

Abstract
The synthetic biology toolbox lacks extendable and conformationally controllable yet easy-to-synthesize building blocks that are long enough to span membranes. To meet this need, an iterative synthesis of α-aminoisobutyric acid (Aib) oligomers was used to create a library of homologous rigid-rod 310-helical foldamers, which have incrementally increasing lengths and functionalizable N- and C-termini. This library was used to probe the inter-relationship of foldamer length, self-association strength, and ionophoric ability, which is poorly understood. Although foldamer self-association in nonpolar chloroform increased with length, with a ∼14-fold increase in dimerization constant from Aib6 to Aib11, ionophoric activity in bilayers showed a stronger length dependence, with the observed rate constant for Aib11 ∼70-fold greater than that of Aib6. The strongest ionophoric activity was observed for foldamers with >10 Aib residues, which have end-to-end distances greater than the hydrophobic width of the bilayers used (∼2.8 nm); X-ray crystallography showed that Aib11 is 2.93 nm long. These studies suggest that being long enough to span the membrane is more important for good ionophoric activity than strong self-association in the bilayer. Planar bilayer conductance measurements showed that Aib11 and Aib13, but not Aib7, could form pores. This pore-forming behavior is strong evidence that Aibm (m ≥ 10) building blocks can span bilayers.
Introduction
Foldamers are synthetic oligomers that can mimic some of the structural complexity of proteins and peptides without the constraints imposed by natural biopolymers. Like many peptides, in solution these oligomers can fold into conformationally stable structures which have large and structurally well-defined surfaces that are able to interact with individual biopolymers or biomolecular assemblies. For example, foldamers have been used to replicate protein–protein interactions1 and to form foldamer–DNA complexes for gene delivery,2 suggesting a role in a number of biomedical applications.
In recent years there have been a number of reports of foldamers displaying membrane activity, for example mimicking cell-penetrating peptides3 and antimicrobial agents,4 with the latter implying that foldamers could address the growing problem of antibiotic resistance.5 Like the antimicrobial peptides (AMPs), these foldamers may produce pores after spanning the membrane, although membrane disruption is another mechanism suggested to be behind AMP antibacterial activity.6 However, for many AMPs and foldamers7 the mechanisms behind cell toxicity are poorly understood.
Peptide foldamers containing high proportions of α-aminoisobutyric acid (Aib) have a number of attractive features as membrane-spanning building blocks, including high hydrophobicity and a propensity to adopt rigid-rod 310-helical secondary structures in a variety of solvents. Moreover, extended sequences of Aib are found in peptaibols, a class of naturally occurring AMPs produced by Trichoderma fungi. These AMPs are mostly between 11 and 21 residues long, often with a high Aib content, and are typically terminated by an N-terminal acyl group and a C-terminal 1,2-aminoalcohol residue.8 Since Aib residues stabilize 310 helices,9 shown schematically in Figure 1a, such peptaibols can have 310-helical and/or α-helical secondary structures.10
Figure 1.

(a) 310-Helical Aib foldamers 1–9. (b) Schematic representation of Aib foldamer helices interacting with a bilayer to form a pore with activity determined by foldamer length (a, black arrows) and foldamer–foldamer interactions (b, white arrows).
One well-studied peptaibol is alamethicin, which can adopt membrane-spanning orientations in membranes and produce ion channels.11,12 The mixed 310- and α-helical conformation of this 19-residue peptide13 results in sufficient length to span the 2.0–2.8 Å hydrophobic section of the phospholipid bilayer at the center of the cell membrane.13a,14 Alamethicin acts through a “barrel-stave” mechanism in which multimeric channels are formed by the self-association of 3–12 helical monomers, yielding parallel bundles with a central hydrophilic pore.11,12 The number of alamethicin peptides in a bundle is dynamic, with different conductance levels observed for bundles of different molecularity.15 Nonetheless, shorter peptaibols that cannot span a bilayer, such as the trichogins,16 are also membrane-active, including 6-residue trichodecinin I17 and 4-residue peptaibolin,18 showing that the relationship between peptaibol length and ionophoric activity is unclear.19 A family of foldamers with incrementally increasing lengths may throw light on this relationship, with such a study also identifying the minimum foldamer lengths needed to span different phospholipid bilayers.
There have been a handful of investigations into the relationship between ionophore length and membrane activity, with most using conformationally flexible systems. Although ionophoric activity was not assessed, a small family of homologous lipopeptaibols structurally related to the trichogins was assayed for membrane disruption. Release of dye from vesicles occurred upon addition of 11-, 15-, and 19-residue peptides, with the 15-mer most active, but not the 7-residue homologue.20 Sakai and Matile reported that a rigid-rod tetriphenyl exhibited no membrane activity, but homologous rigid-rod sexiphenyls (2.6 nm long) and octiphenyls (3.4 nm long) acted as ionophores; the latter was three times more active than the former.21 Gokel et al. investigated flexible channel-forming ionophores, which comprised three 4,13-diaza-18-crown-6 macrocycles connected by incrementally longer alkyl chains (−(CH2CH2)n–, n = 4, 5, 6, 7, 8). The rate constant for sodium ion transport across vesicle membranes increased 200-fold from n = 4 to n = 6, and then halved for n = 8.22 Interestingly, these compounds had antibiotic activity, with the n = 6 compound 13 times more effective at killing ampicillin-resistant E. coli than the n = 4 homologue.23
Although these studies imply that longer compounds should be more membrane-active than shorter ones and hint that an optimal length may exist, several questions remain unanswered. For example, the relationship between length and self-association strength is poorly understood, which is especially important for the multimeric barrel-stave channels formed by oligophenyls or peptaibols. Ionophore flexibility also complicates the analysis of length-dependent behavior, as long, flexible compounds might still adopt conformations with high membrane activity. The ideal system for a systematic study would be based on conformationally defined structures that can be extended in small increments. Such a study would produce a nanoscale scaffold optimized for spanning a given bilayer and useable as a potential building block for truly synthetic biology in a membrane or new generation antibiotics. Furthermore, any building block will need chemical functionality at both ends that allows it to be integrated into more complex constructs.
We now describe the synthesis and analysis of a series of 310-helical foldamers built exclusively from Aib residues, ranging from 5 to 13 residues in length (Figure 1a). A flexible strategy was developed for the synthesis of the “Aibm” oligomers that gave incrementally extendable mimics of the pore-forming peptaibols, structures with chemical functionality at both termini and without complicating side chain functionality. The ionophoric activity of each of these compounds was assessed and used to probe how the activity of helical peptides in bilayer membranes depended on both self-association strength and chain length (Figure 1b), with the adoption of membrane-spanning conformations confirmed using planar bilayer conductance assays.
