TNF-α.131
By contrast, exacerbations consistently lack these defining ecological features of infection. Culture-independent studies have compared bacterial communities at baseline and during exacerbations in the airways of patients with COPD,132,133 cystic fibrosis,134–138 or bronchiectasis.139 With remarkable consistency, all studies report no increase in bacterial burden and no decrease in community diversity during exacerbations. By any conventional or modern definition, therefore, exacerbations are not acute bacterial infections of the airways.
Nor do exacerbations behave clinically like true acute respiratory infections, such as pneumonia. Whereas invitro bacterial sensitivity to antibiotics is crucial in the management of pneumonia, there is no detectable relation between antibiotic susceptibility of cultured organisms and clinical response to therapy in exacerbations, even in cystic fibrosis.140,141 Antibiotics are unquestionably useful in the treatment of pneumonia, but in respiratory exacerbations views on their use range from controversial (COPD) to useless (asthma). Additionally, whereas pneumonia is the most common cause of sepsis, exacerbations rarely or never provoke a septic response.
Although exacerbations are not bacterial infections, the microbiome is clearly involved in the pathogenesis of exacerbations. Baseline differences in airway microbiota are predictive of subsequent exacerbation frequency.142 The intervention most consistently proven to decrease exacerbation frequency (in COPD,143 cystic fibrosis144 and bronchiectasis145) is azithromycin, a macrolide antibiotic. In exacerbation states, membership of the lung bacterial community shifts, often towards enrichment of the Proteobacteria phylum,133,146 which contains clinically relevant Gram-negative rods, such as Pseudomonas spp and Haemophilus spp. As opposed to infections, therefore, exacerbations are more accurately described as respiratory dysbiosis: disorder of the respiratory ecosystem coupled with a dysregulated host immune response. Airway inflammation leads to altered microbial growth conditions and the resulting disordered bacterial community further drives airway inflammation.126 This self-perpetuating positive-feedback loop might explain why clinical exacerbations can last weeks longer than the presence of their triggers, and why macrolides (which have antimicrobial and immunomodulatory effects147) have such consistently demonstrated preventive benefits across diseases.143–145
Important clinical lessons and areas for further study
With virtually every treatment used in intensive care, the patient’s microbiota are knowingly or unknowingly manipulated (tables 1, 2). In view of the clear relevance of the microbiome to outcomes in critically ill patients, the ecological effects of interventions must be studied rigorously. In instances in which the effects are known, they should be taken seriously. For instance, proton-pump inhibitors decrease elimination of gastric microbiota38 and increase immigration of bacteria into the lungs, which increases the risk of pneumonia.148 Maddeningly, however, proton-pump inhibitors are commonly included in treatment bundles purported to prevent ventilator-associated pneumonia, and are prescribed indiscriminately to critically ill patients. Other common interventions need to be reconsidered from an ecological perspective. Raising of the head of the patient’s bed decreases immigration to the lungs of gastric microbiota compared with supine positioning,149 but this practice also compromises microbial elimination from the lungs, which is gravitationally dependent in critically ill patients.59 Lowering the head of the bed might be more protective than raising it,59 but has not been studied in clinical trials. Historically, the composition of enteral nutrition has been tailored to meet the perceived metabolic needs of the host, without taking into account its effects on the microbiome. This approach, however, might overlook the most direct means of shaping environmental growth conditions within the gut microbiome.41 Observational human studies alone cannot disentangle the effects of critical illness from the effects of its treatment (eg, antibiotics). Thus future investigation of the microbiome’s role in critical illness will require the use of animal studies and prospective, controlled human trials.
The microbiome can be manipulated therapeutically, as has been shown by the success of faecal microbiota transplantation in the treatment of refractory Clostridium difficile infection. Evidence of therapeutic manipulation of the microbiome in critical illness is promising.106 SDD is the most thoroughly studied intervention in critical care research, and has unambiguous benefits in the prevention of infections, multiorgan failure, and death.91,96 Early intensive-care studies of probiotics suggest that they decrease the risk of pneumonia and shorten the length of stay in the intensive-care unit for ventilated patients150 and decrease systemic infections in high-risk postoperative patients.151 Improved survival has been reported in a mouse model of sepsis.152 These blunt and broad interventions, with one-size-fits-all cocktails of antibiotics or probiotics, however, represent the opposite of targeted therapy. With the advent of culture-independent microbiology, the means are at last available to identify specific features of the microbiome that promote and disrupt homoeostasis in critically ill patients. At the current pace of development, point-of-care community sequencing and identification of pathogens will be available and affordable within years rather than decades.62,144 Improved understanding of what constitutes a healthy microbiome is urgently needed in this population so that rational therapies to restore and maintain it can be developed.
The microbiome is central to the biology of critical illness and, therefore, should be included in any discussion of disease phenotyping in intensive care. Most studies and reviews of precision medicine in critical illness, however, focus on host genetics, immune responses, and exposures.153–155 None of these accounts for the differences in outcomes attributable solely to differences in patients’ microbiota (figure 2). Before tailored therapy can be provided to patients, how the microbiota informs prognosis and response to treatment needs to be understood. All clinical trials in critical illness should consider assessment of the microbiome, in the gut and the lungs, as an important secondary outcome, as both a mediator of disease and as a modifier of therapy.
