

Abstract
Recent studies indicate that cancer cells express elevated levels of type II transglutaminase (TG2), and that expression is further highly enriched in cancer stem cells derived from these cancers. Moreover, elevated TG2 expression is associated with enhanced cancer stem cell marker expression, survival signaling, proliferation, migration, invasion, integrin-mediated adhesion, epithelial–mesenchymal transition, and drug resistance. TG2 expression is also associated with formation of aggressive and metastatic tumors that are resistant to conventional therapeutic intervention. This review summarizes the role of TG2 as a cancer cell survival factor in a range of tumor types, and as a target for preventive and therapeutic intervention. The literature supports the idea that TG2, in the closed/GTP-binding/signaling conformation, drives cancer cell and cancer stem cell survival, and that TG2, in the open/crosslinking conformation, is associated with cell death.
Keywords: cancer stem cells, epithelial–mesenchymal transition, EMT, transglutaminase, squamous cell carcinoma, epidermal cancer stem cells, ovarian cancer, pancreatic cancer, prostate cancer, glioma, breast cancer, drug resistance
Introduction
Tumor survival requires that the cancer cells circumvent normal cell death processes. This is often associated with mutation or overexpression of specific oncogenes that drive cancer cell survival, and/or silencing of tumor suppressor genes leading to enhanced cell division [1]. The fact that tumor cells proliferate at a higher rate than normal cells led to the design of cancer therapies that target rapidly proliferating cells. However, this approach has not been entirely satisfactory, as the cells often escape and become resistant. In this context, it has been realized that normal body tissues are derived from organ-specific stem cells that display a capacity to self-renew and to differentiate into the cell types that comprise the organ [2]. The cancer stem cell theory proposes that a small population of slow cycling, long-lived cancer cells, derived by mutation of normal stem cells, exist in tumors and are required for tumor maintenance. This theory further suggests that the formation of a mutated stem cell is an early event in tumor formation. Increasing evidence suggests the cancer stem cells facilitate tumor formation, cancer recurrence, and metastasis [3–8], and resistance to conventional anti-cancer therapy [9].
An important recent goal in cancer biology is identification of therapeutic and preventive treatments that reduce cancer stem cell survival [10,11]. A key strategy in this context is identifying cancer stem cell survival proteins, that are either upregulated or display enhanced activity in cancer stem cells, as targets for anti-cancer prevention and therapy. In the present review, we discuss type II transglutaminase (TG2) as a marker of cancer development, as a cancer stem cell-survival protein, and as a potential anti-cancer stem cell prevention and therapy target.
TG2 Structure and Activity
TG2 is predominantly a cytosolic protein, but is also present in the nucleus, at the plasma membrane and in the extracellular environment [12,13]. As shown in Figure 1A, the TG2 sequence encodes an integrin- and fibronectin-binding N-terminal β-sandwich domain, a catalytic core domain which includes the catalytic triad (Cys277, His335, and Asp358) that mediates TG2 crosslinking (transamidase) activity, and two C-terminal β-barrel domains. The guanine nucleotide binding site, which encompasses part of β-barrel1 and residues in the catalytic domain, is required for TG2-related signal transduction [14,15]. The TG2 GTP binding and the crosslinking functions have been heavily studied. In intact cells, where GTP/GDP levels are high and free calcium levels are low, TG2 exists in the GTP/GDP-bound closed/folded (signaling) conformation [12,16–19] (Figure 1B). If intracellular calcium levels rise, during cell death or in response to extracellular stimuli, calcium binding shifts TG2 to an open/extended crosslinking conformation which exposes the catalytic triad and activates protein–protein crosslinking (transamidase) activity [20]. This calcium-dependent change in conformation is associated with loss of GTP/GDP binding and related signaling (Figure 1B) [21–25]. The crosslinking activity of TG2 is allosterically activated by Ca2+ and inhibited by GTP, GDP, and GMP [26,27] (Figure 1B). Thus, the TG2 GTP-binding folded/closed (signaling) structure, and the open/extended (crosslinking) structure, are mutually exclusive. An additional mode of regulation involves oxidation of TG2 which converts the open/extended crosslinking-active form to the open crosslinking-inactive form, an event that is associated with oxidative conditions, particularly in the extracellular environment. We will argue that the TG2 closed (signaling) form is a major driver of cancer cell, and cancer stem cell survival. In addition, we suggest that the open (crosslinking) conformation can, in some contexts, enhance cancer cell survival, but that generally suppresses cell survival. We will review what is presently known in a range of cancer types.
Figure 1.
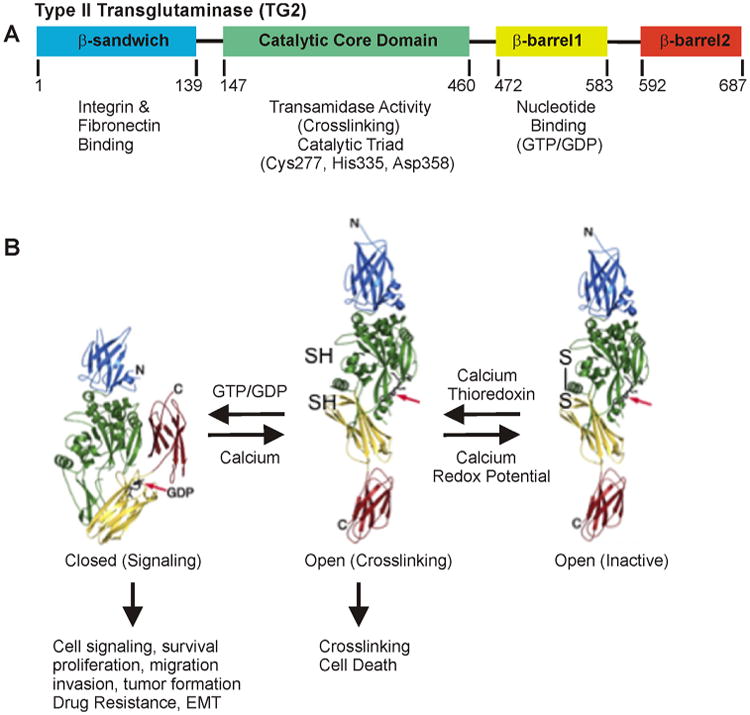
TG2 structure and function. A: Schematic of TG2 showing the β-sandwich, catalytic core, β-barrel1, and β-barrel2 domains, and the biological functions associated with each domain. Nucleotide binding (GTP/GDP) is mainly to residues from the first and last strands (amino acids 476–482 and 580–583) of β-barrel 1, and two core domain residues (Lys-173 and Phe-174) [147–150]. B: Guanine nucleotide (GTP/GDP)-bound TG2 is folded/closed. This form of TG2 is involved in intracellular signal transduction, survival, proliferation, migration, invasion, tumor formation, drug resistance, and EMT. It is also enriched in cancer stem cells and functions as a cancer stem cell survival factor. Open TG2 does not bind GTP/GDP. Instead, the transamidation (crosslinking) site is accessible to substrate and open TG2 catalyzes crosslinking of intracellular proteins and is associated with cancer cell apoptosis. Oxidation of the open form of TG2 converts it to an inactive form. The GTP/GDP binding site is indicated by a red arrow. Only closed TG2 binds GTP/GDP [19,22,151].
TG2 Role in Various Cancer Types
TG2 in Ovarian Cancer
Epithelial ovarian cancer arises from the epithelial layer covering the ovaries. Since the epithelial surface faces the peritoneal cavity, shedding of transformed cells leads to the characteristic formation of ascites fluid. These cancer cells are interesting as they have dual mesenchymal and epithelial characteristics and can convert to either phenotype in response to changes in the microenvironment. The mesenchymal characteristics favor release of cells from the tumor into the peritoneal cavity where they form metastatic implants in the intraperitoneal space [28,29]. TG2 is required for ovarian cancer cell adhesion to surfaces, as TG2 knockdown diminishes ovarian tumor cell dissemination and adhesion to the peritoneal matrix [30,31]. This is thought to involve TG2 interaction with and maintenance of β1 integrin level [30]. The TG2/β1– integrin complex interacts with fibronectin to stabilize adhesion [30]. An inhibitor, ITP-79, reduces TG2 interaction with and stabilization of fibronectin and reduces adhesion [31]. This is consistent with the finding that TG2 siRNA treatment reduces tumor cell adhesion in the peritoneal cavity leading to reduced invasion [32,33]. In addition, TG2 knockdown enhances docetaxel-induced ovarian cancer cell death, indicating that TG2 expression fosters drug resistance [32].
