

Abstract
The conserved pterin dithiolene ligand that coordinates molybdenum (Mo) in the cofactor (Moco) of mononuclear Mo enzymes can exist in both a tricyclic pyranopterin dithiolene form and as a bicyclic pterin-dithiolene form as observed in protein crystal structures of several bacterial molybdoenzymes. Interconversion between the tricyclic and bicyclic forms via pyran scission and cyclization has been hypothesized to play a role in the catalytic mechanism of Moco. Therefore understanding the interconversion between the tricyclic and bicyclic forms, a type of ring-chain tautomerism, is an important aspect of study in order to understand its role in catalysis. In this study, equilibrium constants (Keq) as well as enthalpy, entropy, and free energy values are obtained for pyran ring tautomerism exhibited by two Moco model complexes, (Et4N)[Tp*Mo(O)(S2BMOPP)] (1) and (Et4N)[Tp*Mo(O)(S2PEOPP)] (2), as a solvent-dependent equilibrium process. Keq values obtained from 1H NMR data in seven deuterated solvents show a correlation between solvent polarity and tautomer form, where solvents with higher polarity parameters favor the pyran form.
Graphical abstract
Introduction
Mononuclear molybdenum (Mo) and tungsten (W) enzymes form a diverse group of enzymes whose roles in biological metabolic and catabolic processes primarily involve transfer of oxygen atoms in simple reactions having critical importance to the health of the organism. These metalloenzymes have at their active site a unique and structurally-complex cofactor that contains either one or two conserved pyranopterin dithiolene ligands commonly known as “molybdopterin” (MPT).1 The chemical structure of MPT was largely gleaned from chemical degradation studies by Rajagopalan et al. and has been defined in more detail in protein crystal structures.2,3 These X-ray structures depict MPT in the majority of molybdenum enzymes as a triheterocyclic dithiolate chelate (Figure 1a) containing a pterin moiety and a pyran ring, both attached to a dithiolene group that serves as the chelating unit coordinated to the Mo or W center.
Figure 1.
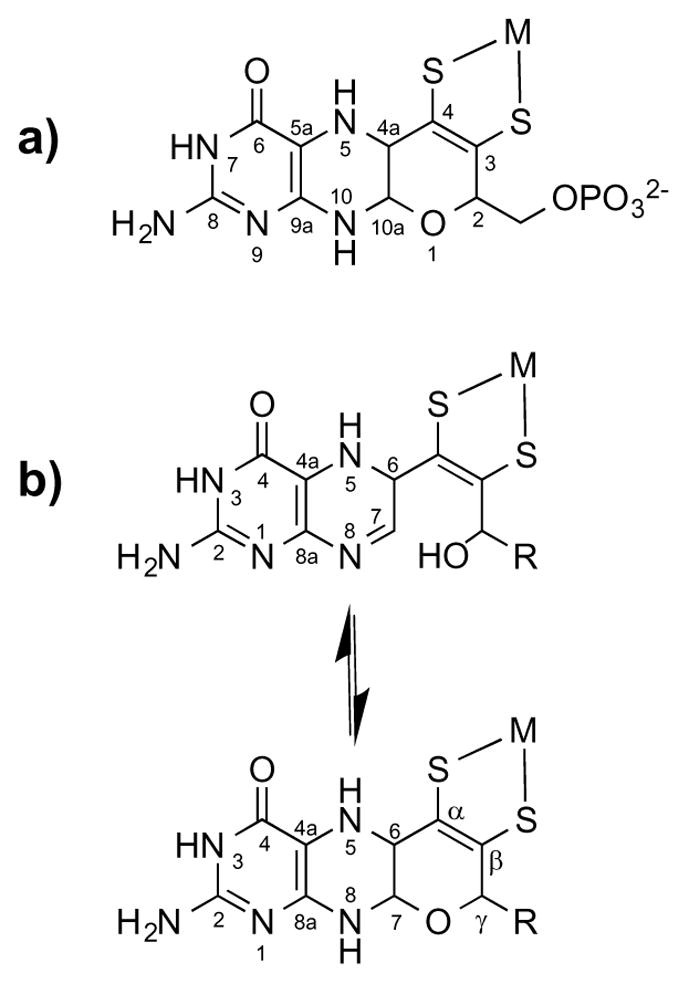
(a) Chemical structure and IUPAC numbering of MPT in the tricyclic pyranopterin form; (b) pyran cyclization equilibrium with the numbering system used in this paper.4 It must be noted that, although the pterin IUPAC numbering system changes upon cyclization, the uncyclized numbering system (in (b), top structure) will be used throughout this paper for simplicity. M = Mo or W; R = phosphate or a dinucleotide.