Neonates represent an important and understudied population as they are highly vulnerable to alterations in the developing microbiome and to life-threatening critical illnesses. Premature neonates are subjected to innumerable microbiome-altering exposures (eg, antibiotics and formula feeding) and lack mature innate and adaptive immune responses. In multiple studies, the composition of the early gut microbiome has been predictive of neonatal sepsis,70,156,157 which can be plausibly explained by either enteric harbouring of potential pathogens or systemic immune derangements provoked by intestinal dysbiosis. Experimental data suggest that early exposure to a diverse intestinal microbiome is essential for the development of an intact immune response: newborn mice with antibiotic-suppressed microbiota have increased susceptibility to pulmonary infections158 and bacterial sepsis.159 Necrotising enterocolitis, a devastating and idiopathic disease of neonates, has been linked to intestinal dysbiosis in animal160 and human studies,161 and randomised controlled trials support a protective role of probiotics.162,163 The acute and chronic consequences of dysbiosis in neonates are worthy of immediate clinical and experimental study.
Finally, although this Review has focused on the causes and consequences of acute perturbations of the microbiome in critical illness, the research into intensive-care outcomes in the past decade has convincingly shown that the sequelae of critical illness persist long after patients are extubated and discharged. Survivors of ARDS and sepsis have chronic deficits in cognitive function and functional status, and are at high risk of re-admission in the months after discharge,164 disproportionately so for infection-related events. The mechanisms underlying this so-called postintensive-care syndrome are poorly understood, but the contribution of a persistently altered microbiome should be explored. Derangements of the microbiome persist for weeks and months after even a short antibiotic course,66 and how quickly or completely the microbiome recovers after the insults and disruptions of critical illness are unknown. Research is needed to define the natural history of microbiome recovery after critical illness, to determine whether recovery can be accelerated (eg, via probiotics or faecal microbiota transplantation), and whether this recovery improves long-term outcomes for patients. In patients recovering from multiorgan failure, it may be that microbiome is the last organ to recover.
Conclusions
Although the importance of the microbiome in critical illness has been established for a half century, the revolution in culture-independent microbiology has at last yielded tools capable of determining its contribution to the pathogenesis of sepsis, ARDS, and multiorgan failure. Continuing clinical and experimental trials will explore how the microbiome is altered in disease, and in turn how its disturbance perpetuates organ injury. The microbiome represents a key therapeutic target for the prevention and treatment of critical illness, and should be included in any discussion of precision medicine in the intensive care unit.
Search strategy and selection criteria.
I searched MEDLINE and Web of Science without date or language restrictions, with the initial search terms of “([microbiota] OR [flora]) AND ([sepsis] OR [shock] OR [acute respiratory distress syndrome] OR [multiorgan failure]).”
I manually screened titles and abstracts to exclude unrelated studies. I read all relevant articles, and identified additional relevant articles via citations. Due to space limitations, only references with immediate relevance to topics discussed in the Review are included.
Key messages.
The microbial ecosystems of the gut and the lungs change substantially in critically ill patients, resulting in dramatic changes to bacterial communities
In animal studies of shock, the microbial contents of the gut determine the severity of multiorgan failure and the risk of death, an observation supported by trials of selective manipulation of the gut microbiome in human beings
The mechanisms that drive gut-derived sepsis are incompletely understood and multifactorial, offering numerous unexplored therapeutic targets
During lung injury, the bacterial ecosystem of the alveolus shifts to a state of abundance in nutrients and growth-promoting host stress signals, leading to a positive feedback loop of inflammation and dysbiosis
The microbiome is a key therapeutic target for the prevention and treatment of critical illness, and it should be included in any discussion of precision medicine in the intensive care unit
Acknowledgments
I am supported by the National Institutes for Health (UL1TR000433) via a grant provided by the Michigan Institute for Clinical & Health Research, the Host Microbiome Initiative of the University of Michigan, and the University of Michigan Center for Integrative Research in Critical Care. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of interests
I declare no competing interests.
References
- 1.Adhikari NK, Fowler RA, Bhagwanjee S, Rubenfeld GD. Critical care and the global burden of critical illness in adults. Lancet. 2010;376:1339–46. doi: 10.1016/S0140-6736(10)60446-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7:245–57. doi: 10.1586/ers.13.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuczynski J, Lauber CL, Walters WA, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johanson WG, Pierce AK, Sanford JP. Changing pharyngeal bacterial flora of hospitalized patients. Emergence of gram-negative bacilli. N Engl J Med. 1969;281:1137–40. doi: 10.1056/NEJM196911202812101. [DOI] [PubMed] [Google Scholar]