Results and Discussion
Design and Synthesis of Aib Foldamers 1–9
A homologous series of foldamers was synthesized that varied only in their length (Scheme 1). As Aib is an achiral residue, these oligomers exist as an equal ratio of M and P 310-helical conformers that interconvert on a submillisecond time scale at room temperature.24 The shortest Aib foldamer 1 was designed to have sufficient length to give one turn of a 310 helix (>4 residues) in solution,25 and this core unit was extended by an iterative synthetic strategy (Scheme 1). By this method, a family of foldamers was created with lengths predicted to be up to and beyond the thickness of a typical bilayer. Groups were incorporated at the termini (azido at the N-terminus and 2-(trimethylsilyl)ethyl, CH2CH2SiMe3, at the C-terminus) which not only facilitated this iterative synthetic procedure but also minimized end-to-end intermolecular interactions.25 These terminal groups will permit the synthesis of functionalized derivatives, either after deprotection or through “click” chemistry. In a 310-helical conformation with a typical rise-per-residue of 1.94 Å, the lengths of compounds 1–9 were anticipated to lie between 1.8 and 3.4 nm.26 These foldamers are all relatively hydrophobic, facilitating partitioning into phospholipid bilayers from the buffer.27
Scheme 1. Synthesis of Foldamers 1–9.

(i) N3AibCl, Et3N, CH2Cl2, RT (room temperature). (ii) Pd/C, MeOH or EtOH, H2, RT. (iii) TFA (trifluoroacetic acid), CH2Cl2, RT. (iv) Ac2O, 120 °C or EDC·HCl (EDC = 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide), CH2Cl2, RT then HAibmOR (R = t-Bu or (CH2)2TMS; TMS = trimethylsilyl), CH3CN, 80 °C. (v) NaN3, DMF, RT. (vi) 2-(Trimethylsilyl)ethanol, EDC·HCl, DMAP (4-(dimethylamino)pyridine), CH2Cl2, RT. (vii) HCl in Et2O.
Solid State Structures
Crystal structures were obtained for N3Aib7OCH2CH2TMS 3, N3Aib8OCH2CH2TMS 4, and N3Aib11OCH2CH2TMS 7, which confirmed the adoption of 310-helical conformations stabilized by intramolecular hydrogen bonds (Figure 2a–c).
Figure 2.
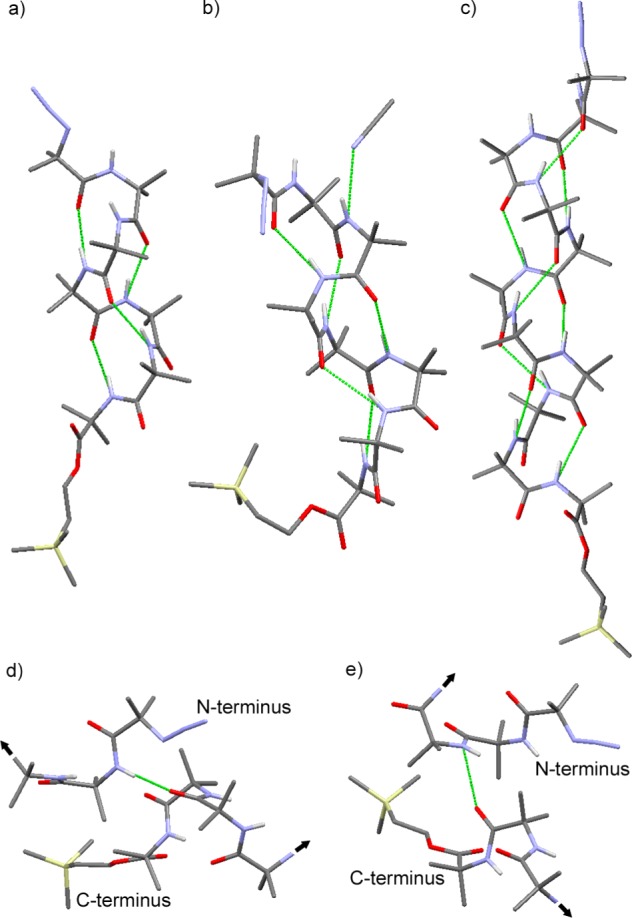
X-ray crystal structures of (a) N3Aib7OCH2CH2TMS 3, (b) N3Aib8OCH2CH2TMS 4 with CH3CN, and (c) N3Aib11OCH2CH2TMS 7. Foldamer–foldamer interactions of N3Aib7OCH2CH2TMS 3 in the solid state, involving hydrogen bonds from (d) the first and (e) the second N-terminal Aib residue to the C-terminus of a neighboring foldamer. Selected hydrogen bonds are shown in green; black arrows indicate the continuation of the foldamer chains.
The extended conformations adopted by 3 and 7 gave head-to-tail distances (CH3 to N3) of 2.25 and 2.93 nm. These values compare well to anticipated lengths for these compounds, calculated as 2.24 and 3.01 nm, respectively, using 0.194 nm per Aib in a 310 helix, with 0.24 nm added for the azido group and 0.64 nm added for the OCH2CH2SiMe3 group. All compounds were found to pack in offset head-to-tail arrangements (see Supporting Information).25,28 The unit cell also revealed side-to-side packing of the helices, a geometry important for the formation of membrane-spanning pores/channels. Heptamer 3 contained head-to-tail intermolecular hydrogen bonds between foldamers involving the NH of the first N-terminal Aib and carbonyl of the penultimate amide link at the C-terminus (NH···O distance 3.038 Å, Figure 2d) and the NH of the second N-terminal Aib and the final amide link at the C-terminus (NH···O distance 2.941 Å, Figure 2e). Octamer 4 only showed intermolecular hydrogen bonding to acetonitrile of crystallization (Figure 2b, NH···N distance 3.067 Å), and unlike 3 and 7 both the azido group and CH2CH2TMS tail folded back onto the helix. Undecamer 7 showed intermolecular hydrogen bonds analogous to those observed for 3, involving the first and second N-terminal Aib residues and the C-terminus of a neighboring foldamer (i.e., NH···O distances 2.999 and 3.130 Å, respectively).
Foldamer Self-Association
The self-association of Aib oligomers in CDCl3 may be quantified by fitting the concentration dependence of NH chemical shifts to either dimerization or isodesmic self-association (with equal K values) models.29 Such self-association in nonpolar environments is a key aspect of most proposed mechanisms of action of peptaibols. However, we and others have found that aggregates larger than dimers do not form to a significant extent for similar Aib oligomers in the millimolar concentration range in chloroform.30 After fitting data to a dimerization model, we have found that longer Aib foldamers self-associate more strongly in nonpolar solvents, as well as displaying lower solubility.25
As found during this previous work, compounds 1–9 showed concentration-dependent NH chemical shifts in CDCl3 but not in CD3OD or other hydrogen bond donor/acceptor solvents.25 These shifts in CDCl3, a solvent that replicates the low polarity at the center of the bilayer,31 allowed the dimerization constant K of 1, 2, 4, 7–9 to be quantified by 1H NMR spectroscopy (Table 1, corresponding dissociation constants, Kd, and curve fitting errors also shown). The NH resonances of these foldamers were monitored as the solutions were diluted, with marked changes in chemical shift observed for some NHs in the longer foldamers even at relatively low concentrations. For example, a 0.4 ppm shift for the N-terminal NH of Aib117 was measured upon dilution from 40 to 1 mM, compared to a 0.05 ppm shift for the N-terminal NH proton of Aib51 over the same concentration range. From these data the dimerization constant for each compound was calculated by standard iterative curve fitting using different minimization algorithms (see the Supporting Information).25
Table 1. Experimental NMR Dimerization Constants K (Dissociation Constants, Kd) for Foldamers 1, 2, 4, 7–9 in CDCl3 at 298 K.