TG2 impacts several signaling cascades in ovarian cancer cells. Under normal conditions, NFκB signaling is inhibited by NFκB association with IκBα. The NFκB/IκBα complex sequesters NFκB in the cytoplasm which reduces nuclear NFκB level leading to reduced signaling [34]. Ovarian cancer cell survival is associated with a TG2-mediated reduction in IκBα subunit expression leading to increased NFκB activity [35], and that increased NFκB signaling enhances cell adhesion [33,36]. CREB transcription factors also have a role in ovarian cancer, as TG2 knockdown or inhibition with KCC009, a TG2 inhibitor, reduces CREB transcription factor phosphorylation leading to reduced MMP-2 expression and activity [36]. As noted above, epithelial ovarian cancer cells have a dual mesenchymal/epithelial phenotype. TG2 expression is associated with a shift to the mesenchymal phenotype via transcriptional activation of mesenchymal transcription factor, Zeb1 [37]. TG2 expression is also associated with increased β-catenin level, and β-catenin stimulation of cyclin D1 and c-myc expression [38]. TG2 physically associates with and recruits c-src, which then phosphorylates β-catenin leading to its activation [38]. Thus, TG2 enhances ovarian cancer cell survival by enhancing NFκB and CREB signaling. In addition, TG2-associated NFκB signaling is important for ovarian cancer cell resistance to cisplatin [32,35].
The mechanisms that control TG2 level have also been studied. TG2 level is regulated by TGFβ. TGFβ, acting via SMADs and TGFβ-activated kinase 1 stimulation of NFκB, increases TG2 expression to enhance EMT and spheroid formation [39]. These studies suggest the existence of a NFκB/TG2 positive feedback loop that maintains NFκB and TG2 expression.
TG2 in Breast Cancer
TG2 has been shown to be an important breast cancer cell survival factor, and overexpression in the tumor-surrounding stroma is associated with high risk of cancer recurrence [40,41]. TG2 is highly expressed in advanced breast cancer and in drug-resistant breast cancer cells [42], and TG2 knockdown restores drug sensitivity [42]. In addition, epigenetic silencing of TG2 expression has been observed in drug sensitive MCF-7 cells [43]. TG2 interacts with β2- and β5-integrin MCF-7 cells, and plating these cells on fibronectin-coated plates strongly activates focal adhesion kinase (FAK)-related signaling leading to an apoptosis-resistant phenotype [44]. Consistent with this, TG2 knockdown reduces fibronectin-mediated cell adhesion and survival [44,45].
TG2 regulates several pro-survival signaling cascades in breast cancer cells. It constitutively activates NFκB signaling leading to enhanced cell survival [46,47]. Moreover, TG2 and IκBα co-precipitate, suggesting that NFκB activation is due to loss of interaction with IκBα [46,48]. This TG2 interaction with NFκB is also observed in ovarian cancer cells [34]. In addition, a TG2/NFκB positive feedback loop maintains TG2 expression and constitutive NFκB activation [46,48,49]. Phosphorylation of the Ser-216 site on TG2 appears to be required for TG2 activation of NFκB and Akt signaling and down-regulation of PTEN (phosphatase and tensin homology, an inhibitor of Akt activity) [50]. Expression of a TG2 mutant in which Ser-216 phosphorylation site is converted to alanine, results in reduced epidermal growth factor (EGFR) level leading to reduced EGFR signaling and reduced cancer cell proliferation [51]. It has also been reported that TG2 facilitates breast cancer metastasis by increasing IL-6 production, and that TG2 level in tumor cells correlates directly with IL-6 level [52]. The authors conclude that TG2 is important for IL-6 mediated tumor aggressiveness in metastatic tumors [52].
TG2 expression is associated with breast cancer cell drug resistance, increased invasiveness, and increased epithelial–mesenchymal transition (EMT) [53]. The EMT response includes loss of E-cadherin, and increased expression of Snail1, Zeb1, Zeb2, and Twist1 [53]. TG2 also increases the Warburg effect in breast cancer cells via NFκB induction of HIF-1α leading to enhanced glucose uptake, with the effect being abolished following knockdown of TG2, NFκB, or HIF-1α [54]. Studies using TG2 mutants show that GTP binding domain is required for TG2-stimulated breast cancer cell survival and EMT [27,55].
Several studies have explored the role of TG2 crosslinking activity (Figure 1B). TG2 crosslinking activity can be increased by treating cells with ionophore to enhance intracellular calcium level. Calcium binds to TG2 to produce the open/crosslinking form of TG2, and this is associated with reduced breast cancer cell survival [56]. In addition, expression of a 55 kDa mutant form of TG2, which lacks GTP binding activity but retains crosslinking activity, increases apoptosis-mediated breast cancer cell death. This suggests that intracellular activation of TG2 crosslinking can kill breast cancer cells [57]. Moreover, increased crosslinking activity of matrix has been reported to suppress breast cancer cell migration and metastasis [58]. These findings suggest that the closed GTP-binding signaling form of TG2 (Figure 1B) stimulates breast cancer cell signaling to drive cancer cell survival. In contrast, it also appears that a high calcium environment can convert TG2 to the open “crosslinking” form which then crosslinks intracellular and extracellular proteins to reduce cancer cell survival and migration [57].
S100A4 is a known regulator of cell migration and knockdown of TG2 attenuates the ability of S100A4 to drive migration [59]. Treatment of MDA-MB-231 cells with R294, a cell impermeable TG2 inhibitor, inhibits migration and reduces S100A4 polymer formation. It is proposed that TG2 crosslinking of S100A4, via a mechanism that involves syndecan-4 and α5β1 integrin signaling, leads to enhanced migration [59]. TG2 has also been shown to phosphorylate IGFBP-3 on plasma membranes in breast cancer cells, but the impact of this has not been studied [60].
Thus, the preponderance of the data suggests that under normal intracellular conditions of high intracellular GTP/GDP and low calcium, TG2 is present in a closed (signaling) conformation that functions to enhance breast cancer cell survival signaling.
TG2 in Pancreatic Cancer
TG2 is enriched in pancreatic cancer cells and serves as a therapy target and diagnostic marker [61–63], and is an important activator of pro-survival signaling in this cancer type. Elevated TG2 level in pancreatic cancer cells is associated with increased NFκB signaling activity, related to TG2 interaction with IκBα to release free and activated NFκB which then moves to the nucleus to activate transcription [46]. The relationship between these two proteins is confirmed by data showing TG2 and active NFκB co-distribution in pancreatic ductal carcinoma samples [46]. Elevated TG2 expression, or expression of a crosslinking inactive TG2 mutant, TG2(C277S), increases invasive potential, FAK activation, metastasis, and resistance to gemcitabine [64]. This is consistent with the finding that TG2 expression is associated with FAK activation, and TG2 knockdown reduces FAK activity [64]. TG2 also promotes degradation of PTEN (phosphatase and tensin homology), an AKT/FAK inhibitor, via an ubquitin-dependent mechanism, leading to constitutive activation of FAK/AKT in pancreatic cancer cells [65]. TG2 also regulates autophagy in pancreatic cancer cells, as PKCδ activation increases TG2 level leading to suppression of autophagy, and inhibition of PKCδ or knockdown of TG2 is associated with increased autophagy [66–68]. TG2 has also been shown to active JNK-phosphorylation and to co-precipitate with keratin 8 and JNK. It is proposed that TG2 may modulate keratin filament organization and thereby increase JNK activity and cell survival [69–71]. TG2 also stimulates pancreatic cancer tumor formation, and TG2 knockdown reduces pancreatic cancer tumor formation in xenograft models [72,73].
Although TG2 GTP/GDP binding activity is required for pancreatic cancer cell survival and EMT [64], activation of TG2 crosslinking by cell treatment with calcium ionophore to increase intracellular calcium level, leads to TG2-dependent intracellular protein–protein crosslink formation leading to cell apoptosis [74]. Likewise, treatment of AsPC-1 pancreatic cancer cells with high temperature results in apoptotic cell death associated with enhanced TG2-dependent protein crosslinking [75]. Treatment of pancreatic cancer cells with 1–10 μM resveratrol, a diet-derived cancer prevention agent, is associated with accumulation of a slower mobility (open, crosslinking) form of TG2 which leads to reduced cell survival [76]. Thus, folded/closed (signaling) TG2 is associated with enhanced cancer cell survival, while open/extended (crosslinking) TG2 is associated with cell death.
Eukaryotic elongation factor-2 kinase (eEF-2K), an atypical kinase, is increased in pancreatic cancer cells. Recent studies show that suppression of eEF-2K with siRNA impairs pancreatic cancer cell migration/invasion and that this is associated with reduced TG2 level and reduced EMT [77]. Thus, as described for other cancer cell types, loss of TG2 is associated with reduced EMT [77].
TG2 in Prostate Cancer
TG2 is also expressed in prostate cancer cells [78]. Advanced prostate cancer is often associated with reduced androgen receptor expression leading to castration-resistant cancer [79]. It has recently been reported that TG2 levels are elevated in advanced prostate cancer which is associated with reduced androgen receptor expression, and that TG2 negatively regulates androgen receptor level [78]. These studies also showed that TG2 activation of NFκB expression induces NFκB binding to DNA elements in the androgen receptor gene to reduce androgen receptor gene expression [78].