- 5.Baas Becking LGM. Geobiologie, of Inleiding Tot de Milieukunde: Met Literatuurlijst en Ind. Van Stockum; The Hague: 1934. [Google Scholar]
- 6.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bahrani-Mougeot FK, Paster BJ, Coleman S, et al. Molecular analysis of oral and respiratory bacterial species associated with ventilator-associated pneumonia. J Clin Microbiol. 2007;45:1588–93. doi: 10.1128/JCM.01963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Björnsson ES, Urbanavicius V, Eliasson B, Attvall S, Smith U, Abrahamsson H. Effects of hyperglycemia on interdigestive gastrointestinal motility in humans. Scand J Gastroenterol. 1994;29:1096–104. doi: 10.3109/00365529409094894. [DOI] [PubMed] [Google Scholar]
- 9.Lowman RM. The potassium depletion states and postoperative ileus. The role of the potassium ion. Radiology. 1971;98:691–94. doi: 10.1148/98.3.691. [DOI] [PubMed] [Google Scholar]
- 10.O’Boyle CJ, MacFie J, Mitchell CJ, Johnstone D, Sagar PM, Sedman PC. Microbiology of bacterial translocation in humans. Gut. 1998;42:29–35. doi: 10.1136/gut.42.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacFie J, Reddy BS, Gatt M, Jain PK, Sowdi R, Mitchell CJ. Bacterial translocation studied in 927 patients over 13 years. Br J Surg. 2006;93:87–93. doi: 10.1002/bjs.5184. [DOI] [PubMed] [Google Scholar]
- 12.Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. 2012;10:350–56. doi: 10.1016/j.surge.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winter SE, Winter MG, Xavier MN, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–11. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lupp C, Robertson ML, Wickham ME, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. cell Host Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol. 2012;30:759–95. doi: 10.1146/annurev-immunol-020711-074937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grootjans J, Lenaerts K, Derikx JP, et al. Human intestinal ischemia-reperfusion-induced inflammation characterized: experiences from a new translational model. Am J Pathol. 2010;176:2283–91. doi: 10.2353/ajpath.2010.091069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Vree JM, Romijn JA, Mok KS, et al. Lack of enteral nutrition during critical illness is associated with profound decrements in biliary lipid concentrations. Am J Clin Nutr. 1999;70:70–77. doi: 10.1093/ajcn/70.1.70. [DOI] [PubMed] [Google Scholar]
- 18.Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29:625–51. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Zaborina O, Lepine F, Xiao G, et al. Dynorphin activates quorum sensing quinolone signaling in Pseudomonas aeruginosa. PLoS Pathog. 2007;3:e35. doi: 10.1371/journal.ppat.0030035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaborin A, Smith D, Garfield K, et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. MBio. 2014;5:e01361–14. doi: 10.1128/mBio.01361-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dive A, Foret F, Jamart J, Bulpa P, Installé E. Effect of dopamine on gastrointestinal motility during critical illness. Intensive Care Med. 2000;26:901–07. doi: 10.1007/s001340051279. [DOI] [PubMed] [Google Scholar]
- 22.Alverdy J, Holbrook C, Rocha F, et al. Gut-derived sepsis occurs when the right pathogen with the right virulence genes meets the right host: evidence for in vivo virulence expression in Pseudomonas aeruginosa. Ann Surg. 2000;232:480–89. doi: 10.1097/00000658-200010000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meduri GU, Kanangat S, Stefan J, Tolley E, Schaberg D. Cytokines IL-1beta, IL-6, and TNF-alpha enhance in vitro growth of bacteria. Am J Respir Crit Care Med. 1999;160:961–67. doi: 10.1164/ajrccm.160.3.9807080. [DOI] [PubMed] [Google Scholar]
- 24.Kanangat S, Meduri GU, Tolley EA, et al. Effects of cytokines and endotoxin on the intracellular growth of bacteria. Infect Immun. 1999;67:2834–40. doi: 10.1128/iai.67.6.2834-2840.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q, Xu DZ, Sharpe S, et al. The anatomic sites of disruption of the mucus layer directly correlate with areas of trauma/hemorrhagic shock-induced gut injury. J Trauma. 2011;70:630–35. doi: 10.1097/TA.0b013e3181e1221b. [DOI] [PubMed] [Google Scholar]
- 26.Rupani B, Caputo FJ, Watkins AC, et al. Relationship between disruption of the unstirred mucus layer and intestinal restitution in loss of gut barrier function after trauma hemorrhagic shock. Surgery. 2007;141:481–89. doi: 10.1016/j.surg.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 27.Katayama M, Xu D, Specian RD, Deitch EA. Role of bacterial adherence and the mucus barrier on bacterial translocation: effects of protein malnutrition and endotoxin in rats. Ann Surg. 1997;225:317–26. doi: 10.1097/00000658-199703000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morehouse JL, Specian RD, Stewart JJ, Berg RD. Translocation of indigenous bacteria from the gastrointestinal tract of mice after oral ricinoleic acid treatment. Gastroenterology. 1986;91:673–82. doi: 10.1016/0016-5085(86)90638-4. [DOI] [PubMed] [Google Scholar]
- 29.Albenberg L, Esipova TV, Judge CP, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;147:1055–63.e8. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer-Hoffert U, Hornef MW, Henriques-Normark B, et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut. 2008;57:764–71. doi: 10.1136/gut.2007.141481. [DOI] [PubMed] [Google Scholar]
