

Abstract
Oxygen is an essential participant in the acid-base chemistry that takes place within many enzyme active sites, yet has remained virtually silent as a probe in NMR spectroscopy. Here, we demonstrate the first use of solution-state 17O quadrupole central transition NMR spectroscopy to characterize enzymatic intermediates under conditions of active catalysis. In the 143 kDa pyridoxal-5’-phosphate-dependent enzyme tryptophan synthase, reactions of the α-aminoacrylate intermediate with the nucleophiles indoline and 2-aminophenol correlate with an upfield shift of the substrate carboxylate oxygen resonances. First principles calculations suggest that the increased shieldings for these quinonoid intermediates result from the net increase in the charge density of the substrate-cofactor π bonding network, particularly at the adjacent alpha-carbon site.
Keywords: oxygen-17, NMR spectroscopy, proteins, pyridoxal-5'-phosphate, ligases
Enzymatic reactions
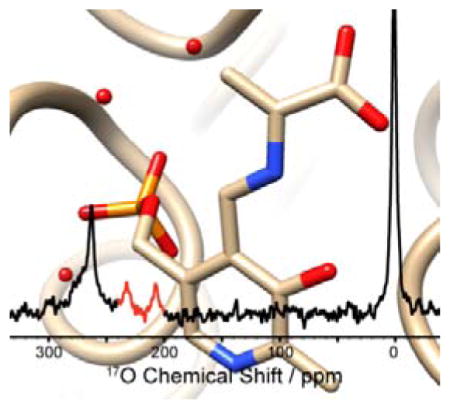
Several intermediates in the catalytic cycle of tryptophan synthase were characterized by 17O quadrupole central transition NMR spectroscopy. The picture shows the 17O solution-state NMR spectrum of the kinetically-competent α-aminoacrylate intermediate superimposed on the β-subunit active site.
Pyridoxal-5’-phosphate (PLP) plays a critical role as cofactor in a large family of enzymes involved in the metabolism of amino acids and other amine-containing biomolecules. PLP acts as an “electron sink,” stabilizing negative charge buildup at multiple steps during the catalytic cycle by means of a highly conjugated π-bond system.[1] A particularly remarkable aspect of PLP is that this single cofactor is capable of participating in a diverse array of chemical transformations, including racemization, transamination, decarboxylation, and β/γ-elimination and substitution.[1] Delineating the factors that fine-tune PLP for a particular reaction remains of great interest.[1–2] Studies of PLP in model compounds have demonstrated the important roles that protonation states and tautomerization play in directing reaction specificity.[3]
NMR is a powerful tool for probing tautomerization and acid-base chemistry at atomic resolution. Tracking the equilibrium involved in tautomerization can be accomplished by interrogating the heteronuclei involved in the proton exchange, yet to date this has largely been limited to 15N chemical shift measurements.[2, 4] Oxygen, the other key atomic species often involved in tautomerism, is not yet a standard nuclear probe in biological NMR spectroscopy, despite the potential wealth of chemical information it can provide. Here we report the first application of 17O quadrupole central transition (QCT) NMR to interrogate kinetically-competent species under conditions of active catalysis, probing two quasi-stable intermediates in the catalytic cycle of the 143 kDa, PLP-dependent enzyme tryptophan synthase (TS). This work highlights the promise of 17O QCT NMR to mechanistic enzymology.