TG2 in Glioma
TG2 is expressed in glioblastoma [80] and may serve as a prognostic marker [81]. In glioblastoma, TG2 is elevated in tumor regions where fibronectin assembles in the extracellular matrix, and knockdown of TG2 results in a loss of this assembly [82]. Treatment with KCC009, a TG2 inhibitor, reduces matrix remodeling both in vitro and in vivo, and sensitives glioma tumors to N,N′-bis(2-chloroethyl)-N-nitrosourea treatment [82,83]. TG2 knockdown also results in enhanced sensitivity of glioma cells to other agents, including ERW1227B [84]. Moreover, a GTP-binding competent/crosslinking-defective TG2 mutant confers resistance to doxorubicin, suggesting that GTP binding activity, but not TG2 crosslinking activity, is required for this response [85]. Thus, TG2 GTP binding activity is required for drug resistance [85]. Expression of TG2 in TG2-null astrocytoma cells increases cAMP accumulation in response to treatment with the β-adrenergic receptor agonist, isoproterenol, or the adenylate cyclase activator, forskolin, and this is reversed by treatment with ERW1069, a TG2 inhibitor [86].
TG2 has also been reported to increase epidermal growth factor receptor (EGFR) level in glioblastoma [87]. Delivery of TG2 to glioblastoma cell lines results in increased EGFR level and signaling via a mechanism that involves TG2 blocking of c-Cbl-catalyzed EGFR ubiquitylation via TG2 adoption of a conformation that enables its interaction with c-Cbl [87]. In rat C6 glioma cells which express endogenous TG2, cAMP accumulation, induced by isoproterenol or forskolin, is significantly inhibited by overexpression of TG2-C277V, a mutant that lacks transglutaminase activity, but is not inhibited by TG2-S171E, a TG2 mutant that cannot bind GTP/GDP [86]. The authors propose that TG2 activates cAMP production by covalent pentylamine modification of adenylylcyclase 8 [86]. Finally, one study shows that in some brain primary glioma samples, the TG25′-flanking region is hypermethylated and TG2 is not expressed [85]. Thus, TG2 gene methylation status may explain why TG2 levels vary in glioma tumor samples [85].
TG2 in Epidermal Squamous Cell Carcinoma
Epidermal squamous cell carcinoma tumors are comprised of phenotypically diverse populations of cells that display varying potential for proliferation and differentiation. We have shown that epidermal cancer stem cells (ECS cells), which comprise 0.15% of the tumor cell population, can be selected by culture on ultralow attachment plates with stem cell medium [88]. These spheroid-selected cells form fast growing, aggressive, invasive, and highly vascularized tumors in immune-compromised mice. This is in contrast to non-stem cancer cells, which form small, non-vascularized, and circumscribed tumors [88]. Moreover, spheroid-selected ECS cells give rise to tumors following injection of as few as 100 cells, suggesting that these cells have clonal tumor-forming potential expected of cancer stem cells. Moreover, cells derived from ECS cell-derived tumors retain enhanced ability to grow as spheroids in non-attached culture conditions [88]. Detailed analysis reveals that spheroid-selected cultures are highly enriched for expression of epidermal stem cell and embryonic stem cell markers, including aldehyde dehydrogenase 1, keratin 15, CD200, keratin 19, Oct4, Bmi-1, Ezh2, and trimethylated histone H3. These studies indicate that a subpopulation of cells that possess stem cell-like properties and express stem cell markers can be derived from human epidermal cancer cells and that these cells display enhanced ability to drive tumor formation [88].
TG2 is highly elevated in ECS cells and TG2 knockdown or suppression of TG2 function with the irreversible TG2 inhibitor, NC9, reduces ECS cell survival, spheroid formation, matrigel invasion and migration, and enhances apoptosis [89]. Mechanistic studies, using TG2 mutants, indicate that GTP-binding activity is required for maintenance of ECS cell growth and survival [89]. However, in contrast to ovarian, breast and prostate cancer, TG2 does not activate NFκB in this cancer [89,90]. These studies further show that TG2 knockdown, or treatment with NC9, results in reduced EMT marker expression. Analysis of a series of TG2 mutants reveals that TG2 GTP binding activity, but not the transamidase activity, is required for expression of EMT markers (Twist, Snail, Slug, vimentin, fibronectin, N-cadherin, and HIF-1α), and increased ECS cell invasion and migration [89]. These studies suggest that the folded/closed (signaling) form of TG2 has an important role in maintaining ECS cell survival, invasive and metastatic behavior, and that TG2 is an important therapeutic and prevention target to reduce survival of cancer stem cells in epidermal squamous cell carcinoma.
TG2 in Melanoma
Human malignant melanoma is a highly aggressive form of cancer with a 5 year survival rate of below 5% for patients with stage III or IV disease. In patients with metastatic melanoma, systemic therapy becomes ineffective because of the high resistance of melanoma cells to anticancer therapy [91]. TG2 expression is correlated with increased melanoma cell growth [92], cell adhesion [93], and metastatic potential [94]. Although it has been suggested that TG2-dependent melanoma cell adhesion is due to TG2 dependent crosslinking of the cells to the substrate [93], it seems more likely in light of recent findings [30,44,45,95,96], that the increased adhesion is due to TG2 interaction with integrins. For example, TG2 interacts with the α9β1 and α5β1 integrins in G-361 human melanoma cells and this is associated with enhanced cell adhesion [97].
It has been reported that hyaluronic acid treatment of melanoma cells increases NFκB signaling to increase TG2 level [98]. This is associated with increased Rac1 and FAK activity, and knockdown of this signaling cascade reduces TG2 level. Elevated TG2 is also associated with increased cell motility which is reduced by TG2 inhibitor treatment [98]. These authors conclude that hyaluronic acid induces melanoma cell motility through activation of Rac1, FAK, and induction of TG2.
Mehta and colleagues identified elevated TG2 expression in metastatic melanoma cell lines as compared to radial growth phase (less aggressive) melanoma cell lines [95]. A fraction of TG2 in A375 melanoma cells is associated with β1 and β5 integrins, leading to strong attachment of these cells to fibronectin, resulting in increased cell survival. Knockdown of TG2 results in reduced cell attachment [95]. These studies suggest that TG2 expression contributes to the development of chemoresistance in malignant melanoma cells by exploiting integrin-mediated cell survival signaling pathways [95].
In contrast, it has been reported that treatment with a host of agents that increase TG2 level, reduce melanoma cell proliferation, survival, and migration [99–104]. For example, treatment of human melanoma cells with polyamine causes melanoma cell apoptosis via a mechanism that is associated with increased TG2 crosslinking activity [101]. Atrial natriuretic peptide treatment of murine B16-F10 or human SK-MEL110 melanoma cells results in increased TG2 crosslinking activity that is associated with reduced polyamine levels and reduced cell growth [105]. Treatment with nimesulide, a nonsteroidal anti-inflammatory agent, reduces B16-F10 cell proliferation and this is associated with increased TG2 [102,103]. A similar reduction in B16-F10 melanoma cell migration, adhesion, and invasion is associated with quinizarin induction of TG2 [104]. These are interesting studies, which show slowed growth associated with increased TG2 level which is associated with intracellular activation of TG2 crosslinking. Consistent with the idea that activation of intracellular TG2-mediated protein crosslinking kills cells, activation of endogenous TG2 crosslinking activity by treatment with calcium ionophore induces a rapid apoptotic response in A375 human melanoma cells which can be blocked by TG2 inhibitors [95]. Studies in B16-F10 murine melanoma cells in xenograft conditions in TG2−/− mice suggest that absence of host TG2 favors melanoma tumor formation and metastasis [106]; however, this study does not address the role of melanoma tumor cell endogenous TG2 activity on survival.
GPR56 is a G protein-coupled adhesion receptor that reduces melanoma cancer cell survival [107,108]. It has been proposed that GPR56 forms a complex with Gap and CD81 (a tetraspanin protein) and that this complex then interacts with TG2 [108,109]. Ultimately, TG2 is internalized and degraded as part of this complex [110], leading to reduced TG2-dependent melanoma cell survival that is associated with reduced deposition of fibronectin. It is interesting that TG2 tumor promoting activity in this model appears to require TG2 crosslinking activity [107,110], as TG2 mutants (C277S and W241A) that lack crosslinking activity, display reduced ability to restore tumor formation when tested in TG2/GPR56 knockout cells [110].