| oligomer | K/M–1 (Kd/mM) |
|---|---|
| Aib51 | <1 (>1000) |
| Aib62 | 1.2 ± 0.2 (830 ± 140) |
| Aib84 | 3.0 ± 1.3 (330 ± 140) |
| Aib11 7 | 17.3 ± 3.1 (58 ± 10) |
| Aib12 8 | 13.6 ± 0.6 (74 ± 3) |
| Aib13 9 | 16.9 ± 0.4 (59 ± 1) |
The dimerization constants were <3 M–1 for the shorter foldamers 1, 2, and 4, but 14–17 M–1 for the longer foldamers 7–9. These values, equivalent to ΔG = −7 kJ mol–1 for dimerization of Aib139,32 are consistent with previous findings that Aib foldamers dimerize more strongly in chloroform as oligomer length increases.25,30 Although these dimerization constants are low, partitioning into the membrane can lead to very large effective concentrations, as the volume of the membrane is much less than the total volume of the sample.33 For instance, 1 mol % foldamer in the membrane is equivalent to ∼8 mM foldamer within the volume occupied by the bilayer. At these “in membrane” concentrations, an increase in dimerization constant from 3 to 17 M–1 (foldamers Aib84 and Aib139, respectively) would give an approximately proportionate increase in dimerization, with a simple model giving 4.4% of Aib84 present as dimer at 1 mol % foldamer incorporation, whereas 18.2% of Aib139 is present as the dimer at the same membrane loading (see the Supporting Information).
8-Hydroxypyrenetrisulfonate (HPTS) Assays of Ionophoric Activity
Foldamers 1–9 were assessed for either Na+/H+ antiport or Na+/OH– symport by means of standard HPTS assays,34 using 1:4 cholesterol:egg yolk phosphatidylcholine (EYPC) vesicles with an interior pH of 7.4 and an exterior pH of 8.4.35 Aliquots of foldamers Aib51 to Aib139 in methanol were equilibrated with suspensions of large unilamellar vesicles (LUVs) for 180 s, before the addition of the base pulse. The change in HPTS fluorescence was measured for 27 min, followed by addition of Triton X-100 to lyse the vesicles and obtain the maximum change in HPTS fluorescence for data normalization (Figure 3 a).
Figure 3.
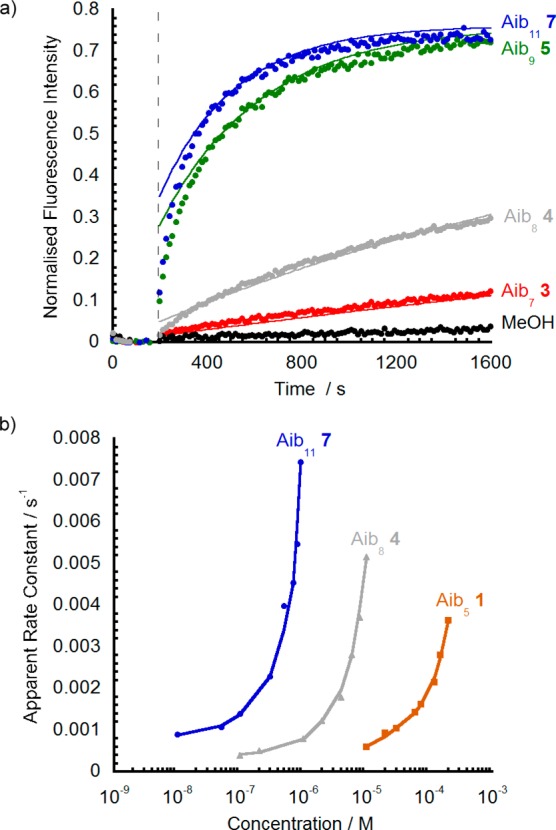
(a) HPTS fluorescence changes for foldamers 7 (Aib11, blue), 5 (Aib9, green), 4 (Aib8, gray), and 3 (Aib7, red) and methanol blank (black). Additions of foldamer solution in methanol (20 μL, 60 μM) to LUVs (20% cholesterol/EYPC, 0.76 mM lipid, 2 mL). Base pulse of NaOH (13 μL, 1 M) at 180 s (dashed line). Data normalized to maximum fluorescence after addition of Triton X-100 [10% v/v in MOPS buffer (MOPS = 3-(N-morpholino)propanesulfonic acid)] at 1800 s. (b) Plot of [foldamer] (log scale) vs pseudo-first-order rate constants of ion leakage from LUVs for foldamers 7 (Aib11, blue), 4 (Aib8, gray), and 1 (Aib5, brown). Curve fits to guide the eye.
These HPTS fluorescence assays showed an initial “burst” of activity (the “burst phase”), due to rapid ion transport through pores/channels that formed in the vesicles before the addition of the base pulse,36 followed by slower pH discharge over the next 1620 s. Foldamers 1–9 could all discharge the transmembrane pH gradient, albeit with strong concentration dependence (data for 1, 4, and 7 shown in Figure 3 b). For the shorter Aibm foldamers (m = 5, 6, 7, 8), a high concentration of 10 μM was required for activity, whereas for the longer Aib m-mers (m = 9, 10, 11, 12, 13) a 0.6 μM concentration was sufficient to show high activity. As these data suggest, the ionophoric activity of these oligomers was markedly length-dependent, with longer molecules significantly more active. For example, 0.08 mol % of undecamer 7 (0.6 μM of Aib11) was able to discharge 70% of the pH gradient within 10 min, but 0.08 mol % heptamer 3 (0.6 μM of Aib7) only discharged 7% over the same time period (Figure 3 a). Shorter oligomers such as pentamer Aib51 only discharged most of the pH gradient at concentrations >200 μM (>26 mol %, see the Supporting Information).
The slower change in fluorescence after the “burst phase” encompasses several molecular level events, including the rate of intervesicle transfer of foldamers.36 Nonetheless, to gauge the relative effectiveness of the compounds as ionophores, all the data was fitted to first-order reaction kinetics as an approximation; this provided consistent observed rate constant values within an error of 5%. Control experiments using methanol only were subtracted from the experimental data to correct for leakage caused by the solvent.