Despite decades of research in molybdoenzymes and model studies of its cofactor Moco, the connection between the highly conserved ligand MPT in Moco and its catalytic function remains obscure.5,6 In part, our lack of knowledge regarding the precise role of MPT in Moco has been limited by its in vitro instability outside of the protein matrix. Yet it is well understood that MPT coordination to Mo in Moco is required for proper enzymatic function, highlighting the importance of MPT as a vital part of Moco.2,7 This fact has spurred a growing interest in elucidating the role MPT plays during catalysis. Since the pterin system is able to achieve different oxidation states through sequential 2e−/2H+ redox processes, it has been speculated that it might participate with the dithiolene in modulating Mo redox potentials.5,8–13 Evidence that pterin may indeed play a role in tuning catalysis emerged from a detailed analysis of pyranopterin conformations in Mo and W protein crystal structures.6 This study revealed a correlation between pterin conformation and enzyme function where the pterin conformation was interpreted as resulting from different oxidation states.6
The pyran ring joining the dithiolene chelate to the pterin in MPT may also have a part in tuning catalysis. Some have speculated that the pyran ring has the capacity to undergo ring scission and produce a bicyclic “open” form as shown in Figure 1b.6,14 This expectation was initially based on reactivity that was documented in the first report of a synthetic pyranopterin.15,16 Pyran ring scission of MPT would disrupt the electronic environment felt by the dithiolene as the tetrahydropyrazine ring (Figure 1a) becomes the 5,6-dihydro system (Figure 1b). Reinforcing this notion that pyran ring scission may occur and have a specific function, two protein crystal structures of bis-MPT sites in bacterial molybdoenzymes exhibit the Moco site as possessing two MPT ligands in different forms, one a tricyclic pyranopterin-dithiolene and one a bicyclic, open pterin-dithiolene, thereby providing structural evidence that reversible pyran cyclization dynamics may be achievable within the active site.17,18
Reports of pyran formation and scission in chemical models of Moco are limited. Joule et al. achieved pyran cyclization in their quinoxaline- and pteridine-containing Moco models after alkylation and subsequent pyrazine reduction.19,20 In a study not directed at modeling Moco, Pfleiderer and Soyka also observed pyran scission in a simple pteridine system.15,16 We subsequently investigated this system in terms of its redox behavior and kinetics under various reductive and oxidative conditions and concluded that the pyran ring protected the reduced pyranopterin from oxidation to neopterin, which requires scission of the pyran ring to occur.21 In a recent communication, we reported the synthesis of the first Moco model complex containing a pyranopterin dithiolene ligand bound to Mo, (Et4N)[Tp*Mo(O)(S2BMOPP)] (1), where Tp* = tris(3,5-dimethylpyrazolyl)hydroborate, and described our initial observations regarding the ability of this model to undergo reversible, solvent-dependent pyran cyclization.22 Here we report a new model, (Et4N)[Tp*Mo(O)(S2PEOPP)] (2), and detailed investigations of the ring-chain tautomerism involving the pyranopterin in both 1 and 2. The chemical structures of both complexes, as well as the pyran cyclization equilibrium they exhibit, are depicted in Figure 2. The incorporation of model 2 in this study was intended to probe the role of sterics in the observed pyran tautomerism. The equilibrium between the open form and the cyclized pyrano form of the pterin-dithiolene chelates S2BMOPP and S2PEOPP was studied by 1H NMR and measured in seven solvents to yield equilibrium constants (Keq) and thermodynamic parameters (ΔH, ΔS, and ΔG) for pyran cyclization in 1 and 2. The set of Keq values reveal that the pyran form is favored with increasing solvent polarity, based on solvent dielectric constant, whereas the open chain, pterin-dithiolene form predominates in low polarity (low dielectric) solvents.
Figure 2.
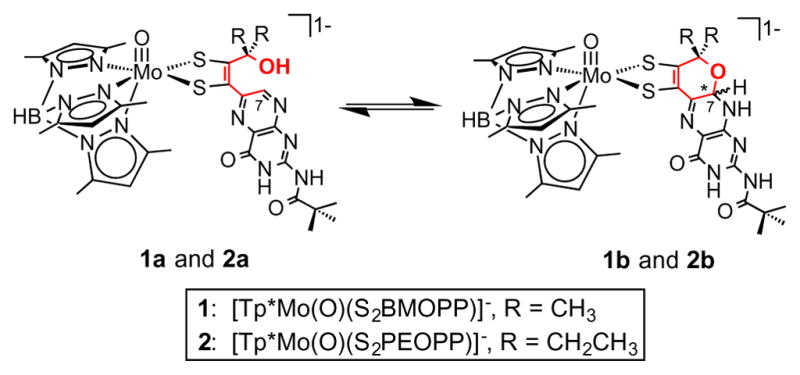
The equilibrium between the open chain forms 1a and 2a and the cyclized pyran forms 1b and 2b. The pyran ring that is involved in the tautomerism is highlighted in red. Cyclization causes the formation of two diastereomers, forming either the R or S isomer at the chiral carbon C7 marked by an asterisk.
Experimental Section
Materials and Methods
The syntheses of (Et4N)[Tp*Mo(S)(S4)], 2-pivaloyl-6-chloropterin, and (Et4N)[Tp*Mo(O)(S2BMOPP)] (1) were performed using previously published procedures.22,23 All other reagents, chemicals, and deuterated solvents were purchased from Sigma-Aldrich and used as received. All solvents for syntheses were purchased from Pharmco-AAPER and were deaerated with N2 gas over activated neutral alumina before use. ESI-MS analyses were performed using a Waters Micromass-ZQ mass spectrometer at Bryn Mawr College via infusion of samples as acetonitrile solutions. All NMR experiments were performed on a Bruker 400 MHz FT-NMR. Infrared spectra were obtained using a PerkinElmer Frontier FT-IR on samples prepared as KBr pellets.
6-(3-Pentynyl-2-ethyl-2-ol)-2-pivaloyl pterin (PEOPP)
2-pivaloyl-6-chloropterin (1.0046 g, 3.5662 mmol), CuI2 (0.1039 g, 0.5456 mmol), Pd(OAc)2 (0.1009 g, 0.4494 mmol), and 1,1′-bis(diphenylphosphino) ferrocene (BDPF) (0.2512 g, 0.4531 mmol) were combined in 30 mL acetonitrile and magnetically stirred for 10 minutes, after which 3-ethyl-1-pentyn-3-ol (2.6 g, 23 mmol) and triethylamine (3.6 g, 35.8 mmol) were added via syringe under N2 gas using standard Schlenk techniques. The resulting brown solution was stirred for 4 hours at 42.5 °C then diluted with dichloromethane (30 mL), chilled for 30 minutes at 0 °C, and then filtered. Diethyl ether (20 mL) was added to the filtrate. The resulting precipitate was subsequently filtered, washed with water (20 mL), and dried under vacuum to produce PEOPP (1.2562 g, 99.0 %) as a tan solid. 1H NMR (CDCl3) δ: 8.87 (1H, s), 1.85 (4H, q), 1.37 (9H, s), 1.15 (6H, t). FT-IR (KBr pellet, cm−1): ν(O–H) 3342, ν(N–H) 3246, 3173, 3112, ν(C≡C) 2221, ν(C=O) 1682, 1612, ν(C=N) 1556, 1478, 1443. Anal. Calcd (%) for C18.5H24N5O3Cl, (PEOPP · 1/2 CH2Cl2): C, 55.57; H, 6.049. Found: C, 55.61; H, 5.69.
