

Abstract
The septins are a conserved family of GTP-binding proteins that, in the baker's yeast, assemble into a highly ordered array of filaments at the mother bud neck. These filaments undergo significant structural rearrangements during the cell cycle. We aimed at identifying key components that are involved in or regulate the transitions of the septins. By combining cell synchronization and quantitative affinity-purification mass-spectrometry, we performed a screen for specific interaction partners of the septins at three distinct stages of the cell cycle. A total of 83 interaction partners of the septins were assigned. Surprisingly, we detected DNA-interacting/nuclear proteins and proteins involved in ribosome biogenesis and protein synthesis predominantly present in alpha-factor arrested that do not display an assembled septin structure. Furthermore, two distinct sets of regulatory proteins that are specific for cells at S-phase with a stable septin collar or at mitosis with split septin rings were identified.
Complementary methods like SPLIFF and immunoprecipitation allowed us to more exactly define the spatial and temporal characteristics of selected hits of the AP-MS screen.
Introduction
The baker’s yeast Saccharomyces cerevisiae undergoes asymmetric cell division by budding.
Establishment and maintenance of cell polarity requires a septin scaffold at the bud neck that attracts different protein complexes during the cell cycle [1]. Yeast cells express five septins during vegetative growth, namely Cdc3, Cdc10, Cdc11, Cdc12 and Shs1. They assemble into hetero-oligomers by association of monomers with the order Cdc11-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Cdc11 with Shs1 sometimes replacing the terminal subunit Cdc11 [2,3].
The septin filaments assemble at the bud neck in an organized array, the so-called septin ring. This septin ring undergoes different cell cycle-dependent architectural transitions [4]. In early G1-phase, the septins are recruited to and accumulate at the presumptive bud site in a patch-like structure. Shortly before bud emergence, the patches are transformed into a ring marking the future site of bud growth and cytokinesis [5]. After bud formation, the septin ring expands into a stable hourglass-shaped collar that is present at the bud neck until the onset of mitosis. Before cytokinesis, the septin collar splits into two distinct rings, one located at the mother and one at the daughter side of the bud neck [4]. Cell separation then occurs between the two rings. After completion of cell separation, the old septin rings are disassembled and septin subunits are partially replaced and recycled for the next round of the cell cycle [6].
The initial recruitment of the septins to the future bud site depends on the small GTPase Cdc42, its effectors Gic1 and Gic2, and the action of the cyclin-dependent kinases Cdc28 and Pho85 [7]. Septin filament assembly is supposed to be mediated by the diffusion-driven annealing of the septin rods at the plasma membrane [8]. The transition of the septin ring into a stable septin collar after bud emergence is associated with the phosphorylation and acetylation of certain subunits [9–12]. Splitting of the septin collar at the onset of cytokinesis is supposed to be initiated by a collective switch in the orientation of the septin filaments from parallel to perpendicular to the growth axis of the cell [13,14]. The switch is accompanied by at least two different modifications. First, the bud neck kinase Gin4 phosphorylates Shs1 at residues different from those being modified in G1-phase [15]. Second, the small ubiquitin-like modifier (SUMO) Smt3 is covalently attached to Cdc3, Cdc11 and Shs1 at the mother side of the bud [16].
A deeper understanding of the regulation of septin structure assembly and subsequent structural transitions requires the identification of all involved molecular components as well as a temporal map of their post-translational modifications and of their dynamic organization into multi-protein complexes. However, systematic interaction screens have yet only been performed for mammalian septins were cDNA libraries were screened for SEPT protein baits using the yeast two hybrid system [17]. The interactome of the yeast septins remains incomplete and rests on numerous targeted studies that aimed at deciphering a certain function or process [18].
We tried to address this shortcoming by systematically screening for specific septin interactors at distinct stages of the cell cycle. Affinity purification followed by mass spectrometry (AP-MS) has evolved as a very efficient tool to identify protein-protein interactions [19,20]. To allow for a spatial and temporal integration of the resulting interaction networks into the context of the cell and its different cell cycle states, we have combined cell synchronization with AP-MS. Synchronization of yeast cells in cell cycle states where septins have a distinct structure (patch—collar—split rings) was followed by affinity purification of tagged septin complexes and SILAC based quantitative mass spectrometry (MS) analysis. This unique combination allowed us to describe the changing pattern of interaction partners of the septins during the cell cycle.
Selected candidates were subjected to a SPLIFF (Split Ubiquitin Fluorophore Fluorophore) analysis [21] to better resolve spatial and temporal aspects of these interactions. The integration of both techniques allowed us to systematically explore the cellular aspects of protein interactions detected by AP-MS.
Methods
Plasmids and strains
The construction of the yeast strains used in this study is described in detail in the Supporting Information.
All yeast strains used in this study are listed in the S3 Table.
Growth of yeast strains and cell synchronization
The Cdc11-TAP strain was grown in standard SD medium and the Cdc11-GFP strain in SILAC-SD medium containing 13C6,15N4-L-arginine and 13C6,15N2-L-lysine (Sigma Aldrich) as only source for arginine and lysine. Incorporation of labeled amino acids was confirmed by analyzing protein extracts from labeled cells by gel-based nano-HPLC-ESI-MS/MS analysis.
Full incorporation of the labeled amino acids was obtained by diluting a preculture grown overnight in SILAC medium into fresh SILAC medium and incubating it at 30°C for 7.5 h (S1 Fig).
An overnight culture of each strain was diluted into 400 ml fresh medium and grown overnight at 30°C to the desired OD600nm for cell synchronization. The synchrony of the cultures was monitored by microscopy and the cells were harvested by centrifugation and immediately used for affinity purification.
Cell synchronization was performed either with alpha-factor (Sigma) (3.7 μM final concentration pre-dissolved in 0.1 M HCl; added to the cell culture at an OD600nm of 1 and incubated at 30°C for 2:40 h) or hydroxyurea (Sigma) (0.2 M final concentration; added to the cell culture at an OD600nm of 1 and incubated at 30°C for 3:30 h). Temperature-sensitive strains were arrested by incubation at 37°C for 3:25 h after the OD600nm reached 0.6.
Affinity purification (AP)
AP was performed separately for the Cdc11-TAP- and the Cdc11-GFP-expressing control strain. The pelleted yeast cells were resuspended in extraction buffer (20 mM Tris pH 7.5, 80 mM NaCl, 1 mM DTT, 8.3 μM antipain, 0.3 μM aprotinin, 1 mM benzamidine, 1 μM bestatin, 10 μM chymostatin, 1.5 μM pepstatin A, 5 μM leupeptin, 1 mM PMSF, 10 mM β-glycerophosphate, 10 mM NaF, 1 mM sodium orthovanadate), acid washed glass beads were added (3-fold wet cell weight) and twelve cycles of bead beating were applied (1 min vortexing followed by 1 min incubation on ice). The resulting extract was clarified by centrifugation for 15 min at 40,000 x g. The protein concentrations were adjusted to 5 mg/ml in extraction buffer and 10% (v/v) glycerol was added. CNBr activated Sepharose (GE Healthcare) was coupled with human IgG (MP Biomedicals) according to the manufacturer's protocol. The obtained HsIgG-coupled Sepharose was equilibrated in extraction buffer and 75 μl slurry per 50 mg protein were added to the protein extract.
The samples were rotated overhead overnight at 4°C and subsequently another equivalent of hsIgG-sepharose was added followed by an incubation for 2 h at 4°C. The Sepharose was washed with a 50-fold hsIgG-Sepharose slurry volume of extraction buffer (containing 10% (v/v) glycerol and lacking aprotinin, benzamidine and NaF) and bound protein was eluted with 6his-AcTEV Protease (Life Technologies). The protease was removed using Ni-NTA agarose. Supernatants containing the cleaved protein complexes from both strains were mixed. Proteins were precipitated by adding the 4-fold volume of ice-cold acetone and stored for at least two days at -20°C. Precipitated proteins were collected by centrifugation, acetone was removed and the pellets were resuspended in a total volume of 40 μl 2x Lämmli buffer. Proteins were separated by SDS-PAGE followed by colloidal Coomassie blue staining.