17O is the only NMR active isotope of oxygen and possesses a quadrupolar, spin-5/2 nucleus with a gyromagnetic ratio comparable to 15N and low natural abundance (0.037%).[5] NMR studies of oxygen in biological macromolecules are rare and until recently had been demonstrated primarily in the solid-state.[6] 17O QCT NMR spectroscopy in solution takes advantage of the unique relaxation properties of the 17O central transition: in the limit of slow isotropic motion the linewidth narrows with increasing rotational correlation time – an unusual feature compared to spin-1/2 nuclei.[7] Applied initially by Lee and Oldfield to carbon monoxide bound to heme-proteins, 17O QCT NMR spectroscopy was recently demonstrated by Zhu and Wu to be applicable even when the quadrupolar coupling constant of the substrate is in the 5–10 MHz range typical of many organic compounds.[7a, 8] An additional feature of QCT NMR is that the peak position of the central transition is perturbed by a 2nd order magnetic-field dependent frequency shift, and to extract the isotropic chemical shift requires measurements at multiple magnetic fields.[9] The field-dependent line shape also allows for the extraction of quadrupole and CSA product parameters, PQ and PSA, that are similarly diagnostic of chemical state.[7a]
Protonation states and tautomerization in the active site of TS have been studied using 13C, 15N, and 31P solid-state NMR for a number of intermediates in the catalytic cycle.[4] TS catalyzes the final two steps in the biosynthesis of L-tryptophan (L-Trp); an abridged mechanism is shown in Scheme 1 and a full description of the chemical transformations it catalyses can be found in the SI. Quasi-stable analogues of the stage 2 quinonoid intermediate, E(Q3), can be formed by supplying the nucleophiles indoline and 2-aminophenol (2AP) in place of the natural substrate indole. These analogues are denoted E(Q3)indoline and E(Q3)2AP, respectively; the former goes on to form the unnatural amino acid dihydroiso-L-tryptophan (DIT), while the latter does not appear to react further.[10] The α-aminoacrylate intermediate, E(A-A), is relatively stable when no nucleophiles are supplied, and its lifetime can be further enhanced by the addition of the inhibitor benzimidazole (BZI), a non-reactive indole analogue.[10a]
Scheme 1.

An abridged catalytic cycle for the TS β-subunit reaction.
Initial SSNMR studies of the E(Q3)indoline intermediate allowed the conclusion to be drawn that an equilibrium involving proton exchange in the pocket adjacent to the Schiff base nitrogen and including the phenolic and nearest carboxylate oxygen is the best description of the chemical state (Scheme 2).[4a] This led to the hypothesis that the acid form of the substrate plays a catalytically-significant role. 17O QCT NMR spectroscopy provides a test of this hypothesis. By supplying the substrate L-serine enriched in 17O at the carboxylic site (synthesis and characterization described in the SI), we are able to explore protonation states in the two quinonoid analogues, E(Q3)indoline and E(Q3)2AP, and in the aminoacrylate intermediates, E(A-A) and E(A-A)(BZI).
Scheme 2.

Exchange between tautomeric forms of the indoline quinonoid intermediate.
Figure 1 shows 17O QCT spectra of the E(Q3)indoline intermediate formed with [17O]-L-Ser and measured at field strengths of 11.7, 14.1, and 16.4 T. The bound resonances are marked with asterisks, and the field-dependent frequency shift is clearly evident. Two resonances for the distinct carboxylic oxygen atoms can be seen at 16.4 and 14.1 T, yet overlap at 11.7 T. Global fitting of the three spectra allows for the extraction of the isotropic chemical shifts and the CSA and quadrupole product parameters for each oxygen site; these are summarized in Table 1. In comparison with shifts measured for amino acids in the solid state,[6] where carboxylates are observed between 250–290 ppm and the carbonyl and hydroxyl oxygens of the acid form range between 300–350 ppm and 165–190 ppm, respectively, the isotropic values of 233 ppm and 243 ppm observed in the E(Q3)indoline intermediate are surprising. To test whether these results are unique to E(Q3)indoline, experiments were carried out on E(Q3)2AP, an even longer-lived quinonoid form.[10b] The resulting spectra (shown in the Supporting Information) reveal two overlapped resonances at all fields. A global fit to the data set gives isotropic shifts of 237 ppm and 239 ppm, again below the carboxylate range. This intermediate gives data of sufficient quality that the PSA and rotational correlation time can be extracted with reasonable precision from the field-dependent line width. The correlation time of 182 ± 26 ns is consistent with estimates of the correlation time for a protein of this size at 3 ° C [11] and is used as a fixed parameter to extract PSA values for the other intermediates in Table 1.
Figure 1.

17O QCT NMR spectra of the E(Q3)indoline intermediate in TS at multiple magnetic fields. The enzyme-bound intermediates are marked with asterisks. Total number of transients and experiment times: at 11.7 T, 2.4 × 106 transients and 22 hours; at 14.1 T, 2.2 × 106 transients and 20 hours; at 16.4 T, 6.4 × 106 transients and 59 hours. The spectra are displayed with 100 Hz line broadening applied. Experiments were conducted at 3±2°C in pH 7.8 buffered aqueous solution. Additional experimental details are provided in the SI.