(Et4N)[Tp*Mo(O)(S2PEOPP)] (2)
Under N2 gas, (Et4N)[Tp*Mo(S)S4] (0.2004 g, 0.2926 mmol) and PEOPP (0.1020 g, 0.2854 mmol) were magnetically stirred in wet acetonitrile (8 mL, 1% H2O v/v) to produce a brown solution that was heated for 2 hours at 63 °C. Addition of diethyl ether precipitated a reddish-brown solid that was subsequently filtered and dried under vacuum, yielding (Et4N)-[Tp*Mo(S)(S2PEOPP)], ESI-MS: [M-] = 848 m/Z, [M + 2TEA+] = 1108 m/Z. After redissolving (Et4N)-[Tp*Mo(S)(S2PEOPP)] in wet acetonitrile (8 mL, 1% H2O v/v), tributylphosphine (0.20 g, 1.0 mmol) was added drop-wise. The brown solution was stirred for 2.5 hours at room temperature. Addition of diethyl ether caused precipitation of a brown solid that was filtered and dried under vacuum to produce 2 (0.0883 g, 60 %). 1H NMR (CDCl3, ppm) δ: 11.63 (2H, s), 9.40 (1H, s), 5.94 (1H, s), 5.88 (1H, s), 5.85 (1H, s), 5.43 (1H, s), 2.76 (4H, s), 2.64 (3H, s), 2.43 (3H, s), 2.23 (3H, s), 2.16 (3H, s), 2.14 (3H, s), 1.42 (6H, q), 1.38 (9H, s), 1.26 (3H, s). FT-IR (KBr pellet, cm−1): 3428 ν(O–H), 3250 ν(N–H), 2546 ν(B–H), 1657 ν(C=O), 1613, 1544 ν(C=N), 1479, 1449, 928 (Mo≡O). ESI-MS: [M-] = 832 m/Z, [M + 2TEA+] = 1092 m/Z.
Computational Methods
Geometry optimization and energy calculations were performed at the density functional level of theory (DFT) using the Gaussian09 collection of software.24 Input files were created and manipulated using Gaussview. Calculations used the B3LYP hybrid functional and applied the Stuttgart/Dresden (SDD) basis set and effective core potential to Mo and the 6-31G* basis set to all other atoms in the molecule. Pterin rotation calculations were performed starting with the optimized structure of 1a but with varying pterin-dithiolene dihedral angles while maintaining the optimized structure for the remaining complex.
Results
Synthesis and Characterization of 2
The overall synthetic pathway for 2 is outlined in Figure 3. In a previous report,23 we established that Mo complexes containing pterin dithiolenes can be achieved by the reaction of a pterinyl alkyne with a molybdenum tetrasulfide complex. The synthesis of 2 employs this same strategy as shown in Figure 3 using the pterinyl alkyne PEOPP. The Sonogashira cross-coupling reaction between 2-pivaloyl-6-chloropterin and 3-ethyl-1-pentyn-3-ol produces the tan-colored PEOPP alkyne in high yield (Figure 3a). The subsequent reaction of PEOPP and (Et4N)[Tp*Mo(S)(S4)] produces (Et4N)[Tp*Mo(S)(S2PEOPP)] (Figure 3b). Ethyl substitution on the alkyne creates steric resistance to coupling of the alkyne and tetrasulfide, hence the reaction requires a higher temperature to achieve dithiolene formation. Subsequent hydrolysis of the sulfido ligand is facilitated by excess (5 equivalents) tributylphosphine in the presence of water and oxygen (Figure 3c). The role of phosphine is presumed to be labilization of the Mo≡S bond towards hydrolysis. Addition of O2 in the reaction flask has been found to expedite this hydrolysis reaction but the details regarding this reaction are not yet known. Care is required since O2 also oxidizes the Mo(IV) metal to Mo(V) at longer reaction times.
Figure 3.

(a) PEOPP alkyne synthesis; (b) reaction of PEOPP and (Et4N)[Tp*Mo(S)(S4)] to produce (Et4N)[Tp*Mo(S)(S2PEOPP)]; (c) hydrolysis of the terminal sulfido ligand on Mo. (i) CuI2, Pd(OAc)2, BDPF, Et3N, MeCN at 42.5 °C; (ii) 1% H2O in MeCN, 2 hrs. at 63 °C; (iii) 5 eq. PBu3, 1% H2O in MeCN, 2.5 hrs. at 25 °C.
Equilibrium Measurements
1H NMR spectroscopy has been documented as a convenient method to obtain Keq constants in systems exhibiting ring-chain equilibria, and this approach was used to determine Keq values for 1 and 2.25–27 In the current study, equilibrium experiments were performed in seven deuterated solvents to study how the solvent environment affected the equilibrium between the cyclized, pyranopterin and open pterin forms of complexes 1 and 2. Keq constants were calculated using the integrations of pertinent resonance signals corresponding to the open or pyran forms. Selected solvents included chlorinated solvents (chloroform-d and dichloromethane-d2) as well as polar aprotic solvents (dimethyl sulfoxide-d6 (DMSO-d6), N,N-dimethylformamide-d7 (DMF-d7), acetonitrile-d3, acetone-d6, and tetrahydrofuran-d8 (THF-d8)). Polar protic solvents such as methanol-d4 were avoided since 1H NMR spectra of 1 and 2 in these solvents typically have broadened signals that complicate the acquisition of integration data.
Signals utilized for integration data were selected based on 13C and HSQC NMR experiments (data not shown) and are shown in Figure 4 for 1 in chloroform-d and acetonitrile-d3 and in all other deuterated solvents in Figure S1 for 1 and in Figure S2 for 2. The integration of the resonance at approximately 9.0 – 9.4 ppm corresponding to the pyrazine proton H7 was used as a measure of the relative amount of the uncyclized, open forms 1a and 2a. During cyclization to the pyrano forms 1b and 2b, H7 remains bound to carbon C7 whose tetrahedral character now produces a more shielded environment causing the H7 resonance to shift upfield between 5.8 – 6.0 ppm in all solvents; pyranopterin 1H and 13C NMR data from the literature corroborate this assignment.15,28,29 Two signals are observed for H7 in the pyrano forms 1b and 2b since C7 becomes a chiral center upon cyclization and may form either the R or S enantiomer as observed from crystal structures of 1 (Figure 4d).22 As a result, two separate signals are found to correspond to the proton in both enantiomers. Although the H7 resonance for 1b in the pyrano form is isolated from any other signals in the 1H NMR spectrum when obtained in acetonitrile-d3, DMF-d7, and DMSO-d6, in the remaining solvents, H7 signals overlap with neighboring resonances corresponding to Tp* pyrazole protons which complicates integration data. To circumvent this problem, integration data for the pyran form in both 1 and 2 were acquired from the proton signals corresponding to the bottom Tp* central pyrazole proton (blue Hb in Figure 4a, Figure S1a, and Figure S2a). Two signals for Tp* Hb corresponding to the R and S enantiomers are observed in all solvents except acetonitrile-d3 for 1b, in which case these signals converge into one peak. Integrations of these two signals were summed to give the total amount of cyclized pyranopterin. We note that no additional resonances for other dihydropyranopterin tautomers other than the 7,8-dihydropyranopterin form of 1b and 2b (Figure 4a, right) were observed.