- 31.Coutinho HB, Robalinho TI, Coutinho VB, et al. Intra-abdominal sepsis: an immunocytochemical study of the small intestine mucosa. J Clin Pathol. 1997;50:294–98. doi: 10.1136/jcp.50.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Book M, Chen Q, Lehmann LE, et al. Inducibility of the endogenous antibiotic peptide beta-defensin 2 is impaired in patients with severe sepsis. Crit Care. 2007;11:R19. doi: 10.1186/cc5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teltschik Z, Wiest R, Beisner J, et al. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology. 2012;55:1154–63. doi: 10.1002/hep.24789. [DOI] [PubMed] [Google Scholar]
- 34.Salzman NH, Hung K, Haribhai D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–39. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Moore JG, Datz FL, Christian PE, Greenberg E, Alazraki N. Effect of body posture on radionuclide measurements of gastric emptying. Dig Dis Sci. 1988;33:1592–95. doi: 10.1007/BF01535951. [DOI] [PubMed] [Google Scholar]
- 37.Cordova-Fraga T, Sosa M, Wiechers C, et al. Effects of anatomical position on esophageal transit time: a biomagnetic diagnostic technique. World J Gastroenterol. 2008;14:5707–11. doi: 10.3748/wjg.14.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol. 2010;8:504–08. doi: 10.1016/j.cgh.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 39.Du Moulin GC, Hedley-Whyte J, Paterson DG, Lisbon A. Aspiration of gastric bacteria in antacid-treated patients: a frequent cause of postoperative colonisation of the airway. Lancet. 1982;319:242–45. doi: 10.1016/s0140-6736(82)90974-6. [DOI] [PubMed] [Google Scholar]
- 40.Hadfield RJ, Sinclair DG, Houldsworth PE, Evans TW. Effects of enteral and parenteral nutrition on gut mucosal permeability in the critically ill. Am J Respir Crit Care Med. 1995;152:1545–48. doi: 10.1164/ajrccm.152.5.7582291. [DOI] [PubMed] [Google Scholar]
- 41.Schneider SM, Le Gall P, Girard-Pipau F, et al. Total artificial nutrition is associated with major changes in the fecal flora. Eur J Nutr. 2000;39:248–55. doi: 10.1007/s003940070003. [DOI] [PubMed] [Google Scholar]
- 42.Alverdy JC, Aoys E, Moss GS. Total parenteral nutrition promotes bacterial translocation from the gut. Surgery. 1988;104:185–90. [PubMed] [Google Scholar]
- 43.Alverdy J, Chi HS, Sheldon GF. The effect of parenteral nutrition on gastrointestinal immunity. The importance of enteral stimulation. Ann Surg. 1985;202:681–84. doi: 10.1097/00000658-198512000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benus RF, Harmsen HJ, Welling GW, et al. Impact of digestive and oropharyngeal decontamination on the intestinal microbiota in ICU patients. Intensive Care Med. 2010;36:1394–402. doi: 10.1007/s00134-010-1826-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Metheny NA, Clouse RE, Chang YH, Stewart BJ, Oliver DA, Kollef MH. Tracheobronchial aspiration of gastric contents in critically ill tube-fed patients: frequency, outcomes, and risk factors. Crit Care Med. 2006;34:1007–15. doi: 10.1097/01.CCM.0000206106.65220.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sekizawa K, Ujiie Y, Itabashi S, Sasaki H, Takishima T. Lack of cough reflex in aspiration pneumonia. Lancet. 1990;335:1228–29. doi: 10.1016/0140-6736(90)92758-a. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa NK, Franchini ML, Driusso P, de Oliveira LR, Saldiva PH, Lorenzi-Filho G. Mucociliary clearance is impaired in acutely ill patients. Chest. 2005;128:2772–77. doi: 10.1378/chest.128.4.2772. [DOI] [PubMed] [Google Scholar]
- 48.Worlitzsch D, Tarran R, Ulrich M, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109:317–25. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt A, Belaaouaj A, Bissinger R, et al. Neutrophil elastase-mediated increase in airway temperature during inflammation. J Cyst Fibros. 2014;13:623–31. doi: 10.1016/j.jcf.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Dickson RP, Erb-Downward JR, Prescott HC, et al. Intraalveolar catecholamines and the human lung microbiome. Am J Respir Crit Care Med. 2015;192:257–59. doi: 10.1164/rccm.201502-0326LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freestone PP, Hirst RA, Sandrini SM, et al. Pseudomonas aeruginosa-catecholamine inotrope interactions: a contributory factor in the development of ventilator-associated pneumonia? Chest. 2012;142:1200–10. doi: 10.1378/chest.11-2614. [DOI] [PubMed] [Google Scholar]
- 52.Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. cell. 2006;124:767–82. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 53.Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370:786–96. doi: 10.1016/S0140-6736(07)61382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7:1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Günther A, Siebert C, Schmidt R, et al. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med. 1996;153:176–84. doi: 10.1164/ajrccm.153.1.8542113. [DOI] [PubMed] [Google Scholar]
- 56.Poroyko V, Meng F, Meliton A, et al. Alterations of lung microbiota in a mouse model of LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;309:L76–83. doi: 10.1152/ajplung.00061.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu H, Kuzmenko A, Wan S, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589–602. doi: 10.1172/JCI16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torres A, Serra-Batlles J, Ros E, et al. Pulmonary aspiration of gastric contents in patients receiving mechanical ventilation: the effect of body position. Ann Intern Med. 1992;116:540–43. doi: 10.7326/0003-4819-116-7-540. [DOI] [PubMed] [Google Scholar]
- 59.Li Bassi G, Zanella A, Cressoni M, Stylianou M, Kolobow T. Following tracheal intubation, mucus flow is reversed in the semirecumbent position: possible role in the pathogenesis of ventilator-associated pneumonia. Crit Care Med. 2008;36:518–25. doi: 10.1097/01.CCM.0000299741.32078.E9. [DOI] [PubMed] [Google Scholar]