These observed rate constants (kobs, Figure 4) confirmed that the longer oligomers, such as Aib117 (kobs = (3.3 ± 0.4) × 10–3 s–1), show very powerful activity. Under the same conditions (0.6 μM peptide in EYPC/cholesterol), an HPTS assay showed the natural Aib-containing peptaibol alamethicin had kobs = (2.7 ± 0.2) × 10–3 s–1, similar to that of foldamer Aib9, 5 (kobs = (2.4 ± 0.3) × 10–3 s–1), and smaller than that of dodecamer Aib128 (kobs = (5.0 ± 0.2) × 10–3 s–1). With a reported37 hydrophobic width of EYPC/cholesterol bilayers in the region of 2.8 nm, the length mismatch between the foldamer and bilayer decreases from 1 to 9 (1.8 to 3.4 nm), with the 310-helical decamer Aib106 closest in length to the hydrophobic length estimated for α-helical 19-residue alamethicin (ca. 2.8 nm).13,14
Figure 4.

Plot of apparent first-order rate constants for Na+ transport vs number of Aib residues for foldamers (a) Aib51 to Aib84 at 10 μM, and (b) Aib84 to Aib139 at 0.6 μM. Additions of foldamer stock solution in methanol (20 μL) to 0.76 mM LUVs (20% cholesterol/EYPC, 2 mL). (c) Estimation of relative rate constants with foldamer length, at 10 μM (1−4) and 0.6 μM (4−9), normalized to relative rate of Aib84 at both concentrations (krel = 1).
The activities of the shorter foldamers 1–3 could not be distinguished from the background level at the lower 0.6 μM concentration, but their activities could be differentiated at 10 μM. The octamer Aib84 was then analyzed at both 10 and 0.6 μM to bridge between the two data sets. Setting the activity of the octamer as 1.0 allowed the activity of all the other Aibm foldamers to be approximately ranked (Figure 4).38 The relative activities were calculated to be 0.04 (m = 5, 10 μM), 0.16 (m = 6, 10 μM), 0.28 (m = 7, 10 μM), 1.0 (m = 8), 9 (m = 9), 14 (m = 10), 11 (m = 11), 17 (m = 12), 8 (m = 13), revealing an approximately 400-fold difference in relative activity between the most and least active foldamers. The sharpest increase in activity (9-fold) occurs for the change from octamer to nonamer. This suggests that the length of Aib95 (estimated as 2.62 nm, calculated as described earlier) represents the minimum length able to span the hydrophobic core of the bilayer (ca. 2.8 nm for EYPC, see the Supporting Information for a representation of 310-helical Aib117 in an idealized bilayer).39 Beyond foldamer Aib106, there appears to be a leveling off or even a diminution of activity with further increases in length. Interestingly it is clear that there is no sudden appearance of ionophoric activity when these incremental changes in foldamer length produce compounds long enough to span the bilayer. The activity of the shorter foldamers and the absence of a step-change in ionophoric activity at a certain length of foldamer suggest the mechanism for foldamer activity may be more nuanced than simply the formation of the “barrel-stave” ion channels suggested to explain the activity of alamethicin.11,12
The HPTS assay cannot easily discriminate between ion channel/pore,11,12 membrane disruption,40 and ion carrier mechanisms,41 all of which have been suggested to occur for the structurally related peptaibols. An ion carrier mechanism for Na+ transport by this family of foldamers was discounted as no activity was observed over 20 h for 1, 7, and 8 in simple U-tube assays (see the Supporting Information). To test for membrane disruption, the dye 5/6-carboxyfluorescein (5/6-CF) was encapsulated within vesicles at self-quenching concentrations. If an added compound disrupts the bilayer and/or forms pores greater than the size of 5/6-CF (ca. 10 Å for 5/6-CF42 compared to ca. 2 Å for sodium43), the dye will escape from the vesicle lumen and give a recovery in emission at 517 nm (λex 492 nm). As with the HPTS assay, the addition of Triton X-100 releases all entrapped 5/6-CF and allows normalization of the data. When Aib foldamers Aib73, Aib117, and Aib139 were tested for 5/6-CF release from EYPC/cholesterol vesicles, it was clear that very little dye release occurred; foldamers 3, 7, and 9 caused only 3%, 3%, and 5% leakage of 5/6-CF over 1200 s at concentrations of 10, 0.6, and 0.6 μM, respectively. The same compounds gave ionophoric activities of 60–75% at the same concentrations under analogous conditions (Figure 5). This observation shows that membrane disruption by these compounds is not significant under these conditions and suggests the majority of ion channels or pores formed by 3, 7, and 9 at these membrane loadings are not large enough to allow 5/6-CF release.44
Figure 5.
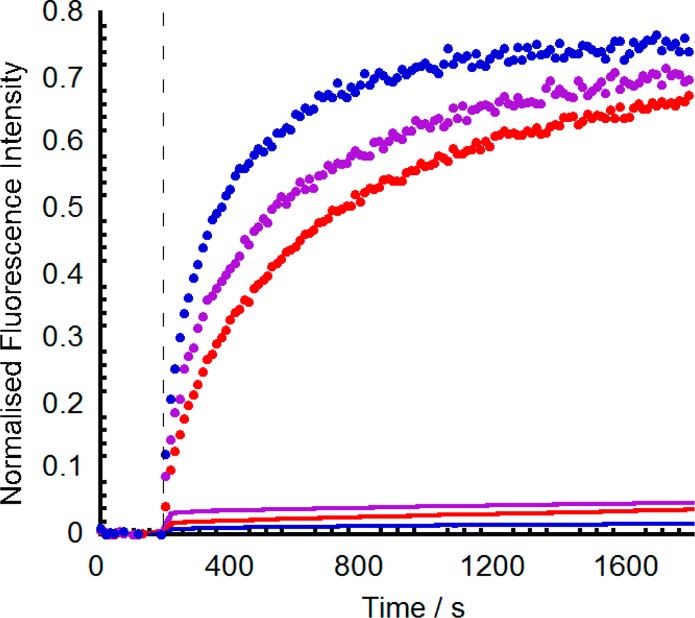
Co-plot of normalized HPTS ion transport data (●) and 5/6-CF release data (—) for 3 (Aib7, 10 μM, red), 7 (Aib11, 0.6 μM, blue), and 9 (Aib13, 0.6 μM purple). Additions of foldamer solution in methanol to LUVs (20% cholesterol/EYPC, 0.76 mM lipid, 2 mL) at 0 s (HPTS data, base pulse at 180 s) or 180 s (5/6-CF data).
Planar Bilayer Conductance (PBC) Assays of Ionophoric Activity
PBC measurements allow ionophoric activity due to pores and/or channels to be identified and the stability of these conducting structures to be assessed, although the mechanism of ion conductance in HPTS vesicle experiments is not necessarily the same under the applied potential used in PBC measurements.45 After application of a potential across the bilayer, if channels or pores form then sudden and intermittent increases in current occur, the length of which correlate to the kinetic stability of an “open” channel.46 Such voltage-clamp techniques can be used to observe single channel activities, multiple conductance states for self-assembled channels, and ensemble conductance from multiple channels.47
Selected foldamers 3, 4, 7, 9 (N3AibmOCH2CH2TMS, m = 7, 8, 11, 13) were assessed for their ability to conduct ions through planar phospholipid bilayer membranes composed of 1:4 cholesterol:EYPC. The shorter foldamers Aib73 and Aib84 at concentrations of 2.5 or 5 μM generally produced irregular and spiky conductances in the current–voltage sweeps (I–V sweeps from +100 mV to −100 mV), which indicates transient membrane disruption by the peptides. At higher concentrations the bilayers became unstable; e.g., 10 μM Aib84 gave multilevel activity after ∼0.5 h at high positive or high negative applied potential, but the shape of the I–V curves suggests progressive membrane weakening until eventually the membranes broke (see Supporting Information). In contrast, current–voltage sweeps from −100 to +100 mV for compounds Aib117 and Aib139 at 0.6 μM displayed a range of ensemble conductances. The flux that passed was seen to gradually increase until a maximum conductance was reached after ca. 2.5 h, after which time the conductance reached an equilibrium value between 50 and 80 pA/0.5–0.8 nS at an applied voltage of 100 mV (see Supporting Information).