Figure 4.

(a) Structures of 1a and 1b highlighting corresponding protons used for integration data from 1H NMR spectra in (b) chloroform-d and (c) acetonitrile-d3; (d) equilibrium between the open form 1a or 2a and the two R and S diastereomers of the pyran forms 1b and 2b; two signals in the 1H NMR spectrum (highlighted in green in acetonitrile-d3) are for H7 in the pyran form, each corresponding to either the R or S isomer.
Keq values obtained from 1H NMR data are listed in Table 1 for complexes 1 and 2. Since sterically bulky substituents near the site of tautomerism tend to push the equilibrium to the cyclized form,26,27 it was thought that the additional steric bulk imposed on the system by the ethyl groups in 2 versus the methyl groups in 1 might cause 2 to have larger Keq values. However, examination of the data seems to show this is not the case: all Keq values for 2, with exception of that for DMSO-d6, are similar in magnitude to those for 1.
Table 1.
Pyran cyclization equilibrium constantsa for 1 and 2 in various deuterated solvents correlated with solvent dielectric constants (ε)
| solvent | Keq (1) | Keq (2) | ε |
|---|---|---|---|
| DMSO-d6 | 14(2) | 8(3) | 46.7 |
| DMF-d7 | 6.9(7) | n/a b | 36.7 |
| acetonitrile-d3 | 6.1(8) | 5(1) | 37.5 |
| acetone-d6 | 2.3(4) | 2.0(6) | 20.7 |
| THF-d8 | 1.6(2) | 1.5(1) | 7.58 |
| dichloromethane-d2 | 1.2(2) | 1.8(4) | 8.93 |
| chloroform-d | 0.41(5) | 0.5(2) | 4.81 |
Keq = [1b]/[1a] or Keq = [2b]/[2a].
Integration data not acquired due to convolution of signals in the 1H NMR spectrum.
Because Keq values in both 1 and 2 vary with solvent, we attempted to correlate physical solvent parameters with Keq data to understand which solvent effects govern the equilibrium between pyranopterin-dithiolene and open chain pterin-dithiolene. Plotting solvent dielectric constant versus Keq (Figure 5) shows that Keq increases as the solvent dielectric constant increases. Solvent dipole moment, the empirical Kosower’s Z constants,30 and the empirical Kamlet-Taft hydrogen bond acceptor basicity scale (βKT)31,32 provided similar trends as dielectric constants but with poorer correlations (Figure S3).
Figure 5.
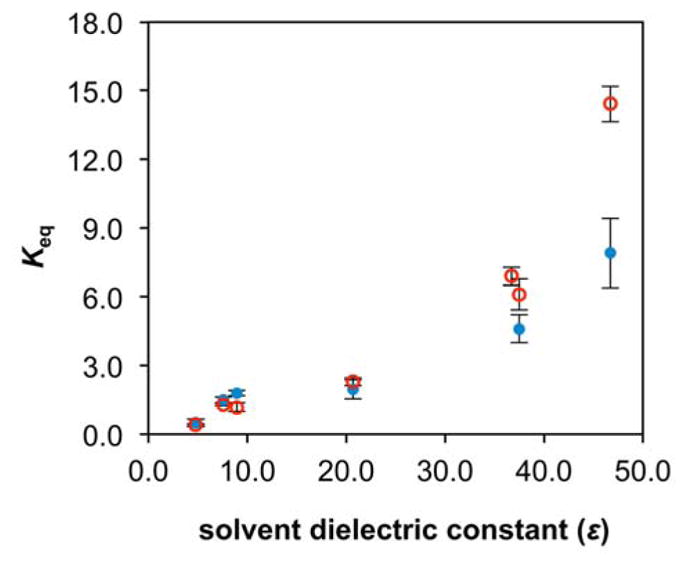
Plot of Keq values versus solvent dielectric constant for 1 (open red circles) and 2 (blue dots).
The trend shown in Figure 5 was compared to studies on ring-chain tautomerism in imine- and carbonyl-containing organic molecules. While the majority of ring-chain tautomerism studies find that increasing solvent polarity generally favors the chain (open) form,26,33–37 a few studies exist whose results mirror ours where polar solvents favor the cyclized form.38–40 However, these studies agree that polar solvents preferentially solubilize and stabilize the more polar form of the molecule undergoing ring-chain tautomerism.27,34,38 Based on the Keq data in Table 1, it may be inferred that the pyran form of 1 and 2 is more polar than the open form.
Other factors contributing to the observed trend in Fig. 5 can be considered. One is that polar solvents could stabilize a charged intermediate between the open and pyran forms, enhancing the thermodynamic accessibility of the pyran form. Another factor is the ability of more polar solvents to act as Brønsted-Lowry acids/bases, aiding proton migration from the hydroxyl oxygen in the open form (1a, 2a) to N8 in the pyran form (1b, 2b).26 It has been argued that hydrogen bonds between polar, aprotic solvents and the –OH group of hydroxyl-containing imines stabilize the open form in those molecules.26,33 Solvent formation of hydrogen bonds will certainly occur with both the pyranopterin tautomers 1b and 2b as well as the open chain tautomers 1a and 2a since all of these have many H-donor and acceptor groups. The wealth of possible H-bonding interactions in pterins makes it difficult to ascertain how H-bonding affects the equilibrium, but the general trend observed between Keq and the βKT scale (Figures S3e and S3f) shows solvents that form stronger hydrogen bonds favor the pyran form. While trace water contamination in higher-polarity solvents can be problematic and could affect the equilibrium, no trend was found between the Keq values and water concentration as observed from 1H NMR spectra in each solvent analyzed (Figure S4). One hydrogen bonding interaction that may provide a significant stabilization of the pyran forms 1b and 2b involves the donation of N–H proton at N8 to basic solvents.
Consideration of possible steric repulsion between H7 and the γ-hydroxyl group in the open chain forms 1a and 2a led us to perform DFT geometry optimizations on both structures. These calculations unanimously resulted in a structure (Figure 6, right and Figure S5) forming an intramolecular hydrogen bond between pterin N5 and the γ-hydroxyl proton made possible through rotation of the pterin group around C6–Cα bond (Figure 6). Optimized H-bonded structures had dihedral angles between the pterin and dithiolene molecular planes (Figure 7) of 149.6° in the optimized structure for 1a and 136.9° for 2a. This rearrangement to the lower energy structure in Figure S5 occurs even when the calculation is initiated with H7 adjacent to the hydroxyl group (per Figure 6, left). Such intramolecular hydrogen bonding has been observed as a stabilizing interaction in some systems exhibiting ring-chain tautomerism.37,41 Rotation of the pterin group in 1a and 2a has precedent in studies of (Et4N)[Tp*Mo(O)(S2BMOQO)] previously reported by us in a model structurally similar to 1 and 2, with a quinoxaline group in place of a pterin. Quinoxaline rotation precedes an intramolecular cyclization process to form a pyrrolo-dithiolene ligand.42,43
Figure 6.