- 60.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369:2126–36. doi: 10.1056/NEJMra1208707. [DOI] [PubMed] [Google Scholar]
- 61.Muscedere J, Rewa O, McKechnie K, Jiang X, Laporta D, Heyland DK. Subglottic secretion drainage for the prevention of ventilator-associated pneumonia: a systematic review and meta-analysis. Crit Care Med. 2011;39:1985–91. doi: 10.1097/CCM.0b013e318218a4d9. [DOI] [PubMed] [Google Scholar]
- 62.Flanagan JL, Brodie EL, Weng L, et al. Loss of bacterial diversity during antibiotic treatment of intubated patients colonized with Pseudomonas aeruginosa. J Clin Microbiol. 2007;45:1954–62. doi: 10.1128/JCM.02187-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simon GL, Gorbach SL. Intestinal flora in health and disease. Gastroenterology. 1984;86:174–93. [PubMed] [Google Scholar]
- 64.Marshall JC, Christou NV, Meakins JL. The gastrointestinal tract. The “undrained abscess” of multiple organ failure. Ann Surg. 1993;218:111–19. doi: 10.1097/00000658-199308000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knoop KA, McDonald KG, Kulkarni DH, Newberry RD. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut. 2015 doi: 10.1136/gutjnl-2014-309059. published online June 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108(suppl 1):4554–61. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marshall JC, Christou NV, Horn R, Meakins JL. The microbiology of multiple organ failure. The proximal gastrointestinal tract as an occult reservoir of pathogens. Arch Surg. 1988;123:309–15. doi: 10.1001/archsurg.1988.01400270043006. [DOI] [PubMed] [Google Scholar]
- 68.de la Cal MA, Rommes JH, van Saene HK, Silvestri L, Zandstra DF. Selective digestive decontamination and bacterial resistance. Lancet Infect Dis. 2013;13:738. doi: 10.1016/S1473-3099(13)70217-2. [DOI] [PubMed] [Google Scholar]
- 69.Shimizu K, Ogura H, Hamasaki T, et al. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis Sci. 2011;56:1171–77. doi: 10.1007/s10620-010-1418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madan JC, Salari RC, Saxena D, et al. Gut microbial colonisation in premature neonates predicts neonatal sepsis. Arch Dis Child Fetal Neonatal Ed. 2012;97:F456–62. doi: 10.1136/fetalneonatal-2011-301373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shimizu K, Ogura H, Goto M, et al. Altered gut flora and environment in patients with severe SIRS. J Trauma. 2006;60:126–33. doi: 10.1097/01.ta.0000197374.99755.fe. [DOI] [PubMed] [Google Scholar]
- 72.Wey SB, Mori M, Pfaller MA, Woolson RF, Wenzel RP. Hospital-acquired candidemia. The attributable mortality and excess length of stay. Arch Intern Med. 1988;148:2642–45. doi: 10.1001/archinte.148.12.2642. [DOI] [PubMed] [Google Scholar]
- 73.Arumugam M, Raes J, Pelletier E, et al. the MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blaser MJ. The microbiome revolution. J Clin Invest. 2014;124:4162–65. doi: 10.1172/JCI78366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Donohoe DR, Garge N, Zhang X, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13:517–26. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 77.Dickson RP, Erb-Downward JR, Freeman CM, et al. Spatial Variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12:821–30. doi: 10.1513/AnnalsATS.201501-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gleeson K, Eggli DF, Maxwell SL. Quantitative aspiration during sleep in normal subjects. Chest. 1997;111:1266–72. doi: 10.1378/chest.111.5.1266. [DOI] [PubMed] [Google Scholar]
- 79.Segal LN, Alekseyenko AV, Clemente JC, et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome. 2013;1:19. doi: 10.1186/2049-2618-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bassis CM, Erb-Downward JR, Dickson RP, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6:e00037. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Munro CL, Grap MJ. Oral health and care in the intensive care unit: state of the science. Am J Crit Care. 2004;13:25–33. [PubMed] [Google Scholar]
- 82.Bousbia S, Papazian L, Saux P, et al. Repertoire of intensive care unit pneumonia microbiota. PLoS One. 2012;7:e32486. doi: 10.1371/journal.pone.0032486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu W, Yu J, Ai Q, Liu D, Song C, Li L. Increased constituent ratios of Klebsiella sp., Acinetobacter sp., and Streptococcus sp. and a decrease in microflora diversity may be indicators of ventilator-associated pneumonia: a prospective study in the respiratory tracts of neonates. PLoS One. 2014;9:e87504. doi: 10.1371/journal.pone.0087504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.May AK, Brady JS, Romano-Keeler J, et al. A pilot study of the noninvasive assessment of the lung microbiota as a potential tool for the early diagnosis of ventilator-associated pneumonia. Chest. 2015;147:1494–502. doi: 10.1378/chest.14-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Senator H. Über einen Fall von Hydrothionämie und über Selbstinfection durch abnorme Verdauungsvorgänge. In: Waldenberg L, editor. Berliner Klinische Wochenschrift. A Hirschwald; Berlin: 1868. pp. 254–56. [Google Scholar]
- 86.Chain E, Florey HW, Gardner AD, et al. Penicillin as a chemotherapeutic agent. Lancet. 1940;236:226–28. [Google Scholar]
- 87.Fine J, Frank H, Schweinburg F, Jacob S, Gordon T. The bacterial factor in traumatic shock. Ann N Y Acad Sci. 1952;55:429–45. doi: 10.1111/j.1749-6632.1952.tb26558.x. [DOI] [PubMed] [Google Scholar]
- 88.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2:319–23. [Google Scholar]
- 89.Cuevas P, De la Maza LM, Gilbert J, Fine J. The lung lesion in four different types of shock in rabbits. Arch Surg. 1972;104:319–22. doi: 10.1001/archsurg.1972.04180030067015. [DOI] [PubMed] [Google Scholar]
