

ABSTRACT
Neisseria meningitidis colonizes the nasopharyngeal mucosa of healthy populations asymptomatically, although the bacterial surface is rich in motifs that activate the host innate immunity. What determines the tolerant host response to this bacterium in asymptomatic carriers is poorly understood. We demonstrated that the conserved meningococcal surface protein NhhA orchestrates monocyte (Mo) differentiation specifically into macrophage-like cells with a CD200Rhi phenotype (NhhA-Mφ). In response to meningococcal stimulation, NhhA-Mφ failed to produce proinflammatory mediators. Instead, they upregulated interleukin-10 (IL-10) and Th2/regulatory T cell (Treg)-attracting chemokines, such as CCL17, CCL18, and CCL22. Moreover, NhhA-Mφ were highly efficient in eliminating bacteria. The in vivo validity of these findings was corroborated using a murine model challenged with N. meningitidis systematically or intranasally. The NhhA-modulated immune response protected mice from septic shock; Mo/Mφ depletion abolished this protective effect. Intranasal administration of NhhA induced an anti-inflammatory response, which was associated with N. meningitidis persistence at the nasopharynx. In vitro studies demonstrated that NhhA-triggered Mo differentiation occurred upon engaged Toll-like receptor 1 (TLR1)/TLR2 signaling and extracellular signal-regulated kinase (ERK) and Jun N-terminal protein kinase (JNK) activation and required endogenously produced IL-10 and tumor necrosis factor alpha (TNF-α). Our findings reveal a strategy that might be adopted by N. meningitidis to maintain asymptomatic nasopharyngeal colonization.
IMPORTANCE
Neisseria meningitidis is an opportunistic human-specific pathogen that colonizes the nasopharyngeal mucosa asymptomatically in approximately 10% of individuals. Very little is known about how this bacterium evades immune activation during the carriage stage. Here, we observed that N. meningitidis, via the conserved surface protein NhhA, skewed monocyte differentiation into macrophages with a CD200Rhi phenotype. Both in vivo and in vitro data demonstrated that these macrophages, upon meningococcal infection, played an important role in forming a homeostatic immune microenvironment through their capacity to eliminate invading bacteria and to generate anti-inflammatory mediators. This work provides novel insight into the mechanisms underlying the commensal persistence of N. meningitidis.
INTRODUCTION
The Gram-negative bacterium Neisseria meningitidis is the leading cause of bacterial meningitis and septicemia. Despite the relatively low incidence rates of the disease, meningococci are obligate commensals in humans and are present in the upper respiratory tract of approximately 10% of healthy individuals asymptomatically (1). The rates of meningococcal carriage are even higher during the teenage years and early adulthood (2) and among semiclosed populations (3). A variety of surface components on meningococci, such as endotoxin lipooligosaccharide (LOS), are potent proinflammatory factors that can activate the host innate immune response (4–6); the strategies used by this bacterium to limit local inflammation and establish the carriage status remain unclear.
Monocytes (Mo) represent a large pool of circulating precursors that can differentiate into macrophages (Mφ) and migrating dendritic cells (DCs) and thereby perform important regulatory functions in innate and adaptive immunity (7). Furthermore, the Mo-derived Mφ exhibit a substantial heterogeneity of phenotypes whose immunomodulatory properties in host defense are dictated by the external inflammatory milieu (8). Classically activated Mφ (referred to as M1Mφ) polarized in response to Th1 cytokines (e.g., gamma interferon [IFN-γ]) are characterized by the production of proinflammatory mediators and cytotoxic effector molecules such as nitric oxide (NO) and reactive oxygen intermediates. Thus, M1Mφ play a dominant role in controlling acute infectious diseases but have the potential to cause tissue damage and septic shock upon aberrant activation. In contrast, Th2 cytokines (e.g., interleukin-4 [IL-4] and IL-13) polarize Mφ toward an alternatively activated phenotype (also known as M2Mφ); these cells produce large amounts of anti-inflammatory mediators and play an important role in counteracting harmful inflammatory responses. In addition, increased expression of arginases (Arg) by M2Mφ is associated with tissue healing. It has been observed that M2Mφ may generate a survival niche that contributes to chronic infections induced by pathogens such as Salmonella enterica serovar Typhimurium (9), Mycobacterium tuberculosis (10), and Helicobacter pylori (11).