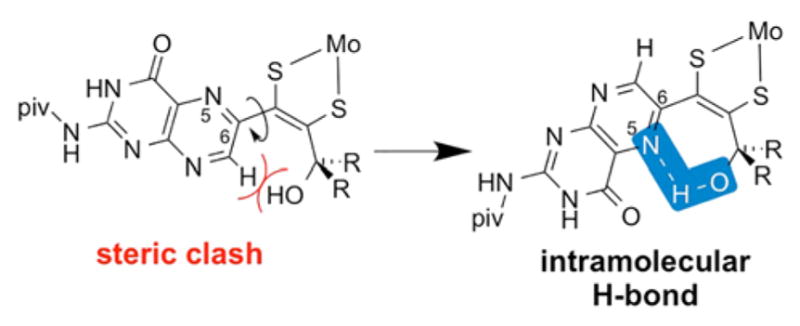
Structures of two possible rotamers of 1a and 2a. A steric clash arises when H7 is oriented towards the hydroxyl group in the co-planar conformation of the pterin and dithiolene units. Pterin rotation around the C6–Cα bond produces a more stable conformation due to possible formation of an intramolecular N5···H–O hydrogen bond (highlighted in blue).
Figure 7.
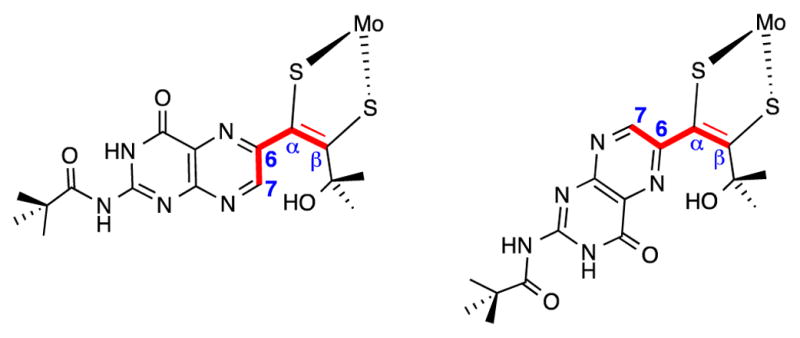
Structures illustrating the dihedral angle C7–C6–Cα–Cβ measured in the theoretical calculations; (left) for co-planar pterin and dithiolene groups, the dihedral angle C7–C6–Cα–Cβ is 0°; (right) for co-planar pterin and dithiolene groups, the dihedral angle C7–C6–Cα–Cβ is 180°.
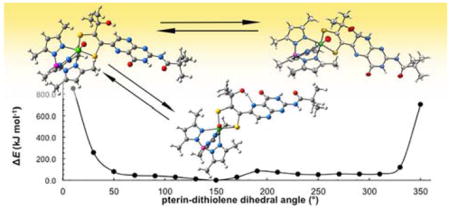
The geometry optimization results above prompted us to further study the energies of different rotamers of 1a. The potential energy diagram in Figure 8 was generated from energy calculations as the pterin/dithiolene dihedral angle around the C6–Cα bond (Figure 7) was varied. The diagram in Figure 8 shows that, although limited pterin rotation is energetically reasonable between the dihedral angles of approximately 40 – 320°, the intramolecular hydrogen bond interaction is optimal near 150°, where this is the most stable rotamer of 1a. Results from 1D NOESY experiments (Figure S6) of 1 in chloroform-d show that 1a exists in solution primarily as the rotamer depicted at 150°, with a smaller population of the 90° and 270° rotamers shown in Figure 6. When the dihedral angle approaches 0° or 360° placing H7 in close in proximity to the hydroxyl group, the energy increases dramatically to 800 kJ/mol. Such an energy barrier must impede full 360° pterin rotation and restricts the pterin rotational motion to dihedral angles between approximately 40 – 320°. Furthermore, this barrier prevents any conformation where the pterin is near coplanarity with dithiolene, such as is implied by the left structure in Figure 6. We conclude that, in solution, the pterin of 1a and 2a are never co-planar with the dithiolene in this fashion.
Figure 8.

Potential energy diagram of 1a with various dihedral angles measured between the pterin and dithiolene moieties. Structures of 1a at select pterin-dithiolene dihedral angles are included above each diagram; the structure best illustrating the N5···H–O hydrogen bond is highlighted in red.
Thermodynamic Parameters
ΔH, ΔS, and ΔG values for the pyran tautomerism in 1 and 2 were calculated from van’t Hoff plots of 1H NMR data collected at varying sample temperatures. Spectra were recorded at 10 °C intervals between 20 and −20 °C, except in DMSO-d6 where 5 °C intervals were used between 25 and 40 °C. Changing sample temperature caused a convergence of signals in some solvents, which complicated the acquisition of integration data. Hence the calculated thermodynamic parameters (Table 2) were limited to select solvents that did not exhibit signal convergence (DMSO-d6, DMF-d7, acetonitrile-d3, THF-d8, and chloroform-d for 1; only DMSO-d6 for 2). As seen in the van’t Hoff plots in Figure S7 and Figure S8, the pyran form is favored as the sample temperature decreases in all solvents analyzed, which is expected based on several studies of systems exhibiting ring-chain equilibria.26,27 The ΔG values for 1 in Table 2 are close to those values (ΔG′) calculated from the equation ΔG′ = −RTln(Keq) at 25 °C using the Keq constants in Table 1, with the exception of data from DMF-d7 and acetonitrile-d3,.
Table 2.