- 90.Souza DG, Vieira AT, Soares AC, et al. The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol. 2004;173:4137–46. doi: 10.4049/jimmunol.173.6.4137. [DOI] [PubMed] [Google Scholar]
- 91.Silvestri L, van Saene HK, Zandstra DF, Marshall JC, Gregori D, Gullo A. Impact of selective decontamination of the digestive tract on multiple organ dysfunction syndrome: systematic review of randomized controlled trials. Crit Care Med. 2010;38:1370–76. doi: 10.1097/CCM.0b013e3181d9db8c. [DOI] [PubMed] [Google Scholar]
- 92.Liberati A, D’Amico R, Pifferi S, Torri V, Brazzi L, Parmelli E. Antibiotic prophylaxis to reduce respiratory tract infections and mortality in adults receiving intensive care. Cochrane Database Syst Rev. 2009;4 doi: 10.1002/14651858.CD000022.pub3. CD000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang D, Chen G, Manwani D, et al. Neutrophil ageing is regulated by the microbiome. Nature. 2015;525:528–32. doi: 10.1038/nature15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rush BF, Jr, Redan JA, Flanagan JJ, Jr, et al. Does the bacteremia observed in hemorrhagic shock have clinical significance? A study in germ-free animals. Ann Surg. 1989;210:342–45. doi: 10.1097/00000658-198909000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Unertl K, Ruckdeschel G, Selbmann HK, et al. Prevention of colonization and respiratory infections in long-term ventilated patients by local antimicrobial prophylaxis. Intensive Care Med. 1987;13:106–13. doi: 10.1007/BF00254795. [DOI] [PubMed] [Google Scholar]
- 96.Silvestri L, de la Cal MA, van Saene HK. Selective decontamination of the digestive tract: the mechanism of action is control of gut overgrowth. Intensive Care Med. 2012;38:1738–50. doi: 10.1007/s00134-012-2690-1. [DOI] [PubMed] [Google Scholar]
- 97.Daneman N, Sarwar S, Fowler RA, Cuthbertson BH, the SuDDICU Canadian Study Group Effect of selective decontamination on antimicrobial resistance in intensive care units: a systematic review and meta-analysis. Lancet Infect Dis. 2013;13:328–41. doi: 10.1016/S1473-3099(12)70322-5. [DOI] [PubMed] [Google Scholar]
- 98.Oostdijk EA, de Smet AM, Blok HE, et al. Ecological effects of selective decontamination on resistant gram-negative bacterial colonization. Am J Respir Crit Care Med. 2010;181:452–57. doi: 10.1164/rccm.200908-1210OC. [DOI] [PubMed] [Google Scholar]
- 99.Prescott HC, Dickson RP, Rogers MA, Langa KM, Iwashyna TJ. Hospitalization type and subsequent severe sepsis. Am J Respir Crit Care Med. 2015;192:581–88. doi: 10.1164/rccm.201503-0483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doig CJ, Sutherland LR, Sandham JD, Fick GH, Verhoef M, Meddings JB. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am J Respir Crit Care Med. 1998;158:444–51. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 101.Moore FA, Moore EE, Poggetti R, et al. Gut bacterial translocation via the portal vein: a clinical perspective with major torso trauma. J Trauma. 1991;31:629–36. doi: 10.1097/00005373-199105000-00006. [DOI] [PubMed] [Google Scholar]
- 102.Deitch EA, Xu D-Z, Lu Q. Gut lymph hypothesis of early shock and trauma-induced multiple organ dysfunction syndrome: a new look at gut origin sepsis. J Organ Dysfunct. 2006;2:70–79. [Google Scholar]
- 103.Sedman PC, Macfie J, Sagar P, et al. The prevalence of gut translocation in humans. Gastroenterology. 1994;107:643–49. doi: 10.1016/0016-5085(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 104.Reino DC, Pisarenko V, Palange D, et al. Trauma hemorrhagic shock-induced lung injury involves a gut-lymph-induced TLR4 pathway in mice. PLoS One. 2011;6:e14829. doi: 10.1371/journal.pone.0014829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alverdy JC, Laughlin RS, Wu L. Influence of the critically ill state on host-pathogen interactions within the intestine: gut-derived sepsis redefined. Crit Care Med. 2003;31:598–607. doi: 10.1097/01.CCM.0000045576.55937.67. [DOI] [PubMed] [Google Scholar]
- 106.Schuijt TJ, van der Poll T, de Vos WM, Wiersinga WJ. The intestinal microbiota and host immune interactions in the critically ill. Trends Microbiol. 2013;21:221–29. doi: 10.1016/j.tim.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 107.Seely AJ, Christou NV. Multiple organ dysfunction syndrome: exploring the paradigm of complex nonlinear systems. Crit Care Med. 2000;28:2193–200. doi: 10.1097/00003246-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 108.Suau A, Bonnet R, Sutren M, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bjarnason I, Williams P, Smethurst P, Peters TJ, Levi AJ. Effect of non-steroidal anti-inflammatory drugs and prostaglandins on the permeability of the human small intestine. Gut. 1986;27:1292–97. doi: 10.1136/gut.27.11.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alverdy J, Aoys E. The effect of glucocorticoid administration on bacterial translocation. Evidence for an acquired mucosal immunodeficient state. Ann Surg. 1991;214:719–23. doi: 10.1097/00000658-199112000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Meng J, Yu H, Ma J, et al. Morphine induces bacterial translocation in mice by compromising intestinal barrier function in a TLR-dependent manner. PLoS One. 2013;8:e54040. doi: 10.1371/journal.pone.0054040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Venkataraman A, Bassis CM, Beck JM, et al. Application of a neutral community model to assess structuring of the human lung microbiome. MBio. 2015;6:e01163–15. doi: 10.1128/mBio.02284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. 2014;2:238–46. doi: 10.1016/S2213-2600(14)70028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bauer TT, Ewig S, Rodloff AC, Müller EE. Acute respiratory distress syndrome and pneumonia: a comprehensive review of clinical data. Clin Infect Dis. 2006;43:748–56. doi: 10.1086/506430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chastre J, Trouillet JL, Vuagnat A, et al. Nosocomial pneumonia in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1998;157:1165–72. doi: 10.1164/ajrccm.157.4.9708057. [DOI] [PubMed] [Google Scholar]