Samples from all steps of the purification were submitted to Western blot analysis using anti-Protein A (Sigma) and anti-GFP (Roche) primary antibodies.
Mass spectrometry and data analysis
Following gel electrophoresis of affinity-purified, differentially SILAC-labeled Cdc11-complexes and colloidal Coomassie blue staining, gel lanes were cut into individual slices. The gel slices were destained and washed as described [22]. Cysteine residues were reduced using 10 mM DTT (30 min at 65°C) and alkylated with 50 mM iodoacetamide (30 min at room temperature in the dark). Proteins were subsequently in-gel digested using trypsin (37°C, overnight). Peptide mixtures of three independent replicates per cell cycle arrest were analyzed by LC/MS using an UltiMate 3000 RSLCnano HPLC system (Thermo Scientific) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Scientific).
Mass spectrometric data were processed using the software MaxQuant [23] (version 1.2.0.18 for alpha-factor and hydroxyurea data or 1.3.0.5 for cdc15-1 data). For protein identification, MS/MS data were searched against the Saccharomyces Genome Database (SGD; www.yeastgenome.org; version of June 2012) using Andromeda [24]. The detailed search engine parameters are given in the Supporting Information. The mass spectrometry proteomics raw data and MaxQuant result files have been deposited to the ProteomeXchange Consortium [25] via the PRIDE partner repository with the dataset identifier PXD002561.
Light-over-heavy (L/H) abundance ratios for proteins were log10-transformed and mean log10 L/H ratios across all three biological replicates of septin complexes purified from yeast cells treated with alpha-factor or hydroxyurea and from cdc15-1 mutant cells as well as p-values (one-sided Student's t-test) were determined. Proteins with a p-value of < 0.05 and a mean L/H ratio of > 4 were considered as putative Cdc11-interacting proteins. All quantified proteins are listed in the S1 Table. Data were visualized by plotting the mean log10 ratios of all proteins quantified in at least two replicates versus the negative value of the corresponding log10-transformed p-value. Candidates were finally sorted into manually defined categories according to their functions that were retrieved from the SGD.
Proteins known to be mislocalized under the respective blocking conditions were excluded from the subsequent candidate selection and evaluation process. See the Supporting Information for details.
Microscopy and SPLIFF analysis
All microscopy experiments were performed on an Axio Observer SD spinning disc confocal microscope (Zeiss) equipped with an Evolve512 EMCCD camera (Photometrics), objectives Plan-Apochromat 63X/1.4 Oil DIC and Plan-Apochromat 100X/1.4 Oil DIC (Zeiss), and 488 nm and 561 nm diode lasers (Zeiss). The microscope was controlled by the ZEN2012 software package (Zeiss). Images were analyzed and created using ZEN2012 or ImageJ64 (version 1.48p).
For microscopy of synchronized yeast cells, a saturated overnight culture of a BY-SILAC strain expressing Shs1-mCherry and a candidate-GFP fusion was diluted into fresh medium and incubated overnight at 30°C until the desired OD600nm was reached. The cell cycle was arrested using the protocols described above. Arrested cells were concentrated by brief centrifugation, loaded onto a microscopy glass slide, fixed with a glass coverslip and directly observed by fluorescence microscopy.
For time lapse mircoscopy, cells from an exponentially growing culture were mounted onto an agar pad prior to microscopy. Microscopy was performed at 30°C and images were acquired every 3 min for up to 5 h. Time resolved SPLIFF analysis was performed as described elsewhere [26].
For steady state SPLIFF analysis, a saturated overnight culture of diploid cells expressing Shs1-CCG and a Nub-fusion was diluted 1:10 into fresh medium containing 0 or 100 μM Cu2+ and grown for at least 4 h at 30°C. Cells were concentrated by brief centrifugation, loaded onto a microscopy glass slide, fixed with a glass coverslip and observed immediately. The microscope settings and data evaluation is described in the Supporting Information.
Pulldown of candidate proteins using purified septin rods
The expression and purification of recombinant SNAP-tagged yeast septin rods was performed as described previously [27]. 6his-SNAP was purified from crude E. coli extracts with immobilized metal affinity chromatography followed by size exclusion chromatography as described for other proteins [27].
As a matrix for pulldown experiments, benzylguanine coupled to Sepharose (BG-Sepharose) was used. Briefly, CNBr-activated Sepharose 4B (GE Healthcare) was functionalized with BG-PEG-NH2 (NE Biolabs) (1 μmol per ml Sepharose) according to the Sepharose' manufacturer's recommendations. Remaining active groups were blocked for 3 h with 1 M ethanolamine pH 8.0.
Extracts of yeast cells expressing the respective TAP-tagged (or GFP tagged Vps1) candidate proteins (50 ml culture volume) were prepared as described above. Pulldown buffer (50 mM Tris pH 8.0, 300 mM NaCl) was used for preparing all subsequent protein solutions and washing steps. Septin rods and SNAP protein were captured onto BG-Sepharose (150 μl of 1 μM SNAP-tagged septin rods or 150 μl of 2 μM 6his-SNAP onto 60 μl BG-Sepharose slurry). The matrix was washed, blocked with 1 mg/ml BSA (1 h under rotation at 8°C) and subsequently incubated with the yeast extract (2 h under rotation at 8°C). The beads were washed five times and captured proteins were eluted by boiling the beads for 5 min in 40 μl of 2x Lämmli buffer. Eluted proteins were detected by Western blot analysis using an anti-protein A (Sigma) or anti-GFP (Roche) primary antibody.
Results
An experimental platform for the identification of cell cycle specific septin interacting proteins
To identify septin interacting proteins at different stages of septin architecture via AP-MS, a robust platform for cell synchronization and complex isolation was implemented in the first place. We constructed a yeast strain expressing the endogenous septin Cdc11 fused at its C-terminus to a TAP tag for the isolation of septin complexes on Protein-A Sepharose. As control strain, we constructed a strain expressing Cdc11-GFP. This strain allows additionally to monitor the state of septin architecture by fluorescence microscopy and to calculate the synchrony of the treated cells.
We chose to investigate the protein composition of three different septins structures: (i) septin patches before their assembly into a distinct structure, (ii) the stable collar and (iii) the split septin rings. The first two states were realized by blocking the cells with alpha-factor (G1 phase) or hydroxyurea (HU) (S1 phase), respectively. Cells bearing the temperature sensitive allele cdc15-1 arrest at the restrictive temperature in late anaphase [28]. Using the Cdc11-GFP strain, we confirmed that the septins are organized in patches at the membrane or are diffusely localized in the cytoplasm in alpha-factor arrested cells (Fig 1: top). HU blocked cells show the stable septin collar at the bud neck (Fig 1: middle), whereas cdc15-1 arrested cells display the split septin rings (Fig 1: bottom). The synchrony of blocked cells was determined by counting the proportion of cells without an assembled septin ring for alpha-factor arrest (94.9 ± 0.6%; n ≥ 409 cells), with a stable septin collar for HU arrest (82.0 ± 5.4%; n ≥ 141 cells) and with split septin rings for cdc15-1 arrest (85.6 ± 2.3%; n ≥ 162 cells). Synchronicity rates were calculated individually for each culture that was subsequently used in AP-MS-experiments. The cells were incubated with the respective blocking compound or at the non-permissive temperature and harvested for AP-MS in the blocked state.
Fig 1. Cell synchronization of Cdc11-GFP and Cdc11-TAP expressing cells.