Thermodynamic parameters ΔH (kJ mol−1), ΔS (J mol−1 K−1), ΔG and ΔG′a (kJ mol−1) for 1 and 2 from van’t Hoff analysis at 25 °C
| solvent | ΔH | ΔS | ΔG | ΔG′a |
|---|---|---|---|---|
| DMSO-d6 | −31.2(7) | −81(2) | −6.98(3) | −6.6(3) |
| DMF-d7 | −30.8(4) | −80(1) | −6.90(2) | −4.8(2) |
| Acetonitrile-d3 | −28(3) | −73(8) | −6.0(5) | −4.5(3) |
| THF-d8 | −14(1) | −40(4) | −1.6(1) | −1.2(4) |
| chloroform-d | −9(2) | −35(9) | 1.5(5) | 2.2(3) |
| DMSO-d6 (2)b | −24(2) | −65(7) | −4.9(1) | −5(2) |
ΔG′ values are calculated from Keq constants in Table 1 at 25 °C.
Parameters for 2 were obtained only in DMSO-d6 due to convolution of signals in all other solvents.
The ΔG values are negative in all solvents except chloroform-d, signifying that pyran cyclization is thermodynamically favored and spontaneous in the polar solvents. This matches the Keq data in Table 1 where the pyran tautomer is favored in THF-d8, acetonitrile-d3, DMF-d7, and DMSO-d6 whereas the open species is favored in chloroform-d. The values of ΔH are negative in all solvents, even for chloroform that has a positive free energy value. The negative enthalpy values are unsurprising since pyran cyclization involves the formation of two new bonds: C7–O (to close the pyran ring) and N8–H. All ΔS values are negative, which is also expected since conversion of the open species to the pyran form inhibits fluxional motion of the pterin moiety via the C6–Cα bond as well as free rotation of the other dithiolene substituent –CR2OH (R = Me in 1, Et in 2), via the Cβ–Cγ bond. It is important to note that all free energy values in Table 2 are small in magnitude, spanning a range of approximately 8.5 kJ mol−1 between the two extreme solvents DMSO and chloroform for 1. In the context of Moco, the small nature of these free energies signify that pyran scission and cyclization are energetically inexpensive, making the interconversion between both forms an energetically facile process. Interestingly, all thermodynamic values become more negative in 1 as solvent polarity increases. The formation of stronger intermolecular associations, such as hydrogen bonds, between solvent molecules and 1 may be a possible explanation for the increasingly negative enthalpy values. More negative entropy terms corroborate this hypothesis because association of solvent molecules solvating 1 in a restricted, more ordered arrangement would decrease the entropy of the system.
During the present study, attempts were made to observe changes in the Mo electronic environment due to pyran cyclization and scission in 1 and 2 using cyclic voltammetry (CV) and solution FT-IR. Unfortunately, results from these analyses were inconclusive due to difficulties posed by the techniques themselves. Electrolyte required for the CV experiment increases the solvent dielectric and thus favors the pyran cyclization state of 1 and 2 in solution. Comparison of vibrational frequencies in different solvents using FT-IR is complicated by overlapping solvent vibrational modes.
Discussion
Several hypotheses in the literature offer speculations on the catalytic role played by the pyranopterin-dithiolene ligand of the molybdenum cofactor (Moco). One idea suggests that reversible pyran ring formation in Moco could be involved in Mo redox tuning.6,14,44–46 Here we present the first experimental results from a Mo-pterin-dithiolene model in support of a feasible, low energy dynamic cyclization process. This report provides a detailed investigation of reversible pyran ring scission and cyclization in two pyranopterin dithiolene models for Moco.
Reversible pyranopterin cyclization may be considered a complicated example of the simpler and much studied organic transformation known as ring-chain tautomerism, exemplified in Figure 9. Ring-chain tautomers exhibit a preference for one tautomer under certain conditions. Most commonly the cyclized ring form predominates in low polarity solvents, while the open, chain structure is favored in higher polarity solvents. Hydrogen bonding of solvents can also influence the tautomer equilibrium.
Figure 9.
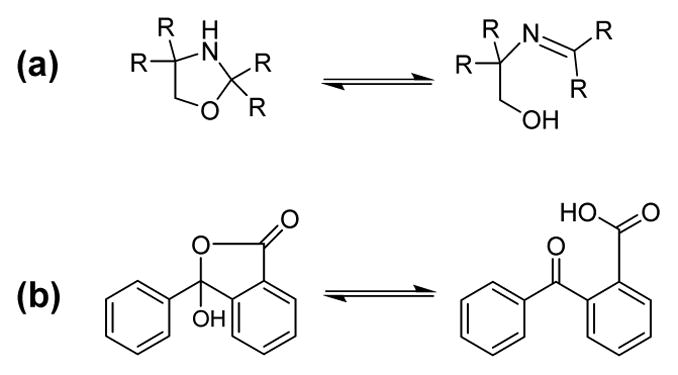
Examples of ring-chain tautomerism in (a) oxazolidines32 and (b) o-benzoylbenzoic acids.38
The Moco model complexes in this study also exhibit a solvent dependent behavior that determines whether the cyclized, pyranopterin form predominates versus the cleaved, open chain pterin form (Figure 2). The models 1 and 2 exhibit the ring form (the pyranopterin form) in high polarity solvents, while the chain form, (the open pterin-dithiolene) predominates in low polarity solvents. Though the behavior of the models differs from the majority of ring-chain tautomers, it is a behavior that has precedence in certain organic ring-chain tautomer systems.38–40 A computational analysis done on our model complexes 1 and 2 revealed that a reason for the preference of the open chain pterin-dithiolene form in low polarity solvent is likely a result of a rotation of the pterin producing a lower energy structure having an intramolecular hydrogen bond (Figure 7). The computational study included calculation of total energy as a function of pterin rotation required to attain the intramolecular hydrogen bond (Figure 8). The energy profile shows that co-planarity of pterin and dithiolene groups is energetically impossible, and that pterin motion is energetically restricted to dihedral angles of 40 – 320° between the pterin and dithiolene planes. Thermodynamic parameters obtained from variable temperature 1H NMR experiments confirm that the free energy difference between the pyranopterin and open-chain pterin forms is small (<10 kJ/mol).