Cytokines endogenously produced by host cells upon activation by microbial components play an essential role in orchestrating the dynamic programming of Mφ. Although both LOS and non-LOS surface components on N. meningitidis have been demonstrated to be able to target monocytes and trigger cytokine production (6, 12), whether N. meningitidis can modulate the functional polarization of Mφ and its impact on bacterial carriage remains unclear.
Neisseria Hia/Hsf homologue A (NhhA) belongs to the family of trimeric autotransporter adhesins and is a conserved surface protein in a number of disease-associated N. meningitidis strains (13). Therefore, NhhA has been considered to be a potential vaccine candidate (14). NhhA plays a variety of roles in promoting bacterial survival, including mediating bacterial interaction with epithelial cells (15) and enhancing bacterial complement resistance via binding to activated vitronectin (16). Our previous studies demonstrated the immunostimulatory properties of NhhA on Mφ (5), which led us to examine its effect on Mo activation and functional differentiation. In this study, we identified a novel function of NhhA in programing Mo differentiation toward Mφ with a CD200Rhi immune homeostatic phenotype (NhhA-Mφ). In vitro and in vivo studies revealed that NhhA-Mφ alleviated inflammatory response upon meningococcal stimulation, which was associated with enhanced bacterial colonization. Overall, modulating Mφ polarization may represent a previously undescribed strategy though which N. meningitidis potentiates host immune tolerance and ensures asymptomatic bacterial carriage at the nasopharyngeal mucosa.
RESULTS
NhhA skews monocyte differentiation toward macrophages but not dendritic cells.
Previously, we observed that NhhA induces the production of cytokines in Mφ (5), which led us to examine its effect on the activation and functional differentiation of monocytes. We found that NhhA is able to induce substantial amounts of inflammatory mediators in human peripheral monocytes in a dose-dependent manner (data not shown). Furthermore, we noticed morphological changes in Mo following NhhA treatment—the cells were clustered, with enhanced adhesion and increased cytoplasmic granularity (Fig. 1A and data not shown). The differentiating action of NhhA was both time and dose dependent, with the strongest effect observed when Mo were treated with NhhA (50 nM) for 3 days (data not shown). In contrast, NhhA subjected to heat treatment failed to induce Mo differentiation (data not shown), ruling out the possibility that the observed effect of NhhA was caused by lipopolysaccharide (LPS) contamination. Prolonged NhhA treatment (5 to 7 days), at a concentration of 50 nM, did not enhance cell maturation but led to increased amounts of dead cells, as observed by dual staining of annexin V (data not shown), likely because of its apoptotic effect on Mφ (17). Based on these observations, NhhA (50 nM) and a 3-day incubation period were subsequently used to induce monocyte differentiation, unless otherwise specified.
FIG 1 .

NhhA induces the differentiation of monocytes (Mo) into macrophages. (A to C) Human peripheral Mo were differentiated with 50 nM NhhA for 3 days. Primary Mo, macrophage colony-stimulating factor (M-CSF)-derived Mφ, and GM-CSF/IL-4-derived dendritic cells (DCs) are shown for comparison. (A) Representative differential interference contrast images of cells (original magnification, ×400). CD14 and CD1a expression of the PI-negative gate was analyzed by flow cytometry. (B) Fluorescence-activated cell sorting plots of one representative experiment. (C) Median fluorescence intensity (MFI) of CD14. Error bars indicate medians with interquartile ranges from three independent experiments. *, P < 0.05 using ANOVA followed by the Bonferroni post hoc test. (D and E) Mo were infected with wild-type (WT) or NhhA-deficient (ΔNhhA) FAM20 meningococcal strains (MOI, 10) for 3 days. Expression of CD14 and CD1a was determined by flow cytometry. (D) Representative fluorescence-activated cell sorting plots. (E) Mean ± standard deviation of the percentages of CD14hi cells from three independent experiments. Untreated Mo were used as the negative control. *, P < 0.05 using ANOVA followed by the Bonferroni post hoc test.