At a set voltage, both 7 and 9 gave intermittent increases in current over time (Figure 6a–c). Both compounds exhibited multiple conductance states and maintained this reproducible behavior for at least 16 h, after which time the bilayer was still intact. Only a few seconds were required for this channel/pore behavior to start appearing, indicating fast diffusion into and across the bilayer. Classical “square-top” traces were not observed, with the traces instead showing flickering conductance events, with poorly defined current levels and short transition lifetimes. Integer steps in the conductance levels were not observed, suggesting that multiple openings of the same type of pore do not occur. Instead, the different conductance states, which range from 1.3 to 1.5 nS (Figure 6a–c), may result from pores with differing molecularities and correspondingly different conductances, similar in behavior to the amphiphilic heptapeptides reported by Ferdani and Gokel.48 This voltage-dependent formation of short-lived, multilevel pores/ion channels is also characteristic for some neutral and many cationic toroidal or α-barrel-forming peptides like melittin and oligo(Ala-Aib-Ala-Aib-Ala).49 Synthetic ion channels typically give conductances between 1 and 100 pS, whereas pores give conductances between 0.1 and 5 nS,50 suggesting that foldamers 7 and 9 form pore-like structures. Very low foldamer concentrations (5–50 nM) were assessed for channel formation using droplet–interface bilayers (DIBs, see the Supporting Information).51 However, DIB studies on 2 (6-mer) and 7 (11-mer) did not indicate that defects of a predefined radius were formed, but at these very low concentrations the foldamers induced points of weakness that nucleated electropores.52
Figure 6.
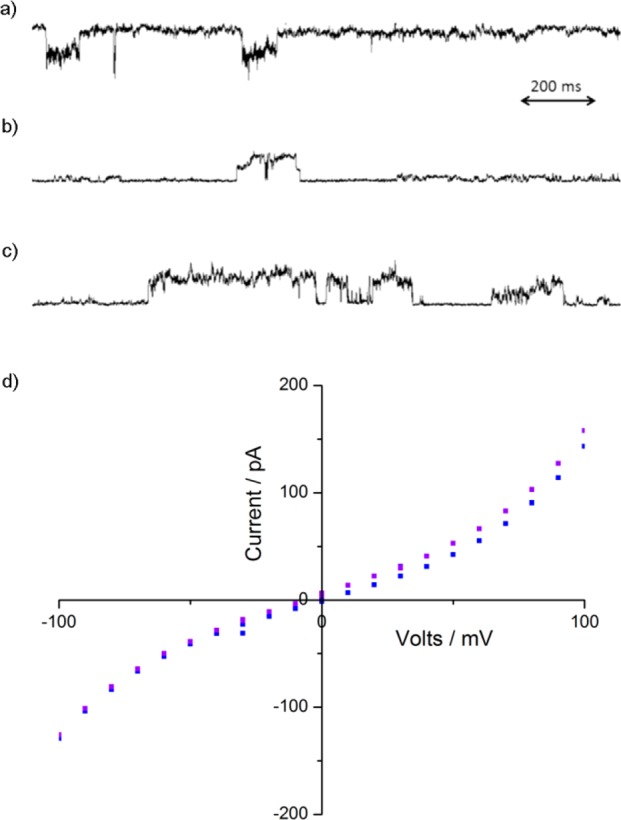
Typical planar bilayer conductance behavior for 7 and 9. (a) 7 (Aib11, 0.6 μM), applied voltage −100 mV, current passed −150 pA; 1.5 nS. (b) 9 (Aib13, 0.6 μM), applied voltage +100 mV, current passed +145 pA, with smaller conducting state also observed at +130 pA; 1.45 or 1.3 nS, respectively. (c) 9 (Aib13), 0.6 μM concentration, applied voltage +100 mV, current passed +145 pA; 1.45 pS. (d) Example I–V traces: 9 (Aib13, purple) and 7 (Aib11, blue), traces approximately 1 h after addition of compound.
The current–voltage sweeps for Aib117 and Aib139 at 0.6 μM showed a nonlinear relationship between current and voltage, with higher conductance at high voltages (Figure 6 d), nonohmic behavior similar to that found for other antimicrobial peptides including alamethicin.12b,15 The increase in conductance at high voltages could be due to a reduction in the energy barrier to pore formation,52 or the interaction of the stronger applied field with the foldamer dipole causing more transmembrane geometries.53 The I–V curves also show asymmetric conductance behavior (see the Supporting Information), behavior that is different from that observed for symmetric pores based on β-sheets (where the opposing backbone orientations produce no net macrodipole) and dipole-free octiphenyl rigid-rod channels.49,54 This behavior suggests a lack of symmetry when interacting with the membrane. Peptide foldamers, such as Aib117 and Aib139, have different termini and intrinsic end-to-end asymmetry that produces a significant dipole moment.55 Since these compounds are added solely to the cis side of the PBC bilayer, easier insertion of the small azido group into the bilayer over the bulky TMS group would produce an asymmetric foldamer alignment in the bilayer, which may either match or oppose the changing applied voltage.56
Conclusions
Simple and versatile synthetic methodology has given access to a homologous family of functionalizable 310-helical Aib foldamers containing from 5 to 13 Aib residues. All members were active as ionophores, but only foldamers that are able to form 310 helices longer than the thickness of the hydrophobic core of the bilayer gave high conductance pores. Aib foldamers that were too short to span the membrane were much less active. They may act through an “amyloid-like” mechanism that has been suggested for other short Aib foldamers,57 where at high concentrations the foldamers assemble in or around the membrane surface, porating and weakening the bilayer. The longer foldamers were remarkably effective ionophores. In the HPTS assay the observed rate constant for transport by dodecamer Aib128 was twice that of the naturally occurring 19-residue antibiotic alamethicin.
Many natural and artificial channel/pore formers are believed to achieve activity by self-associating in the membrane, with “barrel-stave”, “carpet”, and “toroidal-pore”6 mechanisms invoking the formation of defined or ill-defined aggregates. We found that increasing the length of Aib foldamers increased the strength of self-association in nonpolar environments, a principle that may extend to other peptides or foldamers in bilayers. However, the increase in ionophoric activity with length was stronger than the increase in self-association with length; for example, Aib117 was 91 times more membrane-active than Aib62 and 12 times more active than Aib84, whereas K for Aib117 was only 14 times greater than K for Aib62 and 5.8 times greater than K for Aib84. These data suggest a key factor for good ionophoric activity is the ability to achieve a membrane-spanning conformation. Ionophoric activity was similar for foldamers with m = 10 and 13 (lengths from ∼2.8 to ∼3.4 nm), implying that 2.8 nm is the ideal length for these foldamers to span EYPC/cholesterol bilayers (∼2.8 nm hydrophobic width) and further increases in foldamer length have little influence on activity.