TG2 in Lung Cancer
In non-small cell lung cancer patients treated with platinum compounds, disease free survival is longer when tumors have low TG2 levels [111]. Moreover, elevated TG2 level is associated with enhanced TGFβ-stimulated expression of N-cadherin, via a mechanism that involves JNK kinase activation, during EMT in lung cancer cells [112], and inhibition of TG2 sensitizes TRAIL-resistant lung cancer cells though upregulation of the death receptor 5 [113]. Thus, these studies suggest that TG2 expression confers drug resistance on lung cancer cells.
TG2 in Colon Cancer
The role of TG2 in colon cancer is mixed. It has been reported that TG2 levels are lower in colon cancer cells as compared to normal cells, and that increased TG2 in colon cancer cells causes apoptosis and cell growth inhibition [114,115]. However, other studies suggest that TG2 is elevated in colon cancer and may be a useful prognostic indicator [116,117]. It is reported that expression of crosslinking inactive TG2 (Cys277Ser) in CT26 colon carcinoma cells does not alter proliferation, NFκB activation, or sensitivity to doxorubicin [96]. It is also reported that cells transfected with wild-type TG2 expression vector show enhanced β3-integrin expression and TG2/β3-integrin interaction, and that this is associated with reduced cell migration on fibronectin. The authors conclude that TG2 is a potential inhibitor of colon cancer growth [96]. A recent study indicates that SW480 colon cancer cell invasive behavior is enhanced by TG2 knockdown, and the miR-19 expression reduces TG2 level [118]. Thus, these studies suggest that TG2 is an inhibitory factor in colon cancer.
TG2 in Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is a diverse disease associated with differential oncogene expression. Proteomic analysis of AML tissue reveals elevated TG2 expression in transformed cells [119]. Proteomic analysis of 511 AML samples using reverse phase proteomics, demonstrate that TG2 expression is higher in relapsed disease. In addition, elevated TG2 expression is associated with increased expression of numerous adhesion proteins. The authors suggest that increased TG2 level may lead to the protection of the leukemic stem cell due to increased adhesion/reduced motility, and that TG2 expression may define poor outcome in AML patients [119].
TG2 in Cancer Stem Cells
Cancer Stem Cells
Recent evidence suggests that tumors contain a small subpopulation of cells, called cancer stem cells (CSC), which exhibit self-renewal capacity, proliferate infrequently, and are responsible for tumor maintenance and metastasis [120]. Moreover, these cells may be responsible for tumor relapse and resistance to therapy [121–123]. Early evidence for this model came from the study of acute myeloid leukemia [124,125] where a small population of CD34+/CD38− cells was isolated that could recapitulate the original patient tumor phenotype following passage in immune compromised mice [124,126]. Such cancer stem cells were subsequently shown to exist in other tumor types [88,127–134]. Some studies show that injection of just a few cells into immune-compromised mice results in tumor formation [88,127]. It is also recognized that these cells can be selected and grown as cell aggregates, called spheroids, in serum-free medium in ultra-low attachment plates [88,89]. These conditions select for the limited number of cancer stem cells present within the cell population [88,89]. Moreover, reduced spheroid formation in response to treatment with anti-cancer agents provides an assay to study the impact of therapeutic agents on these populations. It has been reported that cancer stem cells are resistant to conventional anti-cancer agents that kill rapidly growing non-stem cancer cells [135]. Since the cancer stem cells appear to give rise to other cells in the tumor, eliminating the stem cell population is required to halt tumor formation and metastasis [135].
An important strategy is identifying genes that are highly overexpressed or activated in cancer stem cells that improve cancer stem cell survival. TG2 level is elevated in pancreatic, breast, colon, non-small cell lung cancer, glioblastoma, melanoma, and epidermal squamous cell carcinoma [30,42,61,81,95,117,136–138], and TG2 level is often further elevated in advanced disease [42,44,64,95,139]. We have proposed that TG2 serves as a cancer stem cell survival factor that regulates selected signaling cascades to enhance survival, epithelial-mesenchymal transition (EMT), invasion, migration, and tumor formation [89,90].
TG2 in Cancer Stem Cells
We have enriched the epidermal cancer stem cells (ECS cell) population in epidermal cancer cells (squamous cell carcinoma) using antibody selection and growth as non-attached spheroids [88–90]. We identified TG2 as highly elevated in ECS cells and we showed that TG2 knockdown or suppression of TG2 function with inhibitors reduces ECS cell survival, spheroid formation, matrigel invasion, and migration. The reduction in survival is associated with activation of apoptosis [89]. Mechanistic studies show that the TG2 GTP-binding activity is required for maintenance of ECS cell growth, survival, invasion, and migration [89,90]. These studies suggest that TG2 has an important role in maintaining cancer stem cell survival, invasive, and metastatic behavior. We also showed that TG2 is required for ECS cell EMT.
A recent study indicates that TG2 is also highly enriched in CD44-high glioma cancer stem cells and that these cells are resistant to therapy [140]. Knockdown of TG2 or treatment with monodansylcadaverine (MDC), a TG2 substrate competitive inhibitor, reduces DNA binding protein 1 level and cell proliferation, and induces apoptosis. This preferentially eliminates CD44-positive glioma cancer stem cells. Moreover, delivery of MDC at 10 or 25 mg/kg body weight/day in an orthotopic model of CD44-positive cells inhibits tumor formation by 60% and 85% [140].
Breast cancer stem cells are a highly tumorigenic subpopulation of breast cancer cells that are characterized by a CD44+/CD24− phenotype and also expression of EMT markers and absence of claudin [47]. Mehta and co-workers showed that TG2-positive breast cancer cells express reduced claudin 1, 4, and 8, and increased expression of EMT markers [47]. These cells are CD44+/CD24− and display an increase ability to grow as spheroids [141,142]. TG2 knockdown results in reduced survival signaling in breast cancer stem cells [47,53,55,141].
TG2 expression has also been linked to the cancer stem cell phenotype in ovarian cancer [39]. Survival of the CD44+/CD117+ ovarian cancer stem cells, which are also positive for EMT markers, is dependent upon TG2 expression. TGFβ signaling, via SMAD and TGFβ-activated kinase 1 activates NFκB to simulate TG2 expression and these cells also aggregate to form spheroids that manifest stem cell properties [39]. TG2 knockdown reduces the number of CD44+/CD117+ cells, demonstrating that TG2 is required for ovarian cancer stem cell survival [39].
Summary
The studies outlined above suggest that TG2 is overexpressed in cancer and is associated with an aggressive cancer phenotype and drug resistance (Table 1). Moreover, in cancers where TG2 has been studied, including epidermal squamous cell carcinoma, breast, glioma, and ovarian cancer, TG2 levels are markedly elevated in cancer stem cells and expression is required for cancer stem cell survival, migration, and invasion. This is associated with TG2 stimulation of a host of signaling cascades leading to EMT marker expression, and enhanced biological indices of EMT. Moreover, in some models, evidence shows that cancer stem cells form highly aggressive tumors, compared to non-stem cancer cells, and TG2 is required for this aggressive phenotype [88]. Additional studies indicate that TG2 activates prosurvival signaling pathways that enhance cell survival and are associated with enhanced cancer cell adhesion to substrate and EMT. A key finding is that the pro-survival activity requires TG2 in a folded/closed (GTP-binding, signaling) conformation (Figure 1). As a GTP/GDP binding protein involved in G-protein signaling, TG2 stimulates activity in a wide range of pro-survival cascades via mechanisms that are only partially understood. In some cases, it interacts directly with transcription factors [34,46,48]. Although additional studies will be required to understand this regulation, drugs exist which can be used to alter TG2 structure and activity [143–146]. An example is NC9, an irreversible inhibitor of TG2 that converts it from a folded/closed (signaling) conformation to an open/extended (non-signaling) configuration. Treatment of cancer stem cells with this agent shows that it efficiently reduces cancer stem cell survival, migration, and invasion [88–90]. Based on these findings, we propose that TG2 is a key cancer stem cell survival protein and mediator of resistance to conventional drug therapy. We further propose that drugs designed to target TG2 will be important for controlling survival of the most dangerous cell type in tumors, the cancer stem cell.