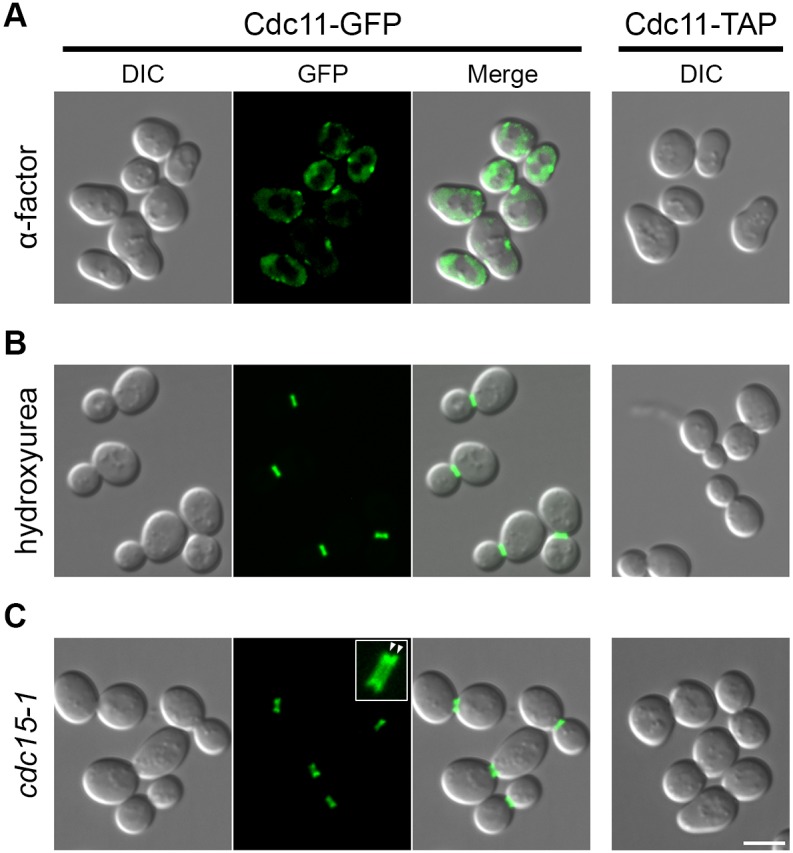
Representative image sections (z-projection) for Cdc11-GFP or Cdc11-TAP expressing strains using the indicated blocking approaches. The structure within the white rectangle represents a zoom in of the respective septin architecture using a 100x objective. White arrowheads indicate split septin rings in cdc15-1-arrested cells. Scale bar 6 μm.
We used a quantitative AP-MS approach to distinguish between genuine septin interacting proteins and contaminants. Stable isoptope labeling of amino acids in cell culture (SILAC) enables to distinguish in one MS experiment between protein complexes isolated from a target strain (here: Cdc11-TAP) from those isolated from a control strain (here: Cdc11-GFP) [29]. Briefly, one of the two strains (here: Cdc11-GFP) is labelled with "heavy", non-radioactive amino acid isotopes. Both strains are mixed and processed jointly for MS analysis. The control strain serves as a reference to identify non-specific binding to any of the used materials. In the case of such an unspecific binding, a contribution of "heavy" peptides from the control strain to an identification hit in the MS data analysis will be detected. Of importance is the stage where both strains are mixed: Mixing the cells or extracts of both strains before complex isolation ("purification after mixing") results in the identification of the stable core complex constituents whereas mixing of the eluates after purification allows also for the identification of transient interaction partners [30,31].
We have chosen a mixing after purification approach to monitor also transient interactions. Equal volumes of the eluates from the AP-MS were combined prior to SDS-PAGE and subsequent LC/MS analysis. The whole workflow is depicted in Fig 2. Affinity purifications were monitored by Western blot analysis of representative samples taken during the purification process (S2 Fig).
Fig 2. Workflow for SILAC-AP-MS.
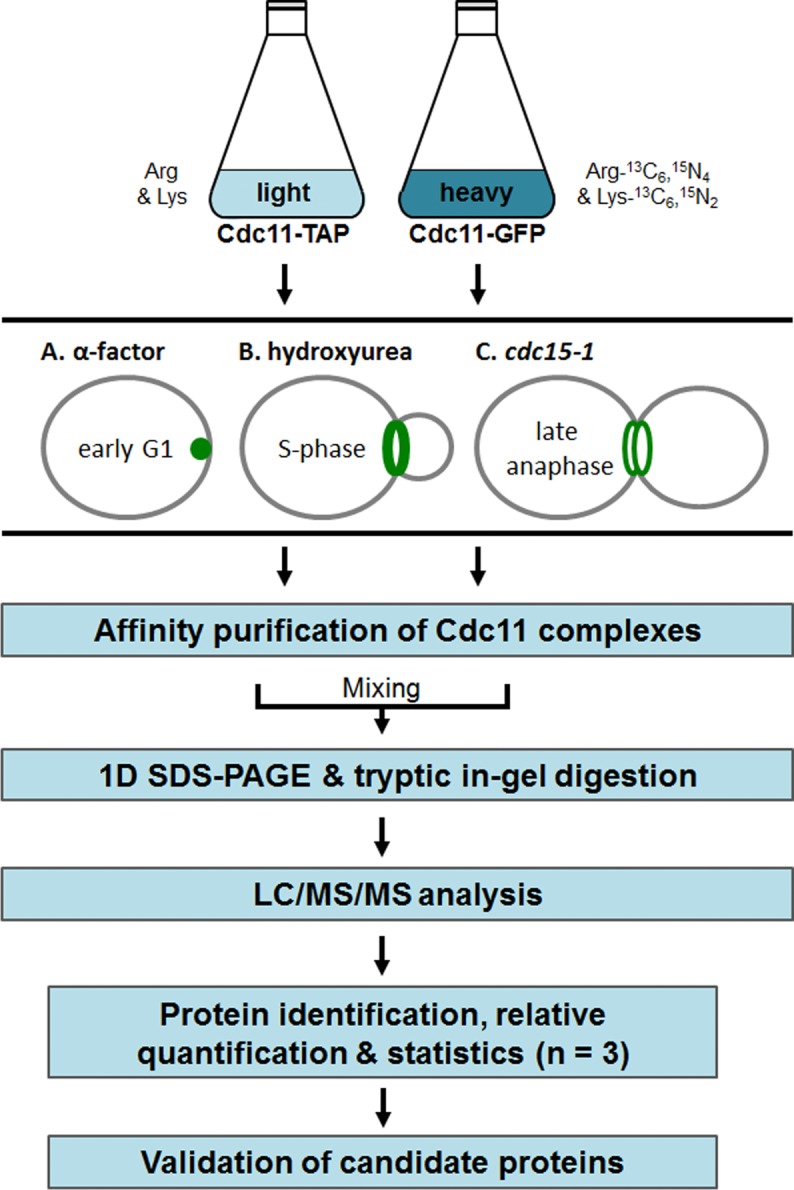
Yeast strains expressing Cdc11-TAP and Cdc11-GFP (metabolically labelled with 13C6,15N4-L-arginine and 13C6,15N2-L-lysine) are blocked with the indicated methods and subjected separately to affinity purification. The eluates are mixed and subjected to separation by SDS-PAGE followed by nano HPLC-ESI-MS/MS analysis and statistical data evaluation.
Identification of septin interaction partners with AP-MS
We performed three independent AP-MS purifications for each synchronization approach. “Light-over-heavy” (L/H) ratios and p-values (using a one-tailed t-test) were determined for each protein. The ratio L/H reflects the enrichment of a protein in the Cdc11-TAP purification (L) in comparison to the Cdc11-GFP purification (H). The negative logarithm of the p-value was plotted against the logarithm of the L/H ratio (Fig 3). Proteins quantified in at least two out of three replicates and exhibiting a ratio ≥ 4 and a p-value < 0.05 were considered as specific interaction partners. All quantified proteins are listed in the S1 Table.
Fig 3. SILAC-AP-MS analysis.

Results of the quantitative MS-analysis. Volcano plots for the MS analysis of cells arrested in G1 phase (alpha facor). (A), S-phase (B) or anaphase (C). The red-dashed lines indicate the thresholds defining a specific interaction. Selected candidate proteins for further validation are named and labeled in blue, septins are marked in red. Specific interactors were grouped into the indicated categories (D). Numbers represent the specific interactors in the respective timepoints for each category.
The specific interaction hits were sorted into the following manually defined categories: Septins, scaffold proteins, regulatory proteins, proteins involved in intracellular trafficking, cell cycle control proteins, DNA interacting proteins, ribosomal proteins, others not fitting in one of these categories.
A total of 1,209 proteins were quantified for alpha-factor synchronized cells. In this dataset, 40 proteins (including the five mitotic septins) fulfill the criteria for specific septin interaction partners (Fig 3A). In alpha-factor arrested cells, the identified septin interactors are predominantly proteins involved in ribosome biogenesis or protein synthesis (13 proteins, 32.5%) and nuclear proteins (10, 25.0%). The other defined categories are illustrated in Fig 3D.