- 116.Futier E, Constantin JM, Paugam-Burtz C, et al. the IMPROVE study group A trial of intraoperative low-tidal-volume ventilation in abdominal surgery. N Engl J Med. 2013;369:428–37. doi: 10.1056/NEJMoa1301082. [DOI] [PubMed] [Google Scholar]
- 117.Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. 1995;108:1303–14. doi: 10.1378/chest.108.5.1303. [DOI] [PubMed] [Google Scholar]
- 118.Freestone P. Communication between bacteria and their hosts. Scientifica (Cairo) 2013;2013:361073. doi: 10.1155/2013/361073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sandrini S, Aldriwesh M, Alruways M, Freestone P. Microbial endocrinology: host-bacteria communication within the gut microbiome. J Endocrinol. 2015;225:R21–34. doi: 10.1530/JOE-14-0615. [DOI] [PubMed] [Google Scholar]
- 120.Flierl MA, Rittirsch D, Nadeau BA, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449:721–25. doi: 10.1038/nature06185. [DOI] [PubMed] [Google Scholar]
- 121.Marks LR, Davidson BA, Knight PR, Hakansson AP. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. MBio. 2013;4:e00438–13. doi: 10.1128/mBio.00438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Donnell PM, Aviles H, Lyte M, Sonnenfeld G. Enhancement of in vitro growth of pathogenic bacteria by norepinephrine: importance of inoculum density and role of transferrin. Appl Environ Microbiol. 2006;72:5097–99. doi: 10.1128/AEM.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Freestone PP, Haigh RD, Williams PH, Lyte M. Stimulation of bacterial growth by heat-stable, norepinephrine-induced autoinducers. FEMS Microbiol Lett. 1999;172:53–60. doi: 10.1111/j.1574-6968.1999.tb13449.x. [DOI] [PubMed] [Google Scholar]
- 124.Kaza SK, McClean S, Callaghan M. IL-8 released from human lung epithelial cells induced by cystic fibrosis pathogens Burkholderia cepacia complex affects the growth and intracellular survival of bacteria. Int J Med Microbiol. 2011;301:26–33. doi: 10.1016/j.ijmm.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 125.Plotkin BJ, Roose RJ, Erikson Q, Viselli SM. Effect of androgens and glucocorticoids on microbial growth and antimicrobial susceptibility. Curr Microbiol. 2003;47:514–20. doi: 10.1007/s00284-003-4080-y. [DOI] [PubMed] [Google Scholar]
- 126.Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. 2014;384:691–702. doi: 10.1016/S0140-6736(14)61136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Flume PA, Mogayzel PJ, Jr, Robinson KA, et al. the clinical practice guidelines for Pulmonary Therapies Committee Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802–08. doi: 10.1164/rccm.200812-1845PP. [DOI] [PubMed] [Google Scholar]
- 128.Chang AB, Bilton D. Exacerbations in cystic fibrosis: 4—non-cystic fibrosis bronchiectasis. Thorax. 2008;63:269–76. doi: 10.1136/thx.2006.060913. [DOI] [PubMed] [Google Scholar]
- 129.Graham V, Lasserson T, Rowe BH. Antibiotics for acute asthma. Cochrane Database Syst Rev. 2001;3 doi: 10.1002/14651858.CD002741. CD002741. [DOI] [PubMed] [Google Scholar]
- 130.Dickson RP, Erb-Downward JR, Prescott HC, et al. Analysis of culture-dependent versus culture-independent techniques for identification of bacteria in clinically obtained bronchoalveolar lavage fluid. J Clin Microbiol. 2014;52:3605–13. doi: 10.1128/JCM.01028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Iwai S, Huang D, Fong S, et al. The lung microbiome of Ugandan HIV-infected pneumonia patients is compositionally and functionally distinct from that of San Franciscan patients. PLoS One. 2014;9:e95726. doi: 10.1371/journal.pone.0095726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:2813–23. doi: 10.1128/JCM.00035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Millares L, Ferrari R, Gallego M, et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis. 2014;33:1101–11. doi: 10.1007/s10096-013-2044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stressmann FA, Rogers GB, Marsh P, et al. Does bacterial density in cystic fibrosis sputum increase prior to pulmonary exacerbation? J Cyst Fibros. 2011;10:357–65. doi: 10.1016/j.jcf.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 135.Zhao J, Schloss PD, Kalikin LM, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–14. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Carmody LA, Zhao J, Schloss PD, et al. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann Am Thorac Soc. 2013;10:179–87. doi: 10.1513/AnnalsATS.201211-107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Price KE, Hampton TH, Gifford AH, et al. Unique microbial communities persist in individual cystic fibrosis patients throughout a clinical exacerbation. Microbiome. 2013;1:27. doi: 10.1186/2049-2618-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carmody LA, Zhao J, Kalikin LM, et al. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome. 2015;3:12. doi: 10.1186/s40168-015-0074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tunney MM, Einarsson GG, Wei L, et al. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med. 2013;187:1118–26. doi: 10.1164/rccm.201210-1937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hurley MN, Ariff AH, Bertenshaw C, Bhatt J, Smyth AR. Results of antibiotic susceptibility testing do not influence clinical outcome in children with cystic fibrosis. J Cyst Fibros. 2012;11:288–92. doi: 10.1016/j.jcf.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Smith AL, Fiel SB, Mayer-Hamblett N, Ramsey B, Burns JL. Susceptibility testing of Pseudomonas aeruginosa isolates and clinical response to parenteral antibiotic administration: lack of association in cystic fibrosis. Chest. 2003;123:1495–502. doi: 10.1378/chest.123.5.1495. [DOI] [PubMed] [Google Scholar]