Table 1.
17O spectroscopic parameters for intermediates in TS
| δiso/ppm | PQ/MHz | PSA/ppm* | |
|---|---|---|---|
| E(Q3)indoline | |||
| O1 | 232.7 ± 0.6 | 7.40 ± 0.05 | 190 ± 26 |
| O2 | 242.7 ± 0.7 | 7.66 ± 0.05 | 226 ± 26 |
| E(Q3)2AP | |||
| O1 | 237.2 ± 0.5 | 7.37 ± 0.04 | 223 ± 25 |
| O2 | 238.7 ± 0.6 | 7.69 ± 0.04 | 196 ± 30 |
| E(A-A)(BZI) | |||
| O1 | 258 ± 2 | 7.2 ± 0.1 | 705 ± 22 |
| O2 | 287 ± 2 | 7.9 ± 0.1 | 686 ± 22 |
| E(A-A) | |||
| O1 | 258 ± 2 | 7.5 ± 0.1 | - |
| O2 | 292 ± 2 | 8.2 ± 0.1 | - |
Extracted using τc = 182 ns.
The E(A-A) intermediate, which precedes E(Q3) in the catalytic cycle, provides context for interpreting these chemical shifts. When [17O]-L-Ser is supplied to the enzyme, E(A-A) remains stable for approximately one day. By also supplying the unreactive indole analogue BZI (which does not covalently bind), the stability of the resulting E(A-A)(BZI) complex is increased to several days, and higher quality spectra can be obtained. Figure 2 shows the spectra of E(A-A)(BZI) measured at 11.7, 14.1 and 16.4 T. Overlap of the resonances for bound substrate with those for free serine and the pyruvate side-reaction product obscure the more downfield peaks at 14.1 and 16.4 T. Taking advantage of the different nutation frequencies of nuclei in the disparate motional regimes, [7a, 12] a triple-echo pulse sequence (detailed in the Supporting Information) was implemented that, when appropriately phase cycled, attenuates the free signals and allows for better resolution of the bound peak (Figure 2, 14.1 and 16.4T). The isotropic chemical shifts extracted for the distinct oxygen sites of E(A-A)(BZI) fall at 258 and 287 ppm. These signals lie convincingly in the range of the carboxylate form.[6] The corresponding shifts for E(A-A) fall at 258 ppm and 292 ppm (spectra and fits in Supporting Information).
Figure 2.

17O quadrupole central transition NMR spectra of the E(AA)(BZI) intermediate in TS at multiple fields. The enzyme-bound intermediates are marked with asterisks. Experiments measured at 14.1 and 16.4 T were acquired using a triple echo-sequence to improve observation of the enzyme-bound peaks by attenuating free substrate/product signal. Total number of transients and experiment times: at 11.7 T, 7.9 × 106 transients and 72 hours; at 14.1 T, 3.3 × 106 transients and 40 hours; at 16.4 T, 12.9 × 106 transients and 156 hours. The spectra are displayed with 100 Hz line broadening applied. Experiments were conducted at 3±2°C in pH 7.8 buffered aqueous solution. Additional experimental details are provided in the SI.
While E(A-A) and E(A-A)(BZI) present 17O NMR chemical shifts consistent with carboxylate forms, both E(Q3)indoline and E(Q3)2AP show upfield shifted resonances that suggest potential acid form character. But the empirical correlation between shift and chemical structure presented earlier would predict both an upfield and a downfield shifted peak for a quinonoid species undergoing fast-exchange between carboxylate and acid forms (assuming, as appears to be the case experimentally, that the carboxylate group is not free to rotate). An alternate hypothesis is that the increased shieldings in the quinonoid intermediates result from changes in the carboxylate hydrogen bonding interactions.[13] However, X-ray crystal structures of the quinonoid intermediates (PDBIDs 3PR2 (indoline) and 4HPJ (2AP)) show no change in the number of hydrogen bond donors compared to the aminoacrylate structures (PDBIDs 4HN4 (A-A) and 4HPX (BZI)) and only one closer contact.[4a, 10b] A third hypothesis is that the increased carboxylate shieldings are due to changes in the electronic structure that are inherent to the quinonoid form, reflecting increased electron density within the π bonding network, which is formally carrying a negative charge for these non-classical quinonoid intermediates.