Table 1. TG2 Impact on Cancer Cell Function.
| Cancer type | TG2 signaling targets | Drug resistance | Impact of TG2/integrin interaction | Signaling impact on TG2 level | Impact on EMT | Impact on tumor formation | Role in cancer stem cells |
|---|---|---|---|---|---|---|---|
| Ovarian | NFκB [35,152], β-catenin, c-src [33] | Docetaxel [32], Cisplatin [35] | TG2/β1-integrin interaction enhances cell adhesion [30] | NFκB signaling increases TG2 level [39] | TG2 expression is associated Zeb1-dependent EMT [37] | Enhances tumor formation [30,37,37] | TG2 is required for survival [39] |
| Breast | TG2 binds and sequesters IκBα to enhance NFκB signaling [46,46-49][51], TG2 maintains EGFR [51], | Doxorubicin [42,43,56] | TG2/β2- and β5-integrin interaction enhances cell adhesion [44,45] | NFκB feedback increases TG2 level in positive feedback loop [46,48,49] | TG2 drives EMT [53] | TG2 enhances tumor formation [52] | TG2 is elevated in breast cancer stem cells and required for survival [47,53,55,141] |
| Pancreatic | TG2 binds and sequesters IκBα to enhance NFκB signaling [153], and also activates FAK kinase [64], TG2 activates JNK [64], TG2 | TG2 is associated with resistance to Gemcitabine [64] | TG2 is required for optimal tumor formation [64,72,73] | ||||
| Prostate | NFkB [78] | TG2 expression is associated with EMT [78] | |||||
| Glioma | TG2 increases EGFR level [87] and cAMP signaling [86] | TG2 confers resistance to doxorubicin [85], TG2 confers resistance to N,N′-bis(2-chloroethyl)-N-nitrosourea treatment [82,83] | TG2 elevated in glioma cancer stem cells and required for survival [140] | ||||
| Epidermal squamous cell carcinoma | TG2 does not alter NFκB signaling [89,154] | TG2 enhances EMT [88,89,154] | TG2 enhances tumor formation [88,89,154] | TG2 overexpressed in epidermal squamous cell carcinoma cancer stem cells and required for survival [88–90] | |||
| Melanoma | TG2 interacts with α9β1 and α5β1 integrins leading to enhanced adhesion [97], TG2 interacts with β1 and β5 integrin to enhance attachment [95], TG2 is internalized by GPR56 leading to reduced adhesion and matrix crosslinking [110] | NFκB signaling increases Rac1 and FAK signaling and TG2 level [98] | TG2 crosslinking activity is required for tumor formation [110] | ||||
| Lung | TG2 confers drug resistance [111,113] | TG2 includes EMT marker expression [112] | |||||
| Colon | TG2 does not alter NFκB signaling [96] | TG2 does not alter sensitivity to doxorubicin [96] | TG2 increases and interacts with β3-integrin to reduce migration [96] |
Acknowledgments
Grant sponsor: This work was supported by National Institutes of Health R01 CA131064 and R01 CA184027 (RLE), an American Cancer Society Investigator Award (CK), and a Research Pilot Award (RLE) from the University of Maryland Greenebaum Cancer Center.
Abbreviations
- TG2
type II transglutaminase
- EMT
epithelial– mesenchymal transition
- ECM
extracellular matrix
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea: A paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 3.Thenappan A, Li Y, Shetty K, Johnson L, Reddy EP, Mishra L. New therapeutics targeting colon cancer stem cells. Curr Colorectal Cancer Rep. 2009;5:209. doi: 10.1007/s11888-009-0029-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpentino JE, Hynes MJ, Appelman HD, et al. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res. 2009;69:8208–8215. doi: 10.1158/0008-5472.CAN-09-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C, Yan Y, Ji W, et al. OCT4 positively regulates Survivin expression to promote cancer cell proliferation and leads to poor prognosis in esophageal squamous cell carcinoma. PLoS ONE. 2012;7:e49693. doi: 10.1371/journal.pone.0049693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol. 2008;26:2806–2812. doi: 10.1200/JCO.2008.16.6702. [DOI] [PubMed] [Google Scholar]
- 8.Saini V, Shoemaker RH. Potential for therapeutic targeting of tumor stem cells. Cancer Sci. 2010;101:16–21. doi: 10.1111/j.1349-7006.2009.01371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu H, Fang X, Luo Q, Ouyang G. Cancer stem cells: A potential target for cancer therapy. Cell Mol Life Sci. 2015 doi: 10.1007/s00018-015-1920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patton JT, Menter DG, Benson DM, Nicolson GL, McIntire LV. Computerized analysis of tumor cells flowing in a parallel plate chamber to determine their adhesion stabilization lag time. Cell Motil Cytoskeleton. 1993;26:88–98. doi: 10.1002/cm.970260109. [DOI] [PubMed] [Google Scholar]
- 11.Yang C, Jin K, Tong Y, Cho WC. Therapeutic potential of cancer stem cells. Med Oncol. 2015;32:619. doi: 10.1007/s12032-015-0619-6. [DOI] [PubMed] [Google Scholar]
- 12.Lorand L, Graham RM. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 13.Lentini A, Abbruzzese A, Provenzano B, Tabolacci C, Beninati S. Transglutaminases: Key regulators of cancer metastasis. Amino Acids. 2013;44:25–32. doi: 10.1007/s00726-012-1229-7. [DOI] [PubMed] [Google Scholar]
- 14.Iismaa SE, Wu MJ, Nanda N, Church WB, Graham RM. GTP binding and signaling by Gh/transglutaminase II involves distinct residues in a unique GTP-binding pocket. J Biol Chem. 2000;275:18259–18265. doi: 10.1074/jbc.M000583200. [DOI] [PubMed] [Google Scholar]
- 15.Iismaa SE, Chung L, Wu MJ, Teller DC, Yee VC, Graham RM. The core domain of the tissue transglutaminase Gh hydrolyzes GTP and ATP. Biochemistry. 1997;36:11655–11664. doi: 10.1021/bi970545e. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pavlyukov MS, Antipova NV, Balashova MV, Shakhparonov MI. Detection of transglutaminase 2 conformational changes in living cell. Biochem Biophys Res Commun. 2012;421:773–779. doi: 10.1016/j.bbrc.2012.04.082. [DOI] [PubMed] [Google Scholar]
- 18.Caron NS, Munsie LN, Keillor JW, Truant R. Using FLIM-FRET to measure conformational changes of transglutaminase type 2 in live cells. PLoS ONE. 2012;7:e44159. doi: 10.1371/journal.pone.0044159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegel M, Strnad P, Watts RE, et al. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS ONE. 2008;3:e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiraly R, Csosz E, Kurtan T, et al. Functional significance of five noncanonical Ca2+-binding sites of human transglutaminase 2 characterized by site-directed mutagenesis. FEBS J. 2009;276:7083–7096. doi: 10.1111/j.1742-4658.2009.07420.x. [DOI] [PubMed] [Google Scholar]
- 21.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285:25402–25409. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang TH, Lee DS, Choi K, et al. Crystal structure of transglutaminase 2 with GTP complex and amino acid sequence evidence of evolution of GTP binding site. PLoS ONE. 2014;9:e107005. doi: 10.1371/journal.pone.0107005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gundemir S, Johnson GV. Intracellular localization and conformational state of transglutaminase 2: Implications for cell death. PLoS ONE. 2009;4:e6123. doi: 10.1371/journal.pone.0006123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: A molecular Swiss army knife. Biochim Biophys Acta. 1823;2012:406–419. doi: 10.1016/j.bbamcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergamini CM. GTP modulates calcium binding and cation-induced conformational changes in erythrocyte transglutaminase. FEBS Lett. 1988;239:255–258. doi: 10.1016/0014-5793(88)80928-1. [DOI] [PubMed] [Google Scholar]
- 27.Eckert RL, Kaartinen MT, Nurminskaya M, et al. Transglutaminase regulation of cell function. Physiol Rev. 2014;94:383–417. doi: 10.1152/physrev.00019.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr Rev. 2001;22:255–288. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 29.Auersperg N, Pan J, Grove BD, et al. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci USA. 1999;96:6249–6254. doi: 10.1073/pnas.96.11.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satpathy M, Cao L, Pincheira R, et al. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007;67:7194–7202. doi: 10.1158/0008-5472.CAN-07-0307. [DOI] [PubMed] [Google Scholar]
- 31.Khanna M, Chelladurai B, Gavini A, et al. Targeting ovarian tumor cell adhesion mediated by tissue transglutaminase. Mol Cancer Ther. 2011;10:626–636. doi: 10.1158/1535-7163.MCT-10-0912. [DOI] [PubMed] [Google Scholar]