- 142.Rogers GB, Zain NM, Bruce KD, et al. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc. 2014;11:496–503. doi: 10.1513/AnnalsATS.201310-335OC. [DOI] [PubMed] [Google Scholar]
- 143.Albert RK, Connett J, Bailey WC, et al. the COPD Clinical Research Network Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689–98. doi: 10.1056/NEJMoa1104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Saiman L, Marshall BC, Mayer-Hamblett N, et al. the Macrolide Study Group Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749–56. doi: 10.1001/jama.290.13.1749. [DOI] [PubMed] [Google Scholar]
- 145.Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet. 2012;380:660–67. doi: 10.1016/S0140-6736(12)60953-2. [DOI] [PubMed] [Google Scholar]
- 146.Molyneaux PL, Mallia P, Cox MJ, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:1224–31. doi: 10.1164/rccm.201302-0341OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Labro MT, Abdelghaffar H. Immunomodulation by macrolide antibiotics. J Chemother. 2001;13:3–8. doi: 10.1179/joc.2001.13.1.3. [DOI] [PubMed] [Google Scholar]
- 148.Herzig SJ, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for hospital-acquired pneumonia. JAMA. 2009;301:2120–28. doi: 10.1001/jama.2009.722. [DOI] [PubMed] [Google Scholar]
- 149.Drakulovic MB, Torres A, Bauer TT, Nicolas JM, Nogué S, Ferrer M. Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial. Lancet. 1999;354:1851–58. doi: 10.1016/S0140-6736(98)12251-1. [DOI] [PubMed] [Google Scholar]
- 150.Siempos II, Ntaidou TK, Falagas ME. Impact of the administration of probiotics on the incidence of ventilator-associated pneumonia: a meta-analysis of randomized controlled trials. Crit Care Med. 2010;38:954–62. doi: 10.1097/CCM.0b013e3181c8fe4b. [DOI] [PubMed] [Google Scholar]
- 151.Pitsouni E, Alexiou V, Saridakis V, Peppas G, Falagas ME. Does the use of probiotics/synbiotics prevent postoperative infections in patients undergoing abdominal surgery? A meta-analysis of randomized controlled trials. Eur J Clin Pharmacol. 2009;65:561–70. doi: 10.1007/s00228-009-0642-7. [DOI] [PubMed] [Google Scholar]
- 152.Khailova L, Frank DN, Dominguez JA, Wischmeyer PE. Probiotic administration reduces mortality and improves intestinal epithelial homeostasis in experimental sepsis. Anesthesiology. 2013;119:166–77. doi: 10.1097/ALN.0b013e318291c2fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Abraham E. It’s all in the genes: moving toward precision medicine in critical illness. Crit Care Med. 2013;41:1363–64. doi: 10.1097/CCM.0b013e31827c02dd. [DOI] [PubMed] [Google Scholar]
- 154.Norris PR, Canter JA, Jenkins JM, Moore JH, Williams AE, Morris JA., Jr Personalized medicine: genetic variation and loss of physiologic complexity are associated with mortality in 644 trauma patients. Ann Surg. 2009;250:524–30. doi: 10.1097/SLA.0b013e3181b8fb1f. [DOI] [PubMed] [Google Scholar]
- 155.Sen A, Yende S. Towards personalized medicine in sepsis: quest for Shangri-La? Crit Care. 2013;17:303. doi: 10.1186/cc12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shaw AG, Sim K, Randell P, et al. Late-onset bloodstream infection and perturbed maturation of the gastrointestinal microbiota in premature infants. PLoS One. 2015;10:e0132923. doi: 10.1371/journal.pone.0132923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mai V, Torrazza RM, Ukhanova M, et al. Distortions in development of intestinal microbiota associated with late onset sepsis in preterm infants. PLoS One. 2013;8:e52876. doi: 10.1371/journal.pone.0052876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Clarke TB. Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via nod-like receptor ligands. Infect Immun. 2014;82:4596–606. doi: 10.1128/IAI.02212-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Deshmukh HS, Liu Y, Menkiti OR, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. 2014;20:524–30. doi: 10.1038/nm.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Azcarate-Peril MA, Foster DM, Cadenas MB, et al. Acute necrotizing enterocolitis of preterm piglets is characterized by dysbiosis of ileal mucosa-associated bacteria. Gut Microbes. 2011;2:234–43. doi: 10.4161/gmic.2.4.16332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sim K, Shaw AG, Randell P, et al. Dysbiosis anticipating necrotizing enterocolitis in very premature infants. Clin Infect Dis. 2015;60:389–97. doi: 10.1093/cid/ciu822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bin-Nun A, Bromiker R, Wilschanski M, et al. Oral probiotics prevent necrotizing enterocolitis in very low birth weight neonates. J Pediatr. 2005;147:192–96. doi: 10.1016/j.jpeds.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 163.Lin HC, Hsu CH, Chen HL, et al. Oral probiotics prevent necrotizing enterocolitis in very low birth weight preterm infants: a multicenter, randomized, controlled trial. Pediatrics. 2008;122:693–700. doi: 10.1542/peds.2007-3007. [DOI] [PubMed] [Google Scholar]
- 164.Prescott HC, Langa KM, Iwashyna TJ. Readmission diagnoses after hospitalization for severe sepsis and other acute medical conditions. JAMA. 2015;313:1055–57. doi: 10.1001/jama.2015.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]