To further test this hypothesis we undertook first principles, quantum chemical calculations of the oxygen chemical shifts within the enzyme active site for the phenolic form of E(A-A) and the three proposed tautomers (Scheme 2) of E(Q3)2AP. Full details are presented in the SI. In brief, a truncated model of the active site was constructed based on the X-ray crystal structures (residues located within 7 Å of the PLP and reacting substrates) for E(A-A) and E(Q3)2AP (Figure 3). The structures of the four intermediates were geometry optimized using DFT with the active site side chains at the periphery of the cluster fixed at their crystallographic locations. Chemical shieldings were calculated for the full clusters as previously reported,[14] converted to chemical shifts, and are summarized in Table 2.
Figure 3.

Model for first principles calculations of chemical shifts in the β subunit enzyme active site of tryptophan synthase. Side chains are shown in wireframe and the PLP-substrate as ball and stick.
Table 2.
First principles 17O chemical shifts for intermediates in TS
| Intermediate | Tautomer | Calc. δiso / ppm | |
|---|---|---|---|
| O1 | O2 | ||
| E(A-A) | phenolic | 267.1 | 303.5 |
| E(Q3)2AP | phenolic | 238.0 | 243.5 |
| E(Q3)2AP | PSB | 228.5 | 233.6 |
| E(Q3)2AP | acid | 171.3 | 113.1 |
| E(Q3)2AP | phenolic // PLP+H | 263.8 | 264.8 |
The cluster models show good agreement between the experimental and predicted chemical shifts for the phenolic form of E(A-A) and both carboxylate forms (phenolic and protonated Schiff base (PSB)) of E(Q3)2AP. It is interesting to note that within the quinonoid models, the transfer of a proton to the carboxylate oxygen is predicted to cause an upfield shift of both oxygen resonances. This is notably different than the behavior observed for amino acids. Quinonoid intermediates with a deprotonated pyridine nitrogen, such as those in TS,[4b] have been proposed to build up increased electron density at Cα to promote protonation at that site.[15] In contrast, the addition of a proton to the pyridine nitrogen gives the canonical quinonoid form with increased electron withdrawing ability that is expected to decrease the charge at Cα. To compare, we also modeled the protonated pyridine form for the E(Q3)2AP phenolic tautomer; the predicted carboxylate shifts (Table 2, phenolic//PLP+H model) fall within the typical carboxylate range. From this we hypothesize that the increased shielding of the quinonoids is mainly effected by the increased electron density within the π bonding network, particularly at the adjacent Cα site.
Finally, we return to the proposed proton exchange equilibrium between the quinonoid tautomers in Scheme 2, which can be viewed with greater acuity in light of the experimental and first principles estimates of the 17O chemical shifts. Two conclusions can be drawn. First, the percentage of acid form is considerably lower than previously proposed; a maximum population of 17% can be placed on the acid form before the predicted and experimental shifts deviate by more than 16 ppm (twice the RMSD of the linear rescaling test set; see the Supporting Information). Second, as the nitrogen chemical shift of the Schiff base indicates that it is (mostly) deprotonated,[4a] the phenolic tautomer must predominate.