The potent ionophoric activity of the longer foldamers suggests they may have rich potential in new generation antibiotics. Much like the Aib-rich peptaibols themselves, they should be resistant to proteases, yet the azido and CH2CH2TMS protecting groups allow simple synthetic modification of the N- and/or C-termini. Simple modifications such as adding chiral28,58 or hydrogen bonding groups to the termini could increase activity by strengthening aggregation in the bilayer.25 Alternatively, specific cell-targeting groups, such as saccharides or biotin, could be added.35,59
The membrane activity reported here for conformationally defined Aib foldamers gives a hint of the wider potential of these nanoscale building blocks. These studies have shown that an Aibm (m = 10–12) core is sufficient achieve a membrane-spanning geometry. Since both the N- and C-termini can be easily modified, these helical transmembrane units could therefore be used as scaffolds for the construction of functional GPCR mimics. For example, foldamers 6–9 exist as racemic mixtures of interconverting right- and left-handed helices, but controlling the helical screw-sense distribution can provide a means of relaying information along the helix.60 Given that, noncovalent reversible switching of chiral control should lead to end-to-end transmission of conformational change, and ultimately information communication across a bilayer.
Acknowledgments
We gratefully acknowledge Dr. J. Sanderson for the “NMR dilution fit” Excel spreadsheet used for dimerization constant determination. This work was supported by the BBSRC (Grant I007962), ESPRC (Grants EP/N009134/1 and EP/K039547), and the ERC (Advanced Grant ROCOCO).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b12057. Additional research data supporting this publication are available from the eScholar repository at DOI: 10.15127/1.294101.
Author Contributions
J.E.J. and V.D. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Nat. Chem. 2013, 5, 161–173. 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Murphy J. E.; Uno T.; Hamer J. D.; Cohen F. E.; Dwarki V.; Zuckermann R. N. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 1517–1522. 10.1073/pnas.95.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bautista A. D.; Craig C. J.; Harker E. A.; Schepartz A. Curr. Opin. Chem. Biol. 2007, 11, 685–692. 10.1016/j.cbpa.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hennig A.; Gabriel G. J.; Tew G. N.; Matile S. J. Am. Chem. Soc. 2008, 130, 10338–10344. 10.1021/ja802587j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Claudon P.; Violette A.; Lamour K.; Decossas M.; Fournel S.; Heurtault B.; Godet J.; Mély Y.; Jamart-Grégoire B.; Averlant-Petit M. C.; Briand J. P.; Duportail G.; Monteil H.; Guichard G. Angew. Chem., Int. Ed. 2010, 49, 333–336. 10.1002/anie.200905591. [DOI] [PubMed] [Google Scholar]; b Tew G. N.; Scott R. W.; Klein M. L.; DeGrado W. F. Acc. Chem. Res. 2010, 43, 30–39. 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schmitt M. A.; Weisblum B.; Gellman S. H. J. Am. Chem. Soc. 2004, 126, 6848–6849. 10.1021/ja048546z. [DOI] [PubMed] [Google Scholar]; d Chongsiriwatana N. P.; Patch J. A.; Czyzewski A. M.; Dohm M. T.; Ivankin A.; Gidalevitz D.; Zuckermann R. N.; Barron A. E. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2794–2799. 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenawy E. R.; Worley S. D.; Broughton R. Biomacromolecules 2007, 8, 1359–1384. 10.1021/bm061150q. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. Nat. Rev. Microbiol. 2005, 3, 238–250. 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- a Hamuro Y.; Schneider J. P.; DeGrado W. F. J. Am. Chem. Soc. 1999, 121, 12200–12201. 10.1021/ja992728p. [DOI] [Google Scholar]; b Tew G. N.; Liu D.; Chen B.; Doerksen R. J.; Kaplan J.; Carroll P. J.; Klein M. L.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5110–5114. 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu D.; Choi S.; Chen B.; Doerksen R. K.; Clements D. J.; Winkler J. D.; Klein M. L.; DeGrado W. F. Angew. Chem., Int. Ed. 2004, 43, 1158–1158. 10.1002/anie.200352791. [DOI] [PubMed] [Google Scholar]; d Choi S.; Isaacs A.; Clements D.; Liu D.; Kim H.; Scott R. W.; Winkler J. D.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6968–6973. 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mensa B.; Kim Y. H.; Choi S.; Scott R.; Caputo G. A.; DeGrado W. F. Antimicrob. Agents Chemother. 2011, 55, 5043–5053. 10.1128/AAC.05009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tew G. N.; Clements D.; Tang H.; Arnt L.; Scott R. W. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1387–1392. 10.1016/j.bbamem.2006.03.001. [DOI] [PubMed] [Google Scholar]; g Chen X.; Tang H.; Even M. A.; Wang J.; Tew G. N.; Chen Z. J. Am. Chem. Soc. 2006, 128, 2711–2714. 10.1021/ja057029t. [DOI] [PubMed] [Google Scholar]; h Arnt L.; Rennie J. R.; Linser S.; Willumeit R.; Tew G. N. J. Phys. Chem. B 2006, 110, 3527–3532. 10.1021/jp054339p. [DOI] [PubMed] [Google Scholar]; i Epand R. F.; Raguse T. L.; Gellman S. H.; Epand R. M. Biochemistry 2004, 43, 9527–9535. 10.1021/bi049414l. [DOI] [PubMed] [Google Scholar]; j Epand R. F.; Schmitt M. A.; Gellman S. H.; Epand R. M. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1343–1350. 10.1016/j.bbamem.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Peptaibols; Toniolo C., Brückner H., Eds.; Wiley-VCH: Weinheim, 2009. [Google Scholar]