- 32.Hwang JY, Mangala LS, Fok JY, et al. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 2008;68:5849–5858. doi: 10.1158/0008-5472.CAN-07-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yakubov B, Chelladurai B, Schmitt J, Emerson R, Turchi JJ, Matei D. Extracellular tissue transglutaminase activates noncanonical NF-kappaB signaling and promotes metastasis in ovarian cancer. Neoplasia. 2013;15:609–619. doi: 10.1593/neo.121878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 35.Cao L, Petrusca DN, Satpathy M, Nakshatri H, Petrache I, Matei D. Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin-induced apoptosis by promoting cell survival signaling. Carcinogenesis. 2008;29:1893–1900. doi: 10.1093/carcin/bgn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satpathy M, Shao M, Emerson R, Donner DB, Matei D. Tissue transglutaminase regulates matrix metalloproteinase-2 in ovarian cancer by modulating cAMP-response element-binding protein activity. J Biol Chem. 2009;284:15390–15399. doi: 10.1074/jbc.M808331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao M, Cao L, Shen C, et al. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009;69:9192–9201. doi: 10.1158/0008-5472.CAN-09-1257. [DOI] [PubMed] [Google Scholar]
- 38.Condello S, Cao L, Matei D. Tissue transglutaminase regulates beta-catenin signaling through a c-Src-dependent mechanism. FASEB J. 2013;27:3100–3112. doi: 10.1096/fj.12-222620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao L, Shao M, Schilder J, Guise T, Mohammad KS, Matei D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene. 2012;31:2521–2534. doi: 10.1038/onc.2011.429. [DOI] [PubMed] [Google Scholar]
- 40.Assi J, Srivastava G, Matta A, Chang MC, Walfish PG, Ralhan R. Transglutaminase 2 overexpression in tumor stroma identifies invasive ductal carcinomas of breast at high risk of recurrence. PLoS ONE. 2013;8:e74437. doi: 10.1371/journal.pone.0074437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bravaccini S, Tumedei MM, Scarpi E, et al. New biomarkers to predict the evolution of in situ breast cancers. Biomed Res Int. 2014;2014:159765. doi: 10.1155/2014/159765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin Cancer Res. 2004;10:8068–8076. doi: 10.1158/1078-0432.CCR-04-1107. [DOI] [PubMed] [Google Scholar]
- 43.Ai L, Kim WJ, Demircan B, et al. The transglutaminase 2 gene (TGM2), a potential molecular marker for chemotherapeutic drug sensitivity, is epigenetically silenced in breast cancer. Carcinogenesis. 2008;29:510–518. doi: 10.1093/carcin/bgm280. [DOI] [PubMed] [Google Scholar]
- 44.Herman JF, Mangala LS, Mehta K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene. 2006;25:3049–3058. doi: 10.1038/sj.onc.1209324. [DOI] [PubMed] [Google Scholar]
- 45.Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene. 2007;26:2459–2470. doi: 10.1038/sj.onc.1210035. [DOI] [PubMed] [Google Scholar]
- 46.Mann AP, Verma A, Sethi G, et al. Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-kappaB in cancer cells: delineation of a novel pathway. Cancer Res. 2006;66:8788–8795. doi: 10.1158/0008-5472.CAN-06-1457. [DOI] [PubMed] [Google Scholar]
- 47.Agnihotri N, Kumar S, Mehta K. Tissue transglutaminase as a central mediator in inflammation-induced progression of breast cancer. Breast Cancer Res. 2013;15:202. doi: 10.1186/bcr3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown KD. Transglutaminase 2 and NF-kappaB: An odd couple that shapes breast cancer phenotype. Breast Cancer Res Treat. 2013;137:329–336. doi: 10.1007/s10549-012-2351-7. [DOI] [PubMed] [Google Scholar]
- 49.Ai L, Skehan RR, Saydi J, Lin T, Brown KD. Ataxia-telangiectasia, mutated (ATM)/nuclear factor kappa light chain enhancer of activated B cells (NFkappaB) signaling controls basal and DNA damage-induced transglutaminase 2 expression. J Biol Chem. 2012;287:18330–18341. doi: 10.1074/jbc.M112.339317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Ande SR, Mishra S. Phosphorylation of transglutaminase 2 (TG2) at serine-216 has a role in TG2 mediated activation of nuclear factor-kappa B and in the downregulation of PTEN. BMC Cancer. 2012;12:277. doi: 10.1186/1471-2407-12-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Ande SR, Mishra S. Overexpression of phospho mutant forms of transglutaminase 2 downregulates epidermal growth factor receptor. Biochem Biophys Res Commun. 2012;417:251–255. doi: 10.1016/j.bbrc.2011.11.094. [DOI] [PubMed] [Google Scholar]
- 52.Oh K, Ko E, Kim HS, et al. Transglutaminase 2 facilitates the distant hematogenous metastasis of breast cancer by modulating interleukin-6 in cancer cells. Breast Cancer Res. 2011;13:R96. doi: 10.1186/bcr3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar A, Xu J, Brady S, et al. Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS ONE. 2010;5:e13390. doi: 10.1371/journal.pone.0013390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar S, Donti TR, Agnihotri N, Mehta K. Transglutaminase 2 reprogramming of glucose metabolism in mammary epithelial cells via activation of inflammatory signaling pathways. Int J Cancer. 2014;134:2798–2807. doi: 10.1002/ijc.28623. [DOI] [PubMed] [Google Scholar]
- 55.Kumar A, Xu J, Sung B, et al. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012;14:R4. doi: 10.1186/bcr3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen JS, Konopleva M, Andreeff M, Multani AS, Pathak S, Mehta K. Drug-resistant breast carcinoma (MCF-7) cells are paradoxically sensitive to apoptosis. J Cell Physiol. 2004;200:223–234. doi: 10.1002/jcp.20014. [DOI] [PubMed] [Google Scholar]
- 57.Fraij BM. The 55 kDa tissue transglutaminase cross-linking active isoform TG induces cell death. Mol Carcinog. 2014 doi: 10.1002/mc.22134. [DOI] [PubMed] [Google Scholar]
- 58.Mangala LS, Arun B, Sahin AA, Mehta K. Tissue transglutaminase-induced alterations in extracellular matrix inhibit tumor invasion. Mol Cancer. 2005;4:33. doi: 10.1186/1476-4598-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Griffin M. The role of TG2 in regulating S100A4-mediated mammary tumour cell migration. PLoS ONE. 2013;8:e57017. doi: 10.1371/journal.pone.0057017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: Identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J Biol Chem. 2004;279:23863–23868. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- 61.Iacobuzio-Donahue CA, Ashfaq R, Maitra A, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: A comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63:8614–8622. [PubMed] [Google Scholar]
- 62.Cheung W, Darfler MM, Alvarez H, et al. Application of a global proteomic approach to archival precursor lesions: Deleted in malignant brain tumors 1 and tissue transglutaminase 2 are upregulated in pancreatic cancer precursors. Pancreatology. 2008;8:608–616. doi: 10.1159/000161012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehta K, Han A. Tissue transglutaminase (TG2)-induced inflammation in initiation, progression, and pathogenesis of pancreatic cancer. Cancers (Basel) 2011;3:897–912. doi: 10.3390/cancers3010897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verma A, Wang H, Manavathi B, et al. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006;66:10525–10533. doi: 10.1158/0008-5472.CAN-06-2387. [DOI] [PubMed] [Google Scholar]
- 65.Verma A, Guha S, Wang H, et al. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin Cancer Res. 2008;14:1997–2005. doi: 10.1158/1078-0432.CCR-07-1533. [DOI] [PubMed] [Google Scholar]
- 66.Akar U, Ozpolat B, Mehta K, Fok J, Kondo Y, Lopez-Berestein G. Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol Cancer Res. 2007;5:241–249. doi: 10.1158/1541-7786.MCR-06-0229. [DOI] [PubMed] [Google Scholar]
- 67.Ozpolat B, Akar U, Mehta K, Lopez-Berestein G. PKC delta and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells. Autophagy. 2007;3:480–483. doi: 10.4161/auto.4349. [DOI] [PubMed] [Google Scholar]
- 68.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park MK, Lee HJ, Shin J, Noh M, Kim SY, Lee CH. Novel participation of transglutaminase-2 through c-Jun N-terminal kinase activation in sphingosylphosphorylcholine-induced keratin reorganization of PANC-1 cells. Biochim Biophys Acta. 1811;2011:1021–1029. doi: 10.1016/j.bbalip.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 70.Byun HJ, Kang KJ, Park MK, et al. Ethacrynic acid inhibits sphingosylphosphorylcholine-induced keratin 8 phosphorylation and reorganization via transglutaminase-2 inhibition. Biomol Ther (Seoul) 2013;21:338–342. doi: 10.4062/biomolther.2013.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee EJ, Park MK, Kim HJ, et al. 12-O-tetradecanoylphorbol-13-acetate induces keratin 8 phosphorylation and reorganization via expression of transglutaminase-2. Biomol Ther (Seoul) 2014;22:122–128. doi: 10.4062/biomolther.2014.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verma A, Guha S, Diagaradjane P, et al. Therapeutic significance of elevated tissue transglutaminase expression in pancreatic cancer. Clin Cancer Res. 2008;14:2476–2483. doi: 10.1158/1078-0432.CCR-07-4529. [DOI] [PubMed] [Google Scholar]