In HU arrested cells, a total of 1,446 unique proteins were quantified. Only 37 of these hits (including the five mitotic septins) fulfill the specificity criteria and were assigned as S-phase specific interactors of the septins (Fig 3B). Among the interactors are eight regulatory proteins such as protein kinases. The number of identified ribosomal and nuclear proteins is significantly reduced. A classification of all interactors is shown in Fig 3D.
A total of 1,278 unique proteins were quantified in cells arrested in late anaphase. Of these, 37 (including the five mitotic septins) were assigned as specific interaction partners (Fig 3C). Besides regulatory and scaffold proteins and proteins involved in processes formerly not associated with the septins (glucose and glycerol metabolism, protein folding, membrane organization), Syp1, a protein involved in endocytosis and septin organization [32] and Mrc1, a protein required for cell cycle control [33] were identified as specific interaction partners of the septins. The classification of all anaphase specific interaction partners is shown in Fig 3D.
The overlaps of the three replicates evaluated for each time point are illustrated in S3 Fig.
Taken together, 88 specific interaction partners of the septins (including the five mitotic septins) were identified (Fig 4A). Only nine proteins (including the five mitotic septins) were present in all three stages (Fig 4A) and each four common interaction partners were identified for alpha factor arrest and S phase and for S phase and anaphase, respectively. None of the identified proteins bound to the septins both in alpha factor arrest and late anaphase.
Fig 4. Overview of the SILAC-AP-MS datasets.

(A) Overlap of the specific hits in alpha-factor arrested cells, S-phase (HU) and late anaphase (cdc15-1). (B) Summary and overlap of the candidate proteins selected for further validation.
Representative candidates were randomly picked from the different functional categories for validation by complementary methods. A total of 26 proteins were selected for validation experiments (Fig 4B).
A variation of the SPLIFF system provides a fast and robust validation platform for MS based interaction hits
The interactions of selected candidates with the septins were analyzed in vivo using a variation of the SPLIFF technique. SPLIFF is a method for the detection of protein-protein interactions in a time- and spatial resolved manner inside a living cell [21,26]. It is based on the split-ubiquitin method [34] and uses two spectrally different fluorophores as interaction signal. A mCherry-Cub-GFP (CCG) module is attached to the C-terminus of a protein of interest, whereas Nub is fused to the N-terminus of a potential interaction partner. An interaction between the two proteins induces a reassociation of the ubiquitin halves Nub and Cub. This native-like ubiquitin is then recognized by a ubiquitin-specific protease that cleaves off the GFP from the Cub. The liberated GFP carries a genetically inserted N-terminal destabilizing amino acid and is rapidly degraded through the ubiquitin-proteasome pathway. The ratio of GFP to mCherry fluorescence indicates the extent of interaction. After examining the effects of tagging all mitotic septins with the CCG module on cell growth and morphology (S4 Fig), we decided to use Shs1-CCG as least disturbing CCG fusion to the septin rod. The acceptor of the label thus differs between the SPLIFF and the AP-MS experiments. We established a “steady-state” SPLIFF as a fast screen for interaction partners of the septins in vivo. A Shs1-CCG strain was mated with strains of the opposite mating type expressing different candidate Nub-fusions under control of the copper-inducible PCUP1-promoter. Resulting diploid cells were grown in the presence and absence of Cu2+ to exponential phase and the interaction was quantified by determining the ratios of GFP over mCherry fluorescence at the bud neck of individual cells. Thresholds for a strong and weak interaction were set to a CCG to CC conversion (SPLIFF signal) of greater than 70% and greater than 30%, respectively. SPLIFF signals lower than 30% were assigned as non-interacting. See the Supplementary Methods (S1 Methods) section for the calculation of the SPLIFF signal and the S2 Table for the corresponding values.
Under PCUP1-inducing conditions of the 26 examined candidate proteins, eight proteins were classified as strong and seven as weak interactors. Seven proteins were assigned as non-interacting with the septins based on the SPLIFF analysis (Fig 5A–5C). Four candidate proteins could not be labeled with Nub. The results of all SPLIFF interaction assays are summarized in Table 1. The result of the SPLIFF analysis for all evaluated candidates under non-inducing conditions is shown in the S5 Fig.
Fig 5. Steady-state SPLIFF measurements.

Each point represents a single cell measurement. The expressions of the Nub -fusion proteins were induced by100 μM Cu2+. The calculated medians and SEMs are shown in red. Nub-fusions are grouped according to their interaction signal. (A) Strong interaction (SPLIFF signal ≥ 70%). (B) Weak interaction (SPLIFF signal ≥ 30%). (C) No interaction (SPLIFF signal ≤ 30%). The negative value for Nap1 might result from a weaker expression of Nap1 than the control Nub. Note a subpopulation of Glc7 cells that have a SPLIFF signal >50%. grey squares: negative control Nub-empty; grey circle: positive control Nub-Cdc11. (D) Comparison between stable collar and split septin rings SPLIFF signals induced by Nub- and -Bud4 with 0 (left) or 100 μM Cu2+ (right).
Table 1. SPLIFF- and alternative evaluation of 26 selected candidates.
| Candidate protein | Category | MS identification point | SPLIFF validation (in vivo interaction with septins) | Pulldown validation (in vitro interaction with septins) | Annotations | Figure reference |
|---|---|---|---|---|---|---|
| Bni5 | scaffold | α-F, S-phase, anaphase | strong interaction | see [36] | Known septin interactor [37,38] | 3A-C, 5A |
| Glc7 | regulatory | α-F, S-phase, anaphase | no interaction | 3A-C, 5C | ||
| Bni4 | regulatory | α-F, S-phase, anaphase | no interaction | Time lapse microscopy: colocalization with septins. Known septin interactor [35] | 3A-C, 5C, 6A | |
| Ppz2 | regulatory (phosphatase) | α-F, S-phase, anaphase | no interaction | 3A-C, 5C | ||
| New1 | ribosome/protein synthesis | α-F, S-phase | strong interaction | n/a ** | 3A, 3B, 5A, S7 | |
| Nip1 | ribosome/protein synthesis | α-F, S-phase | n/a * | n/a ** | Colocalization in arrested cells: no interaction | 3A, 3B, 6B |
| Cse1 | DNA interacting/nucleus | α-F | strong interaction | 3A, 5A | ||
| Nop4 | DNA interacting/nucleus | α-F | weak interaction | 3A, 5B | ||
| Rlp7 | DNA interacting/nucleus | α-F | weak interaction | negative | 3A, 5B, S7 | |
| Pno1 | DNA interacting/nucleus | α-F | n/a * | positive | Colocalization in arrested cells: no interaction | 3A, 6B, S7 |
| Sfb3 | protein sorting/endocytosis | α-F | weak interaction | positive | 3A,5B, S7 | |
| Vps1 | protein sorting/endocytosis | α-F | weak interaction | positive | 3A, 5B, S7 | |
| Sla2 | protein sorting/endocytosis | S-phase | strong interaction | positive | 3B, 5A, S7 | |
| Coy1 | protein sorting/endocytosis | S-phase | no interaction | 3B, 5C | ||
| Syp1 | protein sorting/endocytosis | anaphase | weak interaction | positive | Known septin interactor [32] | 3C, 5B, S7 |
| Gin4 | regulatory (kinase) | S-phase, anaphase | strong interaction | Known septin interactor [15] | 3B, 3C, 5A | |
| Nap1 | regulatory | S-phase, anaphase | no interaction | 3B, 3C, 5C | ||
| Prk1 | regulatory (kinase) | S-phase | no interaction | 3B, 5C | ||
| Hsl1 | regulatory (kinase) | S-phase | strong interaction | Known septin interactor [39] | 3B, 5A | |
| Ste20 | regulatory (kinase) | anaphase | weak interaction | positive | Known septin interactor [40] | 3C, 5B, S7 |
| Sic1 | cell cycle control | anaphase | no interaction | 3C, 5C | ||
| Mrc1 | cell cycle control | anaphase | weak interaction | 3C, 5B | ||
| Smt3 | regulatory | anaphase | n/a * | SPLIFF with expression from plasmid. Known interactor [16] | 3C, S4 | |
| Tub1 | scaffold | anaphase | n/a * | n/a ** | Colocalization in arrested cells: no interaction | 3C, 6B |
| Bud4 | regulatory | S-phase, anaphase | strong interaction | positive | Known septin interactor [41] | 3B, 3C, 5A, 5D, 7C |
| Bud3 | scaffold | anaphase | strong interaction | positive | Known septin interactor [42] | 3C, 5A, 5D, 7C |
(*) a Nub fusion could not be created
(**) proteins bound unspecifically
The SPLIFF analysis classified Bni4 as non-interactor, although the protein had been previously described as septin interacting [35,36]. We could confirm that Bni4-GFP co-localizes with the septins throughout the cell cycle (Fig 6A). Co-localization studies were also performed with three of those proteins (Nip1, Pno1 and Tub1) that did not tolerate the N-terminal Nub-fusion. In none of these cases we observed co-localization with the septins (Fig 6B). The fourth protein, Smt3, represents a known septin interactor [16]. We performed steady-state SPLIFF with Nub-Smt3 expressed from a centromeric plasmid in this case. We observed a mixed population of cells with an enlarged phenotype showing the interaction and of cells with normal size without interaction signal (S6 Fig).