- Marshall G. R.; Hodgkin E. E.; Langs D. A.; Smith G. D.; Zabrocki J.; Leplawy M. T. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 487–491. 10.1073/pnas.87.1.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sansom M. S. P. Q. Rev. Biophys. 1993, 26, 365–421. 10.1017/S0033583500002833. [DOI] [PubMed] [Google Scholar]; b Aravinda S.; Shamala N.; Balaram P. Chem. Biodiversity 2008, 5, 1238–1262. 10.1002/cbdv.200890112. [DOI] [PubMed] [Google Scholar]
- Qian S.; Wang W.; Yang L.; Huang H. W. Biophys. J. 2008, 94, 3512–3522. 10.1529/biophysj.107.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Futaki S.; Noshiro D.; Kiwada T.; Asami K. Acc. Chem. Res. 2013, 46, 2924–2933. 10.1021/ar400051f. [DOI] [PubMed] [Google Scholar]; b Cafiso D. S. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 141–165. 10.1146/annurev.bb.23.060194.001041. [DOI] [PubMed] [Google Scholar]
- a Nagao T.; Mishima D.; Javkhlantugs N.; Wang J.; Ishioka D.; Yokota K.; Norisada K.; Kawamura I.; Ueda K.; Naito A. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 2789–2798. 10.1016/j.bbamem.2015.07.019. [DOI] [PubMed] [Google Scholar]; b Fox R. O.; Richards F. M. Nature 1982, 300, 325–330. 10.1038/300325a0. [DOI] [PubMed] [Google Scholar]
- Marsh D.; Jost M.; Peggion C.; Toniolo C. Biophys. J. 2007, 92, 4002–4011. 10.1529/biophysj.107.104026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriss L. M.; Cronin B.; Thompson J. R.; Wallace M. I. J. Am. Chem. Soc. 2011, 133, 14507–14509. 10.1021/ja204275t. [DOI] [PubMed] [Google Scholar]
- Bobone S.; Gerelli Y.; De Zotti M.; Bocchinfuso G.; Farrotti A.; Orioni B.; Sebastiani F.; Latter E.; Penfold J.; Senesi R.; Formaggio F.; Palleschi A.; Toniolo C.; Fragneto G.; Stella L. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1013–1024. 10.1016/j.bbamem.2012.11.033. [DOI] [PubMed] [Google Scholar]
- Gatto E.; Bocchinfuso G.; Palleschi A.; Oncea A.; De Zotti M.; Formaggio F.; Toniolo C.; Venanzi M. Chem. Biodiversity 2013, 10, 887–903. 10.1002/cbdv.201200388. [DOI] [PubMed] [Google Scholar]
- Crisma M.; Barazza A.; Formaggio F.; Kaptein B.; Broxterman Q. B.; Kamphuis J.; Toniolo C. Tetrahedron 2001, 57, 2813–2825. 10.1016/S0040-4020(01)00124-7. [DOI] [Google Scholar]
- Grigoriev P. A.; Schlegel B.; Kronen M.; Berg A.; Härtl A.; Grafe U. J. Pept. Sci. 2003, 9, 763–768. 10.1002/psc.502. [DOI] [PubMed] [Google Scholar]
- Oancea S.; Hilma G.; Peggion C.; Formaggio F.; Toniolo C. Chem. Biodiversity 2008, 5, 681–692. 10.1002/cbdv.200890065. [DOI] [PubMed] [Google Scholar]
- Sakai N.; Brennan K. C.; Weiss L. A.; Matile S. J. Am. Chem. Soc. 1997, 119, 8726–8727. 10.1021/ja971513h. [DOI] [Google Scholar]
- Murray C. L.; Gokel G. W. Chem. Commun. 1998, 2477–2478. 10.1039/a806317f. [DOI] [Google Scholar]
- Leevy W. M.; Donato G. M.; Ferdani R.; Goldman W. E.; Schlesinger P. H.; Gokel G. W. J. Am. Chem. Soc. 2002, 124, 9022–9023. 10.1021/ja017052o. [DOI] [PubMed] [Google Scholar]
- Hummel R.-P.; Toniolo C.; Jung G. Angew. Chem., Int. Ed. Engl. 1987, 26, 1150–1152. 10.1002/anie.198711501. [DOI] [Google Scholar]
- Pike S. J.; Diemer V.; Raftery J.; Webb S. J.; Clayden J. Chem. - Eur. J. 2014, 20, 15981–15990. 10.1002/chem.201403626. [DOI] [PubMed] [Google Scholar]
- Gessmann R.; Brückner H.; Petratos K. J. Pept. Sci. 2003, 9, 753–762. 10.1002/psc.490. [DOI] [PubMed] [Google Scholar]
- cLog P values have been calculated as 1.94 and 0.52 for N3Aib5O(CH2)2TMS and N3Aib13O(CH2)2TMS, respectively, using OSIRIS Property Explorer.
- a Solà J.; Helliwell M.; Clayden J. Biopolymers 2011, 95, 62–69. 10.1002/bip.21535. [DOI] [PubMed] [Google Scholar]; b Pike S. J.; Raftery J.; Webb S. J.; Clayden J. Org. Biomol. Chem. 2014, 12, 4124–4131. 10.1039/C4OB00268G. [DOI] [PubMed] [Google Scholar]
- The value of Kisodesmic is twice that of Kdimerization. See:Neumann B.; Huber K.; Pollmann P. Phys. Chem. Chem. Phys. 2000, 2, 3687–3695. 10.1039/b004172f. [DOI] [Google Scholar]
- Wilkening R. R.; Stevens E. S.; Bonora G. M.; Toniolo C. J. Am. Chem. Soc. 1983, 105, 2560–2561. 10.1021/ja00347a007. [DOI] [Google Scholar]
- Epand R. M.; Kraayenhof R. Chem. Phys. Lipids 1999, 101, 57–64. 10.1016/S0009-3084(99)00055-9. [DOI] [PubMed] [Google Scholar]
- This free energy for dimerization is similar to that reported for PG1, an octadecameric peptide, in POPC bilayers. See:Buffy J. J.; Waring A. J.; Hong M. J. Am. Chem. Soc. 2005, 127, 4477–4483. 10.1021/ja043621r. [DOI] [PubMed] [Google Scholar]
- Doyle E. L.; Hunter C. A.; Phillips H. C.; Webb S. J.; Williams N. H. J. Am. Chem. Soc. 2003, 125, 4593–4599. 10.1021/ja021048a. [DOI] [PubMed] [Google Scholar]
- Kano K.; Fendler J. H. Biochim. Biophys. Acta, Biomembr. 1978, 509, 289–299. 10.1016/0005-2736(78)90048-2. [DOI] [PubMed] [Google Scholar]
- Wilson C. P.; Boglio C.; Ma L.; Cockroft S. L.; Webb S. J. Chem. - Eur. J. 2011, 17, 3465–3473. 10.1002/chem.201002031. [DOI] [PubMed] [Google Scholar]
- Gorteau V.; Bollot G.; Mareda J.; Matile S. Org. Biomol. Chem. 2007, 5, 3000–3012. 10.1039/b708337h. [DOI] [PubMed] [Google Scholar]
- The thickness of EYPC is thought to be similar to that of DOPC, which has a hydrocarbon core thickness of 2.68 nm. Addition of 20 mol % cholesterol should thicken the bilayers by an estimated 5%. See ref (21) and; a Kučerka N.; Tristram-Nagle S.; Nagle J. F. J. Membr. Biol. 2006, 208, 193–202. 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]; b Gallová J.; Uhríková D.; Kučerka N.; Svorková M.; Funari S. S.; Murugova T. N.; Almásy L.; Mazúr M.; Balgavý P. J. Membr. Biol. 2011, 243, 1–13. 10.1007/s00232-011-9387-1. [DOI] [PubMed] [Google Scholar]
- A low background value of 0.004 s–1, which was not due to methanol, was apparent when fitting the rate data for foldamers 1, 4, and 7 at different concentrations. Above this value, the observed rate constants are approximately proportional to concentration (see the Supporting Information).