17O QCT NMR holds great promise for the characterization of biomolecular systems, and perhaps no field is as well-poised to take advantage of it as chemical enzymology, where proton locations and electrostatic environments are key to understanding mechanism. This is particularly true of the acid-base chemistry that takes place within the active sites of PLP-dependent enzymes such as tryptophan synthase, where proton transfers and changes in electron density could previously only be inferred from the shifts of other nuclei in the active site. What is remarkable is that such measurements can be performed under conditions of active catalysis, catching kinetically competent intermediates in the act of chemical transformation.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01GM097569 (LJM), NSF grant CHE-1213845 (CKL), and NSF GRFP DGE-0813967 (RPY). 16.4 T data were acquired at the UCSD NMR Resource (NIH P41EB002031). Computational rescources were funded by NIH grant S10OD016290 and NSF grant MRI-1429826. The authors thank GJO Beran and JD Hartman for assistance on the 17O chemical shift rescaling and RJ Hooley for discussions on chemical shielding.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Toney MD. Biochim Biophys Acta. 2011;1814:1407–1418. doi: 10.1016/j.bbapap.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hayashi H. J Biochem. 1995;118:463–473. doi: 10.1093/oxfordjournals.jbchem.a124931. [DOI] [PubMed] [Google Scholar]
- 2.Chan-Huot M, Dos A, Zander R, Sharif S, Tolstoy PM, Compton S, Fogle E, Toney MD, Shenderovich I, Denisov GS, Limbach HH. J Am Chem Soc. 2013;135:18160–18175. doi: 10.1021/ja408988z. [DOI] [PubMed] [Google Scholar]
- 3.a) Sharif S, Denisov GS, Toney MD, Limbach HH. J Am Chem Soc. 2007;129:6313–6327. doi: 10.1021/ja070296+. [DOI] [PubMed] [Google Scholar]; b) Chan-Huot M, Sharif S, Tolstoy PM, Toney MD, Limbach HH. Biochemistry. 2010;49:10818–10830. doi: 10.1021/bi101061m. [DOI] [PubMed] [Google Scholar]
- 4.a) Lai JF, Niks D, Wang YC, Domratcheva T, Barends TRM, Schwarz F, Olsen RA, Elliott DW, Fatmi MQ, Chang CEA, Schlichting I, Dunn MF, Mueller LJ. J Am Chem Soc. 2011;133:4–7. doi: 10.1021/ja106555c. [DOI] [PubMed] [Google Scholar]; b) Caulkins BG, Bastin B, Yang C, Neubauer TJ, Young RP, Hilario E, Huang YMM, Chang CEA, Fan L, Dunn MF, Marsella MJ, Mueller LJ. J Am Chem Soc. 2014;136:12824–12827. doi: 10.1021/ja506267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerothanassis IP. Prog Nucl Magn Reson Spectrosc. 2010;56:95–197. doi: 10.1016/j.pnmrs.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Wong A, Poli F. Annu Rep NMR Spectrosc. 2014;83:145–220. [Google Scholar]
- 7.a) Zhu JF, Wu G. J Am Chem Soc. 2011;133:920–932. doi: 10.1021/ja1079207. [DOI] [PubMed] [Google Scholar]; b) Bull TE, Forsen S, Turner DL. J Chem Phys. 1979;70:3106–3111. [Google Scholar]
- 8.Lee HC, Oldfield E. J Am Chem Soc. 1989;111:1584–1590. [Google Scholar]
- 9.a) Werbelow LG. J Chem Phys. 1979;70:5381–5383. [Google Scholar]; b) Butler A, Danzitz MJ, Eckert H. J Am Chem Soc. 1987;109:1864–1865. [Google Scholar]
- 10.a) Roy M, Keblawi S, Dunn MF. Biochemistry. 1988;27:6698–6704. doi: 10.1021/bi00418a009. [DOI] [PubMed] [Google Scholar]; b) Niks D, Hilario E, Dierkers A, Ngo H, Borchardt D, Neubauer TJ, Fan L, Mueller LJ, Dunn MF. Biochemistry. 2013;52:6396–6411. doi: 10.1021/bi400795e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Torre JG, Huertas ML, Carrasco B. J Magn Reson. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 12.Man PP, Klinowski J, Trokiner A, Zanni H, Papon P. Chem Phys Lett. 1988;151:143–150. [Google Scholar]
- 13.Wong A, Pike KJ, Jenkins R, Clarkson GJ, Anupold T, Howes AP, Crout DHG, Samoson A, Dupree R, Smith ME. J Phys Chem A. 2006;110:1824–1835. doi: 10.1021/jp055807y. [DOI] [PubMed] [Google Scholar]
- 14.Hartman JD, Neubauer TJ, Caulkins BG, Mueller LJ, Beran GJ. J Biomol NMR. 2015;62:327–340. doi: 10.1007/s10858-015-9947-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Major DT, Gao JL. J Am Chem Soc. 2006;128:16345–16357. doi: 10.1021/ja066334r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.