Fig 6. Validation of selected candidate proteins by fluorescence microscopy.

(A) Live cell imaging of cells expressing Shs1-mCherry and Bni4-GFP at indicated time points of the cell cycle. Scale bar 5 μm. (B) Fluorescence microscopy images of blocked cells expressing Shs1-mCherry and Nip1-, Pno1-or Tub1-GFP. Scale bar 6 μm.
To substantiate the finding of our SPLIFF analysis, we expressed a random selection of candidates that were positively evaluated in the SPLIFF analysis as TAP- or GFP tagged proteins in S. cerevisiae and performed a pulldown on purified septin rods. We tested also the proteins that did not tolerate the Nub-fusion (Nip1, Pno1, Tub1). Clear signals above background validated Sfb3, Sla2, Ste20, Syp1, Pno1 and Vps1 as specific binding partners (S7A Fig).
The extent of unspecific binding to the matrix excluded New1, Nip1 and Tub1 (S7B Fig, data not shown for the latter two) from the analysis.
Rlp7 showed a weak interaction in the SPLIFF analysis but did not bind to the septin rods in the pulldown analysis (S7C Fig). Bni5 was previously evaluated with the same method as septin interacting protein [37]. All pulldown results are summarized in Table 1 and shown in S7 Fig.
Spatial and temporal refinement of MS defined interactions: The interaction between Bud3 and Bud4 and the septins as a test case
Bud3 and Bud4 are known to interact with each other and with the septins in an interdependent manner [43,44]. They both co-localize at the bud neck well before the onset of mitosis. In agreement with published data [42] we could localize the GFP fusions of both proteins in Shs1-mCherry co-expressing cells to the incipient bud site approximately 20 minutes after the septin patches became detectable at this site (Fig 7A). From this time point on the distributions of both fusions closely match the cellular localization of the septins including the splitting into a double ring during cytokinesis.
Fig 7. Timing of the interaction of Bud3 and Bud4 with the septins.

(A) Time lapse microscopy of Bud3-GFP or Bud4-GFP and Shs1-mCherry expressing cells. The time of the first appearance of the GFP fusion protein after the appearance of Shs1-mCherry at the bud neck was recorded. (B) Pulldown of Bud3-TAP and Bud4-TAP on immobilized septin rods and detection with an anti-Protein A antibody. The asterisk marks the hardly visible Bud3-TAP signal. A cross reaction of the antibody with one of the septins is marked with +. (C) Time resolved SPLIFF analysis (Nub-Bud3 or Nub-Bud4 vs. Shs1-CCG). Imaging time frame 3 min, 100 μM Cu2+. N = 10 for Bud3 and N = 9 for Bud4. The start of the Nub induced conversion of Shs1-CCG is marked with an arrow.
In contrast to the strict co-localization, our MS screen identified Bud4 in S-phase and late anaphase as septin interactor whereas Bud3 was unexpectedly found to be associated with the septins only at mitosis during the splitting of the septin rings (Fig 4). Bud3 and Bud4 were both validated as strong interaction partners of the septins in the steady-state SPLIFF analysis (Fig 5A). We noticed that the SPLIFF signal of Nub-Bud3 and Nub-Bud4 further increases in cells with split septin rings in comparison to cells with a stable septin collar (Fig 5D).
To resolve the discrepancy between co-localization- and protein interaction data we followed the interaction of Bud3 and Bud4 with the septins during the 90 min of a single cell cycle by time resolved SPLIFF analysis using Nub-Bud3 or Nub-Bud4 and Shs1-CCG as the corresponding Split-Ub fusion proteins (Fig 7B).
The interaction between Nub-Bud4 and Shs1-CCG started shortly after the fusion of the Nub-Bud4 and Shs1-CCG expressing yeast cells. The linear conversion of Shs1-CCG lasted till min 45 and paused for 18 min before abruptly continuing during the splitting of the septin rings. The kinetics of the Nub-Bud3 induced conversion of Shs1-CCG was very similar except that initiation and the linear increase were shifted by more than 6 min into the cell cycle. As observed for Nub-Bud4, the Nub-Bud3-induced conversion of Shs1-CCG stopped at min 45. We conclude that Bud4 interacts with the septins immediately upon arrival at the incipient bud site whereas Bud3 is initially recruited by Bud4 and only later able to directly interact with the septins.
We finally asked whether Bud3 and Bud4 are both direct interaction partners of the septins. We purified SNAP tagged septin rods and immobilized them on BG-Sepharose beads. The beads were incubated with extracts from yeast expressing Bud3-TAP or Bud4-TAP. Bud4 was specifically eluted from septin-coated beads whereas Bud3 did only show a weak interaction (Fig 7C).
Discussion
To determine septin-associated interactomes in dependence of the cell cycle, we blocked the cell cycle at three distinct stages of septin architecture: i) in alpha-factor arrested cells (structure before ring assembly), ii) in S-phase (stable septin collar; block by HU), and iii) in late anaphase (split septin rings; cdc15-1 temperature-sensitive mutant).
The cdc15-1-allele has been described to arrest the cell cycle in late anaphase [28], before the splitting of the septin collar. However, we found that cdc15-1-arrested cells displayed split septin rings to a high percentage of about 86%. The reduction of the temperature during the visualization- or sample preparation process probably led to a partial release from the arrest and subsequently promoted the splitting of the “instable” septin collar. The associated interaction partners might have partially accompanied this change in septin structure, and the identified interactome might represent more the split septin architecture than the mitotic septin ring. However, the situation is not as clear-cut as described as we find Bni5 as interaction partner of the split septin ring in cdc15-1 cells whereas Bni5 was shown in wild type cells to leave the septin ring cells shortly before splitting [37,38].
Using Cdc11-TAP as target, our quantitative MS analyses revealed 83 potential new physical interaction partners of Cdc11. It is however important to realize that not Cdc11 alone but the intact rod forms the bait during the affinity precipitations. All found interaction partners are therefore binders of the septin rod. To reveal the identity of the septin subunit that interacts with a certain partner protein would require additional experiments that exceed the scope of this work.
The interaction of 26 selected candidate proteins was further validated in vivo by steady-state SPLIFF measurements. We chose SPLIFF as a truly orthogonal validation technique and deliberately refrained from the commonly used IPs. While IPs represent a well established in vitro validation method, SPLIFF independently measures interactions in their native environment inside the living cell.
SPLIFF, in contrast to the standard Split-Ubiquitin assay, allows the detection of weak interactions because no complete degradation of the Cub-attached reporter is required to obtain an interaction signal [21]. The standard SPLIFF method requires the time-resolved measurement of the interaction and is thereby not suitable for screening a large number of interaction partners. To circumvent this limitation, we established the steady-state SPLIFF. Diploid cells expressing Shs1-CCG and a Nub-candidate fusion protein were grown to exponential phase and subjected directly to fluorescence microscopy. This method benefits from the feature of the septins to form a stable structure at the bud neck during nearly the complete cell cycle that comprises the inherited as well as the newly synthesized Shs1-CCG molecules [45].