- 73.Mehta K. Biological and therapeutic significance of tissue transglutaminase in pancreatic cancer. Amino Acids. 2009;36:709–716. doi: 10.1007/s00726-008-0128-4. [DOI] [PubMed] [Google Scholar]
- 74.Fok JY, Mehta K. Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis. 2007;12:1455–1463. doi: 10.1007/s10495-007-0079-3. [DOI] [PubMed] [Google Scholar]
- 75.Nishimura T, Horino T, Nishiura K, et al. Apoptotic cells of an epithelial cell line, AsPC-1, release monocyte chemotactic S19 ribosomal protein dimer. J Biochem. 2001;129:445–454. doi: 10.1093/oxfordjournals.jbchem.a002876. [DOI] [PubMed] [Google Scholar]
- 76.Kumar A, Hu J, LaVoie HA, Walsh KB, DiPette DJ, Singh US. Conformational changes and translocation of tissue-transglutaminase to the plasma membranes: role in cancer cell migration. BMC Cancer. 2014;14:256. doi: 10.1186/1471-2407-14-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ashour AA, Gurbuz N, Alpay SN, et al. Elongation factor-2 kinase regulates TG2/beta1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J Cell Mol Med. 2014;18:2235–2251. doi: 10.1111/jcmm.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Han AL, Kumar S, Fok JY, Tyagi AK, Mehta K. Tissue transglutaminase expression promotes castration-resistant phenotype and transcriptional repression of androgen receptor. Eur J Cancer. 2014;50:1685–1696. doi: 10.1016/j.ejca.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 79.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 80.Hilton DA, Love S, Barber R. Increased endothelial expression of transglutaminase in glioblastomas. Neuropathol Appl Neurobiol. 1997;23:507–511. doi: 10.1111/j.1365-2990.1997.tb01328.x. [DOI] [PubMed] [Google Scholar]
- 81.Zhang R, Tremblay TL, McDermid A, Thibault P, Stanimirovic D. Identification of differentially expressed proteins in human glioblastoma cell lines and tumors. Glia. 2003;42:194–208. doi: 10.1002/glia.10222. [DOI] [PubMed] [Google Scholar]
- 82.Yuan L, Siegel M, Choi K, et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26:2563–2573. doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- 83.Yuan L, Choi K, Khosla C, et al. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol Cancer Ther. 2005;4:1293–1302. doi: 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- 84.Yuan L, Holmes TC, Watts RE, et al. Novel chemo-sensitizing agent, ERW1227B, impairs cellular motility and enhances cell death in glioblastomas. J Neurooncol. 2011;103:207–219. doi: 10.1007/s11060-010-0379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dyer LM, Schooler KP, Ai L, et al. The transglutaminase 2 gene is aberrantly hypermethylated in glioma. J Neurooncol. 2011;101:429–440. doi: 10.1007/s11060-010-0277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Obara Y, Yanagihata Y, Abe T, Dafik L, Ishii K, Nakahata N. Galpha(h)/transglutaminase-2 activity is required for maximal activation of adenylylcyclase 8 in human and rat glioma cells. Cell Signal. 2013;25:589–597. doi: 10.1016/j.cellsig.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 87.Zhang J, Antonyak MA, Singh G, Cerione RA. A mechanism for the upregulation of EGF receptor levels in glioblastomas. Cell Rep. 2013;3:2008–2020. doi: 10.1016/j.celrep.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adhikary G, Grun D, Kerr C, et al. Identification of a population of epidermal squamous cell carcinoma cells with enhanced potential for tumor formation. PLoS ONE. 2013;8:e84324. doi: 10.1371/journal.pone.0084324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fisher ML, Keillor JW, Xu W, Eckert RL, Kerr C. Transglutaminase is required for epidermal squamous cell carcinoma stem cell survival. Mol Cancer Res. 2015 doi: 10.1158/1541-7786.MCR-14-0685-T. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fisher ML, Adhikary G, Xu W, Kerr C, Keillor JW, Eckert RL. Type II transglutaminase stimulates epidermal cancer stem cell epithelial-mesenchymal transition. Oncotarget. 2015 doi: 10.18632/oncotarget.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spagnolo F, Ghiorzo P, Orgiano L, et al. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015;8:157–168. doi: 10.2147/OTT.S39096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kozlowska K, Cichorek M, Zurawska-Czupa B. Heterogeneity of transplantable melanomas differing in the rate of growth and cellular differentiation in relation to their cell transglutaminase activity. Neoplasma. 1991;38:343–349. [PubMed] [Google Scholar]
- 93.Menter DG, Patton JT, Updyke TV, et al. Transglutaminase stabilizes melanoma adhesion under laminar flow. Cell Biophys. 1991;18:123–143. doi: 10.1007/BF02989810. [DOI] [PubMed] [Google Scholar]
- 94.van Groningen JJ, Klink SL, Bloemers HP, Swart GW. Expression of tissue-type transglutaminase correlates positively with metastatic properties of human melanoma cell lines. Int J Cancer. 1995;60:383–387. doi: 10.1002/ijc.2910600319. [DOI] [PubMed] [Google Scholar]
- 95.Fok JY, Ekmekcioglu S, Mehta K. Implications of tissue transglutaminase expression in malignant melanoma. Mol Cancer Ther. 2006;5:1493–1503. doi: 10.1158/1535-7163.MCT-06-0083. [DOI] [PubMed] [Google Scholar]
- 96.Kotsakis P, Wang Z, Collighan RJ, Griffin M. The role of tissue transglutaminase (TG2) in regulating the tumour progression of the mouse colon carcinoma CT26. Amino Acids. 2011;41:909–921. doi: 10.1007/s00726-010-0790-1. [DOI] [PubMed] [Google Scholar]
- 97.Takahashi H, Isobe T, Horibe S, et al. Tissue transglutaminase, coagulation factor XIII, and the pro-polypeptide of von Willebrand factor are all ligands for the integrins alpha 9beta 1 and alpha 4beta 1. J Biol Chem. 2000;275:23589–23595. doi: 10.1074/jbc.M003526200. [DOI] [PubMed] [Google Scholar]
- 98.Kim Y, Park YW, Lee YS, Jeoung D. Hyaluronic acid induces transglutaminase II to enhance cell motility; role of Rac1 and FAK in the induction of transglutaminase II. Biotechnol Lett. 2008;30:31–39. doi: 10.1007/s10529-007-9496-1. [DOI] [PubMed] [Google Scholar]
- 99.Knight CR, Rees RC, Griffin M. Apoptosis: A potential role for cytosolic transglutaminase and its importance in tumour progression. Biochim Biophys Acta. 1991;1096:312–318. doi: 10.1016/0925-4439(91)90067-j. [DOI] [PubMed] [Google Scholar]
- 100.Beninati S, Abbruzzese A, Cardinali M. Differences in the post-translational modification of proteins by polyamines between weakly and highly metastatic B16 melanoma cells. Int J Cancer. 1993;53:792–797. doi: 10.1002/ijc.2910530515. [DOI] [PubMed] [Google Scholar]
- 101.Facchiano F, D'Arcangelo D, Riccomi A, Lentini A, Beninati S, Capogrossi MC. Transglutaminase activity is involved in polyamine-induced programmed cell death. Exp Cell Res. 2001;271:118–129. doi: 10.1006/excr.2001.5356. [DOI] [PubMed] [Google Scholar]
- 102.Gismondi A, Lentini A, Tabolacci C, Provenzano B, Beninati S. Transglutaminase-dependent antiproliferative and differentiative properties of nimesulide on B16-F10 mouse melanoma cells. Amino Acids. 2010;38:257–262. doi: 10.1007/s00726-009-0244-9. [DOI] [PubMed] [Google Scholar]
- 103.Tabolacci C, Lentini A, Provenzano B, Gismondi A, Rossi S, Beninati S. Similar antineoplastic effects of nimesulide, a selective COX-2 inhibitor, and prostaglandin E1 on B16-F10 murine melanoma cells. Melanoma Res. 2010;20:273–279. doi: 10.1097/CMR.0b013e328339d8ac. [DOI] [PubMed] [Google Scholar]
- 104.Rossi S, Tabolacci C, Lentini A, et al. Anthraquinones danthron and quinizarin exert antiproliferative and antimetastatic activity on murine B16-F10 melanoma cells. Anticancer Res. 2010;30:445–449. [PubMed] [Google Scholar]
- 105.Baldini PM, Lentini A, Mattioli P, et al. Decrease of polyamine levels and enhancement of transglutaminase activity in selective reduction of B16-F10 melanoma cell proliferation induced by atrial natriuretic peptide. Melanoma Res. 2006;16:501–507. doi: 10.1097/01.cmr.0000232296.99160.d7. [DOI] [PubMed] [Google Scholar]
- 106.Di GG, Lentini A, Beninati S, Piacentini M, Rodolfo C. In vivo evaluation of type 2 transglutaminase contribution to the metastasis formation in melanoma. Amino Acids. 2009;36:717–724. doi: 10.1007/s00726-008-0119-5. [DOI] [PubMed] [Google Scholar]