Currently, the only protein crystal structures definitively exhibiting Moco in the open, bicyclic form are of nitrate reductase (NR) from Escherichia coli (1Q16)17 and ethylbenzene dehydrogenase (EBDH) from Aromatoleum aromaticum (2IVF).18 As these enzymes are members of the DMSO reductase family, their active sites consist of a bis-MPT-containing Moco, with one MPT in the pyranopterin form and one MPT in an open pterin-dithiolene structure (Figure 10). In both crystal structures, the MPT proximal to the [4Fe-4S] cluster (P in Figure 10) is in the pyranopterin form while the distal MPT ligand (D in Figure 10) is observed in an uncyclized, open form. The proximal MPT is presumed to be involved in electron transport between the [4Fe-4S] cluster and Mo, whereas the distal MPT has been suggested to modulate Mo oxidation state through pyran cyclization dynamics.6 Both the NR and EBDH structures show a stabilizing interaction of the open chain forms involving multiple H-bonds between protein residues and the –OH group.47 Low polarity groups around the pyran ring of the cyclized forms indicate that the protein environment is being used to selectively stabilize one form. The reported structures of NR and EBDH (Fig. 10) show different dihydropterin tautomers for the uncyclized distal pterin dithiolene, though it must be noted that the electron density maps for EBDH were reported as insufficient for determination of the precise pterin structure.18
Figure 10.
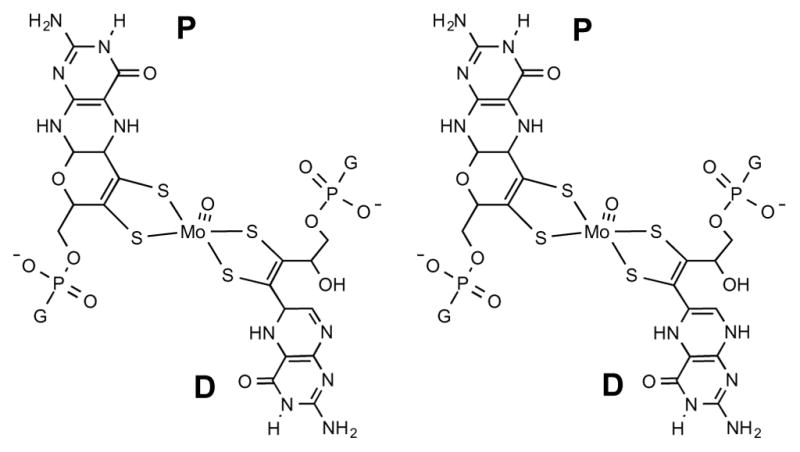
Structure of Moco in NR (left) and EBDH (right), where the proximal (P) pterin dithiolene is in the pyran form while the distal (D) one is in the open form. G = guanine mono-nucleotide.
The results from our work confirm that the proposed reversible pyran scission and cyclization in Mo enzymes is possible. The small energies involved in the tautomerism of our model suggest similar energetically inexpensive costs associated with interconversion of pyran and open chain forms of the pterin-dithiolene in the enzymes that could be compensated by H-bond formation. The results are also consistent with the notion that pyran cyclization dynamics in the active sites of Mo and W enzymes will be controlled in part by local polarities established by neighboring active site residues. The apparent polarity around the pyranopterin and open pterin regions in the enzymes is opposite of that observed in our solubilized model, but a direct comparison of the model to the enzymes is complicated since the model system has considerable flexibility while the pterin-dithiolene in MPT is tightly constrained by multiple H-bonds.
It is anticipated that the electronic environment at Mo in 1 and 2 will change with pyran cyclization and scission. In the X-ray crystal structure22 of 1 in the pyranopterin-dithiolene form, the pyran ring formation locks the pterin into a conformation where the pyrazine and dithiolene are nearly co-planar (dihedral angle = ~7°), allowing electronic communication between the Mo atom, the dithiolene and pterin moieties. However, in the chain or open form, steric repulsion between the –OH and the pterin H7 causes the pterin to rotate out of co-planarity, thus electronically isolating the dithiolene from the pterin group. Similar changes in planarity between pterin and dithiolene groups within Moco could be a mechanism to control the electronic structure in the catalytic reaction.
Further investigation may reveal how pyran ring-chain tautomerism changes the electronic structure in 1 and 2. Here we note that, in addition to pyran scission and cyclization, the Mo reduction potential may be controlled by other pterin modifications, such as protonation or changing the pterin oxidation state, and these may be coupled to pyran cyclization dynamics to significantly affect the Mo reduction potential. Protonation and chemical reduction of models 1 and 2 may provide more information regarding this behavior, and these studies are currently underway in our lab.
Conclusions
In this study we have reported Keq and thermodynamic constants for the solvent-dependent tautomerism between the pyran and open forms of pterin-dithiolene complexes 1 and 2. These values show that the pyran form is more favored in polar solvents and point to a strong correlation with solvent dielectric constant. Polar solvents may stabilize the closed form either through stabilization of a charged intermediate, formation of intermolecular hydrogen bonds, or some combination of both. Stabilization of the open form in less polar solvents is likely to occur by an intramolecular N···H–O hydrogen bond between the hydroxyl group and the pyrazine N5 as predicted by DFT geometry optimization calculations. These Keq trends may offer insight into the mechanism of reversible pyran scission and cyclization in Mo enzymes.
Supplementary Material
Synopsis.
This study addresses the hypothesis that reversible pyran ring formation in the pyranopterin-dithiolene ligand of the molybdenum cofactor (Moco) might have a catalytic role. We present the first experimental results from a Mo-pterin-dithiolene model in support of a feasible, low energy dynamic cyclization process and report equilibrium constants (Keq), enthalpy, entropy, and free energy values obtained for pyran ring tautomerism exhibited by two Moco model complexes.
Acknowledgments
Funding Sources
The authors wish to acknowledge the NIH (GM081848) and the NSF (CHE-0958996) for financial assistance.