We used Shs1 as attachment site for the CCG module as in contrast to the other tested septin subunits, Shs1-CCG looked indistinguishable from the GFP-labeled septins during the cell cycle. However, by restricting the SPLIFF substrate to the terminal Shs1 we had to keep in mind that a) the three-dimensional structure of Shs1 containing septins might differ from the one of Cdc11 containing septins [11] and b) that the analysis will sample only those interactors that locate close the terminal subunits of the septin rods. Proteins that bind to the center of the octamers or exclusively to Cdc11 might thus not be detected. Without knowledge of the nearest subunit neighbors of Shs1 in the different higher-order septin structures it is difficult to discuss why a certain interaction revealed by TAP precipitation is not seen by SPLIFF. Bni4 is a relevant example. This protein comes up as interaction partner in the AP-MS analysis during all three stages but is not revealed by SPLIFF as interaction partners of the septins. Bni4 was shown by Two-Hybrid analysis to interact only with Cdc10 [35,36], the central subunit of the septin rod. The central location of the coupled Nub might thus prevent the generation of a SPLIFF signal with a peripherically located Shs1-CCG. Again, repeating the SPLIFF analysis with all other septin subunits as CCG-fusions could eliminate some of the encountered discrepancies. However, the majority of proteins that were evaluated as positive interactors in the pulldown analysis were also found as positive interactors in the SPLIFF analysis.
In particular the prevalence of nuclear proteins and proteins involved in ribosome biogenesis among the septin interactors is puzzling as the septins were never reported to stay in the nucleus. In alpha-factor treated cells, proteins of these two categories represent the majority of the identified interactors (together 23 proteins, 57.5%). Of these proteins, 20 are described to localize to the nucleus or nucleolus [46] with the majority (17) being involved in ribosomal RNA biosynthesis and processing or biogenesis of ribosomal subunits as annotated in the Saccharomyces genome data base (SGD). Before flatly rejecting these hits as artefacts of abundant and perhaps sticky proteins one has to consider three arguments that support the specificity of the measured interactions. 1. Most of those hits are only reported for alpha-factor-treated cells and not for the other two cell cycle stages. 2. Three proteins involved in ribosomal RNA processing (Nop4, New1 and Rlp7) and one nuclear protein, Cse1, were validated as septin interactors by SPLIFF analysis. 3. The applied SILAC approach is especially good in avoiding these kind of artefacts. Independent of these considerations, the potential cellular significance of these interactions remains enigmatic.
Three endocytic proteins were found to interact with the septins at the three different time points: Vps1 in early alpha factor arrested cells, Sla2 in S-phase and Syp1 in late anaphase.
Of these only Syp1, which is involved in endocytic site formation, was already shown to interact with the septins [32,47]. Furthermore, a role of Syp1 in maintaining septin ring stability has been suggested [48]. The mechanism proposed by Merlini et al. opens the possibility that Syp1 is involved in the coordination of membrane remodeling controlled by mechanical stress at the septin ring.
Overall, the identification of endocytic proteins as septin interactors supports the assumption that septins foster endocytosis and bias the polarized distribution of cargo material in the plasma membrane [49].
The septins are known targets of cell cycle regulated post-translational modifications. Phosphorylations, SUMOylations and N-acetylations of the septins are involved in the transitions of the septin structure during the cell cycle [10]. Our quantitative MS-analysis revealed distinct sets of regulatory proteins that might perform these modifications at different stages of the cell cycle. In particular, the set of regulatory proteins in cells with a stable collar varies considerably from the set of proteins associated with split rings. The kinase Gin4 and the anillin-like protein Bud4 were identified in both time points. Three kinases, Hsl1, Mck1 and Prk1 are specifically associated with the septins in cells with a stable collar. Another kinase (Ste20) and yeast SUMO (Smt3) were only identified as specific septin interactors in cells with split septin rings. The septins Cdc3, Cdc11 and Shs1 are known to be SUMOylated at the G2/M transition and de-SUMOylated shortly before cytokinesis [16]. This timing agrees well with Smt3 being precipitated with the septins only from the extracts of the cdc15-1 arrested cells.
For three of the five identified kinases (Gin4, Hsl1 and Ste20), a physical interaction with the septins is described in literature [39,40,50,51]. Gin4 is recruited together with the septins to the presumptive bud site, co-localizes with the septins at the bud neck for the complete cell cycle and disappears from the bud neck after splitting of the septin collar [52]. This timing is in good agreement with our MS analysis. The Gin4 homologue in C. albicans was recently shown to regulate Nap1 by phosphorylation, a protein being directly involved in septin organization [53]. Gin4 and Nap1 were both identified in this study as interaction partners of the septins in cells with a stable septin collar and split rings. Our data thus support the assumption that Gin4 possesses the same functions in S. cerevisiae as discovered in C. albicans.
Recently, it was shown that Bud4 possesses a crucial role in the assembly of the axial landmark. Bud4 is recruited to the bud neck via an association with the septins and builds up a platform for the assembly of the other components of the axial landmark [41,54].
Bud3 and Bud4 were thought to interdependently and simultaneously bind to the septins after disassembly from this landmark. However, our MS study and subsequent steady state SPLIFF analysis suggested that Bud3 binds to the septins later than Bud4 although both proteins always localize together at the bud neck.
We used a time and spatially resolved SPLIFF analysis to explain this discrepancy. We could show that Bud4 indeed interacts with the septins at least 6 min earlier than Bud3. After this lag time both Nub-fusions convert Shs1-CCG with the same kinetics and cease to interact with Shs1-CCG at exactly the same time. During the splitting of the septins the interactions suddenly resume.
Without further knowledge about the nature of the interactions between the septins and Bud4 and Bud3 these striking kinetic patterns are difficult to explain. However, a simple model can account for this pattern if one assumes that the interaction between the septins and Bud3 and Bud4 are supported by two different post-translational modifications of the septins. The model would than postulate that the first modification precedes and perhaps is required for the second modification and that Bud4 binds preferentially to septins harbouring the first and Bud3 to septins harbouring the second modification.
Supporting Information
Saturated overnight cultures grown in “heavy” SILAC medium were diluted into fresh “heavy” SILAC medium. Cells were incubated at 30°C for 7.5 h and the incorporation of “heavy” amino acids was analyzed by MS. The percentage of all measured peptide pairs was plotted against the percentage of incorporation.
(TIF)
Representative overviews of affinity purifications of Cdc11-fusions from extracts of α-factor- (I), HU- (II) or cdc15-1-arrested cells (III). From left to right: anti-Protein A Western Blots of the purification of Cdc11-TAP (arrowhead: TEV-protease cleaved Protein A), anti-GFP Western Blots of the control Cdc11-GFP, colloidal Coomassie stains of the eluates. The bands were subsequently excised and used for quantitative MS-analyses. Ex: extract, S: supernatant, P: pellet, FT: flow-through, W: wash, E: Elution with TEV-Protease, BB: boiled beads. MAP: "mixing after purification".
(TIF)
Venn diagrams showing the overlap of all identified proteins in the three replicates of (A) G1-phase (alpha factor), (B) S1-phase, (C) anaphase. Specific interactors were grouped into the indicated categories. Percentages of specific interactors for each category are presented.
(TIF)
Fluorescence microscopy of cells expressing Cdc3-CCG, Cdc10-CCG, Cdc11-CCG, Cdc12-CCG and Shs1-CCG. Only cells expressing Shs1-CCG do not display a visible phenotype. Scale bar 5 μm.
(TIF)
Each point represents a single cell measurement. The calculated medians and SEMs are shown in red. Nub-fusions are grouped according to their interaction signal. (A) Strong interaction (SPLIFF signal ≥ 70%). (B) Weak interaction (SPLIFF signal ≥ 30%). (C) No interaction (SPLIFF signal ≤ 30%). grey squares: negative control Nub-empty; grey circle: positive control Nub-Cdc11.
(TIF)
Fluorescence microscopy images of diploid cells expressing Shs1-CCG and Nub-Smt3 (from plasmid) at 0 μM Cu2+ (upper panel) and 100 μM Cu2+. Scale bar 3 μm.