- 107.Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci USA. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang L, Xu L. GPR56 in cancer progression: Current status and future perspective. Future Oncol. 2012;8:431–440. doi: 10.2217/fon.12.27. [DOI] [PubMed] [Google Scholar]
- 109.Xu L, Hynes RO. GPR56 and TG2: Possible roles in suppression of tumor growth by the microenvironment. Cell Cycle. 2007;6:160–165. doi: 10.4161/cc.6.2.3760. [DOI] [PubMed] [Google Scholar]
- 110.Yang L, Friedland S, Corson N, Xu L. GPR56 inhibits melanoma growth by internalizing and degrading its ligand TG2. Cancer Res. 2014;74:1022–1031. doi: 10.1158/0008-5472.CAN-13-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jeong JH, Cho BC, Shim HS, et al. Transglutaminase 2 expression predicts progression free survival in non-small cell lung cancer patients treated with epidermal growth factor receptor tyrosine kinase inhibitor. J Korean Med Sci. 2013;28:1005–1014. doi: 10.3346/jkms.2013.28.7.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Park MK, You HJ, Lee HJ, et al. Transglutaminase-2 induces N-cadherin expression in TGF-beta1-induced epithelial mesenchymal transition via c-Jun-N-terminal kinase activation by protein phosphatase 2A down-regulation. Eur J Cancer. 2013;49:1692–1705. doi: 10.1016/j.ejca.2012.11.036. [DOI] [PubMed] [Google Scholar]
- 113.Frese-Schaper M, Schardt JA, Sakai T, Carboni GL, Schmid RA, Frese S. Inhibition of tissue transglutaminase sensitizes TRAIL-resistant lung cancer cells through upregulation of death receptor 5. FEBS Lett. 2010;584:2867–2871. doi: 10.1016/j.febslet.2010.04.072. [DOI] [PubMed] [Google Scholar]
- 114.Kosa K, Rosenberg MI, Chiantore MV, De Luca LM. TPA induces transglutaminase C and inhibits cell growth in the colon carcinoma cell line SW620. Biochem Biophys Res Commun. 1997;232:737–741. doi: 10.1006/bbrc.1997.6363. [DOI] [PubMed] [Google Scholar]
- 115.Takaku K, Futamura M, Saitoh S, Takeuchi Y. Tissue-type transglutaminase is not a tumor-related marker. J Biochem. 1995;118:1268–1270. doi: 10.1093/oxfordjournals.jbchem.a125017. [DOI] [PubMed] [Google Scholar]
- 116.D'Argenio G, Iovino P, Cosenza V, et al. Transglutaminase in azoxymethane-induced colon cancer in the rat. Dig Dis Sci. 1995;40:685–695. doi: 10.1007/BF02064391. [DOI] [PubMed] [Google Scholar]
- 117.Miyoshi N, Ishii H, Mimori K, et al. TGM2 is a novel marker for prognosis and therapeutic target in colorectal cancer. Ann Surg Oncol. 2010;17:967–972. doi: 10.1245/s10434-009-0865-y. [DOI] [PubMed] [Google Scholar]
- 118.DC KP, SQ HP, et al. MiR-19-mediated inhibition of transglutaminase-2 leads to enhanced invasion and metastasis in colorectal cancer. Mol Cancer Res. 2015 doi: 10.1158/1541-7786.MCR-14-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pierce A, Whetton AD, Meyer S, et al. Transglutaminase 2 expression in acute myeloid leukemia: Association with adhesion molecule expression and leukemic blast motility. Proteomics. 2013;13:2216–2224. doi: 10.1002/pmic.201200471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pincelli C, Marconi A. Keratinocyte stem cells: Friends and foes. J Cell Physiol. 2010;225:310–315. doi: 10.1002/jcp.22275. [DOI] [PubMed] [Google Scholar]
- 121.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sakariassen PO, Immervoll H, Chekenya M. Cancer stem cells as mediators of treatment resistance in brain tumors: Status and controversies. Neoplasia. 2007;9:882–892. doi: 10.1593/neo.07658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Y, Tang L. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacol Sin. 2007;28:1343–1354. doi: 10.1111/j.1745-7254.2007.00679.x. [DOI] [PubMed] [Google Scholar]
- 124.Bednar F, Simeone DM. Pancreatic cancer stem cells and relevance to cancer treatments. J Cell Biochem. 2009;107:40–45. doi: 10.1002/jcb.22093. [DOI] [PubMed] [Google Scholar]
- 125.Ischenko I, Seeliger H, Schaffer M, Jauch KW, Bruns CJ. Cancer stem cells: how can we target them? Curr Med Chem. 2008;15:3171–3184. doi: 10.2174/092986708786848541. [DOI] [PubMed] [Google Scholar]
- 126.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 127.Al-Hajj M, Wicha MS, ito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 129.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 130.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 131.Yang ZF, Ho DW, Ng MN, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 132.Korkaya H, Paulson A, Charafe-Jauffret E, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7:e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 135.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23:7274–7282. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 136.Lin A, Minden A, Martinetto H, et al. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 137.Grigoriev MY, Suspitsin EN, Togo AV, et al. Tissue transglutaminase expression in breast carcinomas. J Exp Clin Cancer Res. 2001;20:265–268. [PubMed] [Google Scholar]
- 138.Martinet N, Bonnard L, Regnault V, et al. In vivo transglutaminase type 1 expression in normal lung, preinvasive bronchial lesions, and lung cancer. Am J Respir Cell Mol Biol. 2003;28:428–435. doi: 10.1165/rcmb.2002-0114OC. [DOI] [PubMed] [Google Scholar]
- 139.Zhang R, Tremblay TL, McDermid A, Thibault P, Stanimirovic D. Identification of differentially expressed proteins in human glioblastoma cell lines and tumors. Glia. 2003;42:194–208. doi: 10.1002/glia.10222. [DOI] [PubMed] [Google Scholar]
- 140.Fu J, Yang QY, Sai K, et al. TGM2 inhibition attenuates ID1 expression in CD44-high glioma-initiating cells. Neuro Oncol. 2013;15:1353–1365. doi: 10.1093/neuonc/not079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kumar A, Gao H, Xu J, Reuben J, Yu D, Mehta K. Evidence that aberrant expression of tissue transglutaminase promotes stem cell characteristics in mammary epithelial cells. PLoS ONE. 2011;6:e20701. doi: 10.1371/journal.pone.0020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kumar A, Xu J, Sung B, et al. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012;14:R4. doi: 10.1186/bcr3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther. 2007;115:232–245. doi: 10.1016/j.pharmthera.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Keillor J, chica R, Chabot N, et al. The bioorganic chemistry of transglutaminase—From mechanism to inhibition and engineering. Can J Chem. 2008;86:271–276. [Google Scholar]
- 145.Griffin M, Mongeot A, Collighan R, et al. Synthesis of potent water-soluble tissue transglutaminase inhibitors. Bioorg Med Chem Lett. 2008;18:5559–5562. doi: 10.1016/j.bmcl.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 146.Badarau E, Collighan RJ, Griffin M. Recent advances in the development of tissue transglutaminase (TG2) inhibitors. Amino Acids. 2013;44:119–127. doi: 10.1007/s00726-011-1188-4. [DOI] [PubMed] [Google Scholar]
- 147.Begg GE, Holman SR, Stokes PH, Matthews JM, Graham RM, Iismaa SE. Mutation of a critical arginine in the GTP-binding site of transglutaminase 2 disinhibits intracellular crosslinking activity. J Biol Chem. 2006;281:12603–12609. doi: 10.1074/jbc.M600146200. [DOI] [PubMed] [Google Scholar]
- 148.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Iismaa SE, Holman S, Wouters MA, Lorand L, Graham RM, Husain A. Evolutionary specialization of a tryptophan indole group for transition-state stabilization by eukaryotic transglutaminases. Proc Natl Acad Sci USA. 2003;100:12636–12641. doi: 10.1073/pnas.1635052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Begg GE, Carrington L, Stokes PH, et al. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc Natl Acad Sci USA. 2006;103:19683–19688. doi: 10.1073/pnas.0609283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dafik L, Albertelli M, Stamnaes J, Sollid LM, Khosla C. Activation and inhibition of transglutaminase 2 in mice. PLoS ONE. 2012;7:e30642. doi: 10.1371/journal.pone.0030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yakubov B, Chen L, Belkin AM, et al. Small molecule inhibitors target the tissue transglutaminase and fibronectin interaction. PLoS ONE. 2014;9:e89285. doi: 10.1371/journal.pone.0089285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Verderio EA, Scarpellini A, Johnson TS. Novel interactions of TG2 with heparan sulfate proteoglycans: Reflection on physiological implications. Amino Acids. 2009;36:671–677. doi: 10.1007/s00726-008-0134-6. [DOI] [PubMed] [Google Scholar]
- 154.Sneddon JB, Werb Z. Location, location, location: the cancer stem cell niche. Cell Stem Cell. 2007;1:607–611. doi: 10.1016/j.stem.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]