Footnotes
Assignments of pertinent 1H NMR spectral regions in all solvents analyzed; plots of equilibrium and thermodynamic constants versus measures of solvent polarity; plots of H2O data from 1H NMR spectra versus Keq; 1D NOESY spectrum of 1a; van’t Hoff plots for 1 and 2; geometry optimization structures of 1a and 2a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rajagopalan KV. Molybdenum Enzymes, Cofactors and Model Systems. Vol. 535. American Chemical Society; Washington, D. C: 1993. Biochemistry of the Molybdenum Cofactors; pp. 38–49. ACS Symposium Series. [Google Scholar]
- 2.Leimkühler S, Wuebbens MM, Rajagopalan KV. Coord Chem Rev. 2011;255:1129–1144. doi: 10.1016/j.ccr.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson JL, Hainline BE, Rajagopalan KV, Arison BH. J Biol Chem. 1984;259:5414–5422. [PubMed] [Google Scholar]
- 4.Fischer B, Enemark JH, Basu P. J Inorg Biochem. 1998;72:13–21. doi: 10.1016/s0162-0134(98)10054-5. [DOI] [PubMed] [Google Scholar]
- 5.Basu P, Burgmayer SJN. Coord Chem Rev. 2011;255:1016–1038. doi: 10.1016/j.ccr.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothery RA, Stein B, Solomonson M, Kirk ML, Weiner JH. P Natl Acad Sci USA. 2012;109:14773–14778. doi: 10.1073/pnas.1200671109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pateman JA, Cove DJ, Rever BM, Roberts DB. Nature. 1964;201:58–60. doi: 10.1038/201058a0. [DOI] [PubMed] [Google Scholar]
- 8.Raghavan R, Dryhurst G. J Electroanal Chem Interfacial Electrochem. 1981;129:189–212. [Google Scholar]
- 9.Ege-Serpkenci D, Dryhurst G. Bioelectrochem Bioenerg. 1982;9:175–195. [Google Scholar]
- 10.Karber LG, Dryhurst G. J Electroanal Chem Interfacial Electrochem. 1982;136:217–289. [Google Scholar]
- 11.Ege-Serpkenci D, Raghavan R, Dryhurst G. Bioelectrochem Bioenerg. 1983;10:357–376. [Google Scholar]
- 12.Karber LG, Dryhurst G. J Electroanal Chem Interfacial Electrochem. 1984;160:141–157. [Google Scholar]
- 13.Diculescu VC, Militaru A, Shah A, Qureshi R, Dryhurst G. J Electroanal Chem Interfacial Electrochem. 2010;647:1–7. [Google Scholar]
- 14.Enemark JH, Garner CD. J Biol Inorg Chem. 1997;2:817–822. [Google Scholar]
- 15.Soyka R, Pfleiderer W. R Prewo Helv Chim Acta. 1990;73:808–826. [Google Scholar]
- 16.Soyka R, Pfleiderer W. Pteridines. 1990;2:63–74. [Google Scholar]
- 17.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NCJ. Nat Struct Biol. 2003;10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 18.Kloer DP, Hagel C, Heider J, Schulz GE. Structure. 2006;14:1377–1388. doi: 10.1016/j.str.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Bradshaw B, Dinsmore A, Collison D, Garner CD, Joule JA. J Chem Soc Perkin Trans. 2001:3232–3238. [Google Scholar]
- 20.Bradshaw B, Collison D, Garner CD, Joule JA. Chem Commun. 2001:123–124. [Google Scholar]
- 21.Burgmayer SJN, Pearsall DL, Blaney SM, Moore EM, Sauk-Schubert C. J Biol Inorg Chem. 2004;9:59–66. doi: 10.1007/s00775-003-0496-x. [DOI] [PubMed] [Google Scholar]
- 22.Williams BR, Fu Y, Yap GPA, Burgmayer SJN. J Am Chem Soc. 2012;134:19584–19587. doi: 10.1021/ja310018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgmayer SJN, Kim M, Petit R, Rothkopf A, Kim A, Bel Hamdounia S, Hou Y, Somogyi A, Habel-Rodriguez D, Williams A, Kirk ML. J Inorg Biochem. 2007;101:1601–1616. doi: 10.1016/j.jinorgbio.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda F, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam MJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 25.Bowden K, Byrne JM. J Chem Soc, Perkin Trans 2. 1997;1:123–127. [Google Scholar]
- 26.Valters RE, Flitsch W. In: Ring-Chain Tautomerism. Katritzky AR, editor. Plenum Press; New York: 1985. p. 169. [Google Scholar]
- 27.Escale R, Verducci J. Bull Soc Chim. 1974:1203–1206. [Google Scholar]
- 28.Santamaria-Araujo JA, Wray V, Schwarz G. J Biol Inorg Chem. 2012;17:113–122. doi: 10.1007/s00775-011-0835-2. [DOI] [PubMed] [Google Scholar]
- 29.Clinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA. J Med Chem. 2013;56:1730–1738. doi: 10.1021/jm301855r. [DOI] [PubMed] [Google Scholar]
- 30.Kosower EM. J Am Chem Soc. 1958;80:3253–3260. [Google Scholar]
- 31.Kamlet MJ, Taft RW. J Am Chem Soc. 1976;98:377–383. [Google Scholar]
- 32.Sandström M, Persson I, Persson P. Acta Chem Scand. 1990;44:653–675. [Google Scholar]
- 33.Paukstelis JV, Hammaker RM. Tetrahedron Lett. 1968:3557–3560. [Google Scholar]
- 34.Dorman LC. J Org Chem. 1967;32:255–260. [Google Scholar]
- 35.Potekhin AA, Zhdanov SL, Gindin VA, Ogloblin KA. Zh Org Khim. 1976;12:2090–2094. [Google Scholar]
- 36.Whitting JE, Edward JT. Can J Chem. 1971;49:3799–3806. [Google Scholar]
- 37.McDonagh AF, Smith HE. J Org Chem. 1968;33:1–8. [Google Scholar]
- 38.Bhatt MV, Kamath KM. J Chem Soc B. 1968:1036–1044. [Google Scholar]
- 39.Bhatt MV, Kamath KM. Tetrahedron Lett. 1966:3885–3890. [Google Scholar]
- 40.Walter W, Rohloff C. Justus Liebigs Ann Chem. 1977:485–490. [Google Scholar]
- 41.McDonagh AF, Smith HE. Chem Commun. 1966:374. [Google Scholar]
- 42.Matz KG, Mtei RP, Leung B, Burgmayer SJN, Kirk ML. J Am Chem Soc. 2010;132:7830–7831. doi: 10.1021/ja100220x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matz KG, Mtei RP, Rothstein R, Kirk ML, Burgmayer SJN. Inorg Chem. 2011;50:9804–9815. doi: 10.1021/ic200783a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacques JGJ, Fourmond V, Arnoux P, Sabaty M, Etienne E, Grosse S, Baiso F, Bertrand P, Pignol D, Léger C, Guigliarelli B, Burlat B. Biochim Biophys Acta. 2014;1837:277–286. doi: 10.1016/j.bbabio.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 45.Aguey-Zinsou KF, Bernhardt PV, Leimkühler S. J Am Chem Soc. 2003;125:15352–15358. doi: 10.1021/ja037940e. [DOI] [PubMed] [Google Scholar]
- 46.McNamara JP, Joule JA, Hillier IH, Garner CD. Chem Commun. 2005:177–179. doi: 10.1039/b415480k. [DOI] [PubMed] [Google Scholar]
- 47.Rothery RA, Weiner JH. J Biol Inorg Chem. 2015;20:349–372. doi: 10.1007/s00775-014-1194-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.