(TIF)
(A) Beads covalently captured with recombinant septin rods or SNAP tag were incubated with extracts from yeast expressing the indicated TAP tagged (Vps1: GFP-tagged) candidate proteins. After washing, specific binders were eluted in Lämmli buffer and monitored via an anti-protein A or anti-GFP antibody. Left lanes: immobilized SNAP tagged septin rods. Right lanes: SNAP tag protein (control). The band at approx. 70 kD results from a cross reaction of the primary antibody with one of the septins. (B) Unspecific binding of New1 to the pulldown matrix. (C) Rlp1 does not bind to immobilized septin rods (left). The loading control including extract (Ex) and Pellet (P) indicates soluble expressio (right). (D) Representative input control: the 6his-tagged septin rod subunit Cdc12 and 6his-SNAP were detected with an anti-his antibody. For each candidate a representative blot out of minimum two independent replicates is shown. The asterisk marks the expected protein size.
(TIF)
(PDF)
Proteins identified in AP-MS analyses of Cdc11-complexes purified from cells arrested woth alpha factor (G1) (A), S1 phase (B) and late anaphase (C).
(XLSX)
(PDF)
(PDF)
Acknowledgments
We thank Bettina Knapp for excellent technical assistance in sample preparation and LC/MS analyses. Jonas Schepper is acknowledged for performing replicates of the pulldowns. Research in the BW laboratory is supported by the Deutsche Forschungsgemeinschaft and the Excellence Initiative of the German Federal & State Governments (EXC 294 BIOSS Centre for Biological Signalling Studies).
CR was supported by a grant of the Bundesministerium für Bildung und Forschung (BMBF Initiative SysTec 0315690B) to NJ.
Deposition of the data to the ProteomeXchange Consortium is supported by the PRIDE Team, EBI.
Data Availability
Our mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD002561. The reviewer account details are as follows: Username: reviewer18280@ebi.ac.uk Password: TgDhlwNj All other relevant data are within the paper and its Supporting Information files.
Funding Statement
Research in the BW laboratory is supported by the Deutsche Forschungsgemeinschaft and the Excellence Initiative of the German Federal & State Governments (EXC 294 BIOSS Centre for Biological Signalling Studies). CR was supported by a grant of the Bundesministerium für Bildung und Forschung (BMBF Initiative SysTec 0315690B) to NJ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Khan A, McQuilken M, Gladfelter AS. Septins and Generation of Asymmetries in Fungal Cells. Annu Rev Microbiol. 2015;69: 487–503. 10.1146/annurev-micro-091014-104250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertin A, McMurray MA, Grob P, Park SS, Garcia G 3rd, Patanwala I, et al. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembl. Proc Natl Acad Sci U S A. 2008;105: 8274–9. 10.1073/pnas.0803330105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finnigan GC, Takagi J, Cho C, Thorner J. Comprehensive Genetic Analysis of Paralogous Terminal Septin Subunits Shs1 and Cdc11 in Saccharomyces cerevisiae. Genetics. 2015;200: 821–841. 10.1534/genetics.115.176495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMurray MA, Thorner J. Septins: molecular partitioning and the generation of cellular asymmetry. Cell Div. 2009;4: 18 10.1186/1747-1028-4-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwase M, Luo J, Nagaraj S, Longtine M, Kim HB, Haarer BK, et al. Role of a Cdc42p effector pathway in recruitment of the yeast septins to the presumptive bud site. Mol Biol Cell. 2006;17: 1110–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMurray MA, Thorner J. Reuse, replace, recycle. Specificity in subunit inheritance and assembly of higher-order septin structures during mitotic and meiotic division in budding yeast. Cell Cycle. 2009;8: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang CS, Reed SI. Phosphorylation of the septin cdc3 in g1 by the cdc28 kinase is essential for efficient septin ring disassembly. Cell Cycle. 2002;1: 42–49. [PubMed] [Google Scholar]
- 8.Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. Septin assemblies form by diffusion-driven annealing on membranes. Proc Natl Acad Sci U S A. 2014;111: 2146–2151. 10.1073/pnas.1314138111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchell L, Lau A, Lambert JP, Zhou H, Fong Y, Couture JF, et al. Regulation of septin dynamics by the Saccharomyces cerevisiae lysine acetyltransferase NuA4. PLoS One. 2011;6: e25336 10.1371/journal.pone.0025336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez-Rodriguez Y, Momany M. Posttranslational modifications and assembly of septin heteropolymers and higher-order structrues. Curr Opin Microbiol. 2012;15: 660–668. 10.1016/j.mib.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 11.Garcia G 3rd, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, et al. Subunit-dependent modulation of septin assembly: Budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol. 2011;195: 993–1004. 10.1083/jcb.201107123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Versele M, Thorner J. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J Cell Biol 2004;164: 701–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMay BS, Bai X, Howard L, Occhipinti P, Meseroll RA, Spiliotis ET, et al. Septin filaments exhibit a dynamic, paired organization that is conserved from yeast to mammals. J Cell Biol 2011;193: 1065–81. 10.1083/jcb.201012143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ong K, Wloka C, Okada S, Svitkina T, Bi E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat Commun. 2014;5: 5698 10.1038/ncomms6698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mortensen EM, McDonald H, Yates J 3rd, Kellogg DR. Cell cycle-dependent assembly of a Gin4-septin complex. Mol Biol Cell. 2002;13: 2091–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson ES, Blobel G. Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J Cell Biol. 1999;147: 981–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakahira M, Macedo JN, Seraphim TV, Cavalcante N, Souza TA, Damalio JC, et al. A draft of the human septin interactome. PLoS One 2010;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh Y, Bi E. Septin structure and function in yeast and beyond. Trends Cell Biol. 2011;21: 141–148. 10.1016/j.tcb.2010.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walther TC, Mann M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 2010;190: 491–500. 10.1083/jcb.201004052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oeljeklaus S, Meyer HE, Warscheid B. New dimensions in the study of protein complexes using quantitative mass spectrometry. FEBS Lett. 2009;583: 1674–83. 10.1016/j.febslet.2009.04.018 [DOI] [PubMed] [Google Scholar]
- 21.Moreno D, Neller J, Kestler HA, Kraus J, Dunkler A, Johnsson N. A fluorescent reporter for mapping cellular protein-protein interactions in time and space. Mol Syst Biol. 2013;9: 647 10.1038/msb.2013.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiese S, Gronemeyer T, Ofman R, Kunze M, Grou CP, Almeida JA, et al. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol Cell Proteomics. 2007;6: 2045–2057. [DOI] [PubMed] [Google Scholar]
- 23.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26: 1367–1372. 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 24.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10: 1794–1805. 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- 25.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32: 223–226. 10.1038/nbt.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunkler A, Rosler R, Kestler HA, Moreno-Andres D, Johnsson N. SPLIFF: A Single-Cell Method to Map Protein-Protein Interactions in Time and Space. Methods Mol Biol. 2015;1346: 151–168. 10.1007/978-1-4939-2987-0_11 [DOI] [PubMed] [Google Scholar]
- 27.Renz C, Johnsson N, Gronemeyer T. An efficient protocol for the purification and labeling of entire yeast septin rods from E. coli for quantitative in vitro experimentation. BMC Biotechnol. 2013;13: 60-6750-13-60. 10.1186/1472-6750-13-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jimenez J, Cid VJ, Cenamor R, Yuste M, Molero G, Nombela C, et al. Morphogenesis beyond cytokinetic arrest in Saccharomyces cerevisiae. J Cell Biol. 1998;143: 1617–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1: 376–386. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Huang L. Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol Cell Proteomics. 2008;7: 46–57. [DOI] [PubMed] [Google Scholar]