- Altering the width of the bilayer using DLPC (∼2.1 nm hydrocarbon core thickness) did not provide useful information as these bilayers were too leaky.
- a Monaco V.; Formaggio F.; Crisma M.; Toniolo C.; Hanson P.; Millhauser G. L. Biopolymers 1999, 50, 239–253. . [DOI] [PubMed] [Google Scholar]; b Eid M.; Rippa S.; Castano S.; Desbat B.; Chopineau J.; Rossi C.; Béven L. J. Biophys. 2010, 2010, 179641. 10.1155/2010/179641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclohier H.; Snook C. F.; Wallace B. A. Biochim. Biophys. Acta, Biomembr. 1998, 1415, 255–260. 10.1016/S0005-2736(98)00184-9. [DOI] [PubMed] [Google Scholar]
- Pajewski R.; Ferdani R.; Pajewska J.; Djedovič N.; Schlesinger P. A.; Gokel G. W. Org. Biomol. Chem. 2005, 3, 619–625. 10.1039/b417009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Lu X.; Wang Y.; Shi Fluid Phase Equilib. 2002, 194–197, 257–27. 10.1016/S0378-3812(01)00694-X. [DOI] [Google Scholar]
- 9.3 μM alamethicin can release 50% of 5/6-CF encapsulated in 1:1 EYPC:cholesterol vesicles after 20 min, although at a much higher membrane loading of 47 mol %. See:Stella L.; Burattini M.; Mazzuca C.; Palleschi A.; Venanzi M.; Coin I.; Peggion C.; Toniolo C.; Pispisa B. Chem. Biodiversity 2007, 4, 1299–1312. 10.1002/cbdv.200790111. [DOI] [PubMed] [Google Scholar]
- Fyles T. M. Chem. Soc. Rev. 2007, 36, 335–347. 10.1039/B603256G. [DOI] [PubMed] [Google Scholar]
- Andersen O. S.; Koeppe R. E.; Roux B.. Gramicidin Channels: Versatile Tools. In Biological Membrane Ion Channels; Chung S. H., Andersen O. S., Krishnamurty V., Eds.; Springer: New York, 2007; pp 36. [Google Scholar]
- Chui J. K. W.; Fyles T. M. Chem. Soc. Rev. 2012, 41, 148–175. 10.1039/C1CS15099E. [DOI] [PubMed] [Google Scholar]
- Ferdani R.; Gokel G. W. Org. Biomol. Chem. 2006, 4, 3746–3750. 10.1039/b609345k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N.; Houdebert D.; Matile S. Chem. - Eur. J. 2003, 9, 223–232. 10.1002/chem.200390016. [DOI] [PubMed] [Google Scholar]
- Sakai N.; Mareda J.; Matile S. Acc. Chem. Res. 2005, 38, 79–87. 10.1021/ar0400802. [DOI] [PubMed] [Google Scholar]
- a Bayley H.; Cronin B.; Heron A.; Holden M. A.; Hwang W. L.; Syeda R.; Thompson J.; Wallace M. Mol. BioSyst. 2008, 4, 1191–1208. 10.1039/b808893d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b 0.5 μM alamethicin has been successfully used in DIBs. See: Harriss L. M.; Cronin B.; Thompson J. R.; Wallace M. I. J. Am. Chem. Soc. 2011, 133, 14507–14509. 10.1021/ja204275t. [DOI] [PubMed] [Google Scholar]
- Heimburg T. Biophys. Chem. 2010, 150, 2–22. 10.1016/j.bpc.2010.02.018. [DOI] [PubMed] [Google Scholar]
- Duclohier H.; Wróblewski H. J. Membr. Biol. 2001, 184, 1–12. 10.1007/s00232-001-0077-2. [DOI] [PubMed] [Google Scholar]
- Thundimadathil J.; Roeske R. W.; Guo L. Biochem. Biophys. Res. Commun. 2005, 330, 585–590. 10.1016/j.bbrc.2005.02.184. [DOI] [PubMed] [Google Scholar]
- Rizzo V.; Schwarz G.; Voges K.-P.; Jung G. Eur. Biophys. J. 1985, 12, 67–73. 10.1007/BF00260429. [DOI] [PubMed] [Google Scholar]
- Fyles T. M.; Loock D.; Zhou X. Can. J. Chem. 1998, 76, 1015–1026. 10.1139/cjc-76-7-1015. [DOI] [Google Scholar]
- Pike S. J.; Jones J. E.; Raftery J.; Clayden J.; Webb S. J. Org. Biomol. Chem. 2015, 13, 9580–9584. 10.1039/C5OB01652E. [DOI] [PubMed] [Google Scholar]
- a Brown R. A.; Marcelli T.; De Poli M.; Solà J.; Clayden J. Angew. Chem., Int. Ed. 2012, 51, 1395–1399. 10.1002/anie.201107583. [DOI] [PubMed] [Google Scholar]; b Orcel U.; De Poli M.; De Zotti M.; Clayden J. Chem. - Eur. J. 2013, 19, 16357–16365. 10.1002/chem.201302648. [DOI] [PubMed] [Google Scholar]; c Pike S. J.; De Poli M.; Zawodny W.; Raftery J.; Webb S. J.; Clayden J. Org. Biomol. Chem. 2013, 11, 3168–3176. 10.1039/c3ob40463c. [DOI] [PubMed] [Google Scholar]; d De Poli M.; Byrne L.; Brown R. A.; Solà J.; Castellanos A.; Boddaert T.; Wechsel R.; Beadle J. D.; Clayden J. J. Org. Chem. 2014, 79, 4659–4675. 10.1021/jo500714b. [DOI] [PubMed] [Google Scholar]; e Byrne L.; Solà J.; Boddaert T.; Marcelli T.; Adams R. W.; Morris G. A.; Clayden J. Angew. Chem., Int. Ed. 2014, 53, 151–155. 10.1002/anie.201308264. [DOI] [PubMed] [Google Scholar]; f De Poli M.; Clayden J. Org. Biomol. Chem. 2014, 12, 836–843. 10.1039/C3OB42167H. [DOI] [PubMed] [Google Scholar]; g Le Bailly B. A. F.; Clayden J. Chem. Commun. 2014, 50, 7949–7952. 10.1039/c4cc03261f. [DOI] [PubMed] [Google Scholar]
- a Coxon T. P.; Fallows T. W.; Gough J. E.; Webb S. J. Org. Biomol. Chem. 2015, 13, 10751–10761. 10.1039/C5OB01591J. [DOI] [PubMed] [Google Scholar]; b de Cogan F.; Gough J. E.; Webb S. J. J. Mater. Sci.: Mater. Med. 2011, 22, 1045. 10.1007/s10856-011-4271-1. [DOI] [PubMed] [Google Scholar]
- Brown R.; Diemer V.; Webb S. J.; Clayden J. Nat. Chem. 2013, 5, 853–860. 10.1038/nchem.1747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.