- 31.Oeljeklaus S, Reinartz BS, Wolf J, Wiese S, Tonillo J, Podwojski K, et al. Identification of core components and transient interactors of the peroxisomal importomer by dual-track stable isotope labeling with amino acids in cell culture analysis. J Proteome Res. 2012;11: 2567–2580. 10.1021/pr3000333 [DOI] [PubMed] [Google Scholar]
- 32.Qiu W, Neo SP, Yu X, Cai M. A novel septin-associated protein, Syp1p, is required for normal cell cycle-dependent septin cytoskeleton dynamics in yeast. Genetics. 2008;180: 1445–1457. 10.1534/genetics.108.091900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, et al. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3: 958–965. [DOI] [PubMed] [Google Scholar]
- 34.Muller J, Johnsson N. Split-Ubiquitin and the Split-Protein Sensors: Chessman for the Endgame. ChemBioChem. 2008;9: 2029–2038. 10.1002/cbic.200800190 [DOI] [PubMed] [Google Scholar]
- 35.DeMarini DJ, Adams AE, Fares H, De Virgilio C, Valle G, Chuang JS, et al. A septin-based hierarchy of proteins required for localized deposition of chitin in the Saccharomyces cerevisiae cell wall. J Cell Biol. 1997;139: 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pablo-Hernando ME, Arnaiz-Pita Y, Tachikawa H, del Rey F, Neiman AM, Vazquez de Aldana CR. Septins localize to microtubules during nutritional limitation in Saccharomyces cerevisiae. BMC Cell Biol. 2008;9: 55 10.1186/1471-2121-9-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider C, Grois J, Renz C, Gronemeyer T, Johnsson N. Septin rings act as a template for myosin higher-order structures and inhibit redundant polarity establishment. J Cell Sci. 2013;126: 3390–3400. 10.1242/jcs.125302 [DOI] [PubMed] [Google Scholar]
- 38.Finnigan GC, Booth EA, Duvalyan A, Liao EN, Thorner J. The Carboxy-Terminal Tails of Septins Cdc11 and Shs1 Recruit Myosin-II Binding Factor Bni5 to the Bud Neck in Saccharomyces cerevisiae. Genetics. 2015;200: 821–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanrahan J, Snyder M. Cytoskeletal activation of a checkpoint kinase. Mol Cell. 2003;12: 663–73. [DOI] [PubMed] [Google Scholar]
- 40.Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438: 679–684. [DOI] [PubMed] [Google Scholar]
- 41.Kang PJ, Hood-DeGrenier JK, Park HO. Coupling of septins to the axial landmark by Bud4 in budding yeast. J Cell Sci. 2013;126: 1218–1226. 10.1242/jcs.118521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chant J, Mischke M, Mitchell E, Herskowitz I, Pringle JR. Role of Bud3p in producing the axial budding pattern of yeast. J Cell Biol. 1995;129: 767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wloka C, Nishihama R, Onishi M, Oh Y, Hanna J, Pringle JR, et al. Evidence that a septin diffusion barrier is dispensable for cytokinesis in budding yeast. Biol Chem. 2011;392: 813–829. 10.1515/BC.2011.083 [DOI] [PubMed] [Google Scholar]
- 44.Sanders SL, Herskowitz I. The BUD4 protein of yeast, required for axial budding, is localized to the mother/BUD neck in a cell cycle-dependent manner. J Cell Biol. 1996;134: 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McMurray MA, Thorner J. Septin stability and recycling during dynamic structural transitions in cell division and development. Curr Biol. 2008;18: 1203–8. 10.1016/j.cub.2008.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425: 686–91. [DOI] [PubMed] [Google Scholar]
- 47.Stimpson HE, Toret CP, Cheng AT, Pauly BS, Drubin DG. Early-arriving Syp1p and Ede1p function in endocytic site placement and formation in budding yeast. Mol Biol Cell. 2009;20: 4640–51. 10.1091/mbc.E09-05-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merlini L, Bolognesi A, Juanes MA, Vandermoere F, Courtellemont T, Pascolutti R, et al. Rho1- and Pkc1-dependent phosphorylation of the F-BAR protein Syp1 contributes to septin ring assembly. Mol Biol Cell. 2015;26: 3245–3262. 10.1091/mbc.E15-06-0366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Layton AT, Savage NS, Howell AS, Carroll SY, Drubin DG, Lew DJ. Modeling vesicle traffic reveals unexpected consequences for Cdc42p-mediated polarity establishment. Curr Biol. 2011;21: 184–194. 10.1016/j.cub.2011.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carroll CW, Altman R, Schieltz D, Yates JR, Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J Cell Biol. 1998;143: 709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13: 176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Longtine MS, Fares H, Pringle JR. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J Cell Biol. 1998;143: 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang ZX, Zhao P, Zeng GS, Wang YM, Sudbery I, Wang Y. Phosphoregulation of Nap1 plays a role in septin ring dynamics and morphogenesis in Candida albicans. MBio. 2014;5: e00915–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang PJ, Angerman E, Jung CH, Park HO. Bud4 mediates the cell-type-specific assembly of the axial landmark in budding yeast. J Cell Sci. 2012;125: 3840–3849. 10.1242/jcs.103697 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Saturated overnight cultures grown in “heavy” SILAC medium were diluted into fresh “heavy” SILAC medium. Cells were incubated at 30°C for 7.5 h and the incorporation of “heavy” amino acids was analyzed by MS. The percentage of all measured peptide pairs was plotted against the percentage of incorporation.
(TIF)
Representative overviews of affinity purifications of Cdc11-fusions from extracts of α-factor- (I), HU- (II) or cdc15-1-arrested cells (III). From left to right: anti-Protein A Western Blots of the purification of Cdc11-TAP (arrowhead: TEV-protease cleaved Protein A), anti-GFP Western Blots of the control Cdc11-GFP, colloidal Coomassie stains of the eluates. The bands were subsequently excised and used for quantitative MS-analyses. Ex: extract, S: supernatant, P: pellet, FT: flow-through, W: wash, E: Elution with TEV-Protease, BB: boiled beads. MAP: "mixing after purification".
(TIF)
Venn diagrams showing the overlap of all identified proteins in the three replicates of (A) G1-phase (alpha factor), (B) S1-phase, (C) anaphase. Specific interactors were grouped into the indicated categories. Percentages of specific interactors for each category are presented.
(TIF)
Fluorescence microscopy of cells expressing Cdc3-CCG, Cdc10-CCG, Cdc11-CCG, Cdc12-CCG and Shs1-CCG. Only cells expressing Shs1-CCG do not display a visible phenotype. Scale bar 5 μm.
(TIF)
Each point represents a single cell measurement. The calculated medians and SEMs are shown in red. Nub-fusions are grouped according to their interaction signal. (A) Strong interaction (SPLIFF signal ≥ 70%). (B) Weak interaction (SPLIFF signal ≥ 30%). (C) No interaction (SPLIFF signal ≤ 30%). grey squares: negative control Nub-empty; grey circle: positive control Nub-Cdc11.
(TIF)
Fluorescence microscopy images of diploid cells expressing Shs1-CCG and Nub-Smt3 (from plasmid) at 0 μM Cu2+ (upper panel) and 100 μM Cu2+. Scale bar 3 μm.
(TIF)
(A) Beads covalently captured with recombinant septin rods or SNAP tag were incubated with extracts from yeast expressing the indicated TAP tagged (Vps1: GFP-tagged) candidate proteins. After washing, specific binders were eluted in Lämmli buffer and monitored via an anti-protein A or anti-GFP antibody. Left lanes: immobilized SNAP tagged septin rods. Right lanes: SNAP tag protein (control). The band at approx. 70 kD results from a cross reaction of the primary antibody with one of the septins. (B) Unspecific binding of New1 to the pulldown matrix. (C) Rlp1 does not bind to immobilized septin rods (left). The loading control including extract (Ex) and Pellet (P) indicates soluble expressio (right). (D) Representative input control: the 6his-tagged septin rod subunit Cdc12 and 6his-SNAP were detected with an anti-his antibody. For each candidate a representative blot out of minimum two independent replicates is shown. The asterisk marks the expected protein size.
(TIF)
(PDF)
Proteins identified in AP-MS analyses of Cdc11-complexes purified from cells arrested woth alpha factor (G1) (A), S1 phase (B) and late anaphase (C).
(XLSX)
(PDF)
(PDF)
Data Availability Statement
Our mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD002561. The reviewer account details are as follows: Username: reviewer18280@ebi.ac.uk Password: TgDhlwNj All other relevant data are within the paper and its Supporting Information files.