Flow cytometric analysis showed that the expression of membrane CD14, but not CD1a (a surrogate marker for DCs), was highly induced on NhhA-differentiated cells (Fig. 1B and C), suggesting an Mφ-like phenotype. To ascertain the endogenous effect of NhhA on Mo-Mφ differentiation, we stimulated Mo with the FAM20 wild-type (WT) strain or an isogenic NhhA-deficient mutant (ΔNhhA) strain. We found that the lack of NhhA significantly attenuated the differentiating effect of the bacteria, as assessed by morphology, intracellular granularity, and attachment (data not shown). At the same time, decreased amounts of CD14hi cells (44.6% ± 5.4% versus 23.4% ± 3.7%, P = 0.007) were observed (Fig. 1D and E). Intriguingly, the expression of CD1a was not affected (Fig. 1D), suggesting that neither the WT nor the ΔNhhA strain stimulated the conversion of Mo to DCs. Together, these data demonstrated a novel immunomodulatory role for NhhA in skewing the differentiation of Mo specifically toward Mφ.
The differentiating effect of NhhA requires the full passenger domain.
The passenger domain of NhhA consists of three regions, in which the middle region, with two conserved modules, contains the binding site for heparan sulfate (18). To map the functional region of NhhA that triggers Mo differentiation, we generated various forms of NhhA resembling the N-terminal, middle, and C-terminal regions of the passenger domain under native conditions and evaluated their potential to trigger differentiation. In contrast to NhhA-FL, the recombinant NhhA containing a full-length passenger domain (5), truncated NhhA proteins could neither bind cells (see Fig. S1A, upper panel, in the supplemental material) nor trigger Mo-Mφ differentiation, as determined by measuring side scatter (SSC) and CD14 expression of cells (see Fig. S1A, lower panel). NhhA trimerization (13) was detected by native polyacrylamide gel electrophoresis (see Fig. S1B), indicating a link between the structural organization of NhhA and its biological activity.
NhhA-triggered Mφ differentiation relies on TLR1/TLR2 activation.
Blocking cell endocytosis with cytochalasin D, a drug that inhibits actin polymerization, had no effect on differentiation, as assessed by measuring CD14 expression and SSC (Fig. 2A), suggesting that the NhhA-triggered Mφ differentiation was mediated by cell surface receptors. Surface Toll-like receptors (TLRs) play a crucial role in sensing microbial stimulation and triggering Mo differentiation (19). By using specific inhibitory antibodies to human TLRs, we found that blocking the activation of either TLR1 or TLR2 could significantly inhibit the action of NhhA. No effect on differentiation could be detected when TLR6 or TLR4 signaling was blocked, although inhibition of TLR4 partially attenuated NhhA-induced CD14 expression (Fig. 2A). Scavenger receptors of Mo/Mφ recognize various surface molecules on N. meningitidis (20) and mediate the nonopsonic uptake of bacteria, whereas heparan sulfate has been demonstrated to interact with NhhA in vitro (21). Nevertheless, Mo pretreated with dextran sulfate (DxSO4, a broad-range antagonist ligand of scavenger receptors) or heparinase III differentiated to a degree similar to that observed for control cells (Fig. 2A), suggesting that none of these factors plays an essential role in NhhA-triggered differentiation. These results indicate that the differentiating effect of NhhA is initiated through TLR1/TLR2 activation.
FIG 2 .
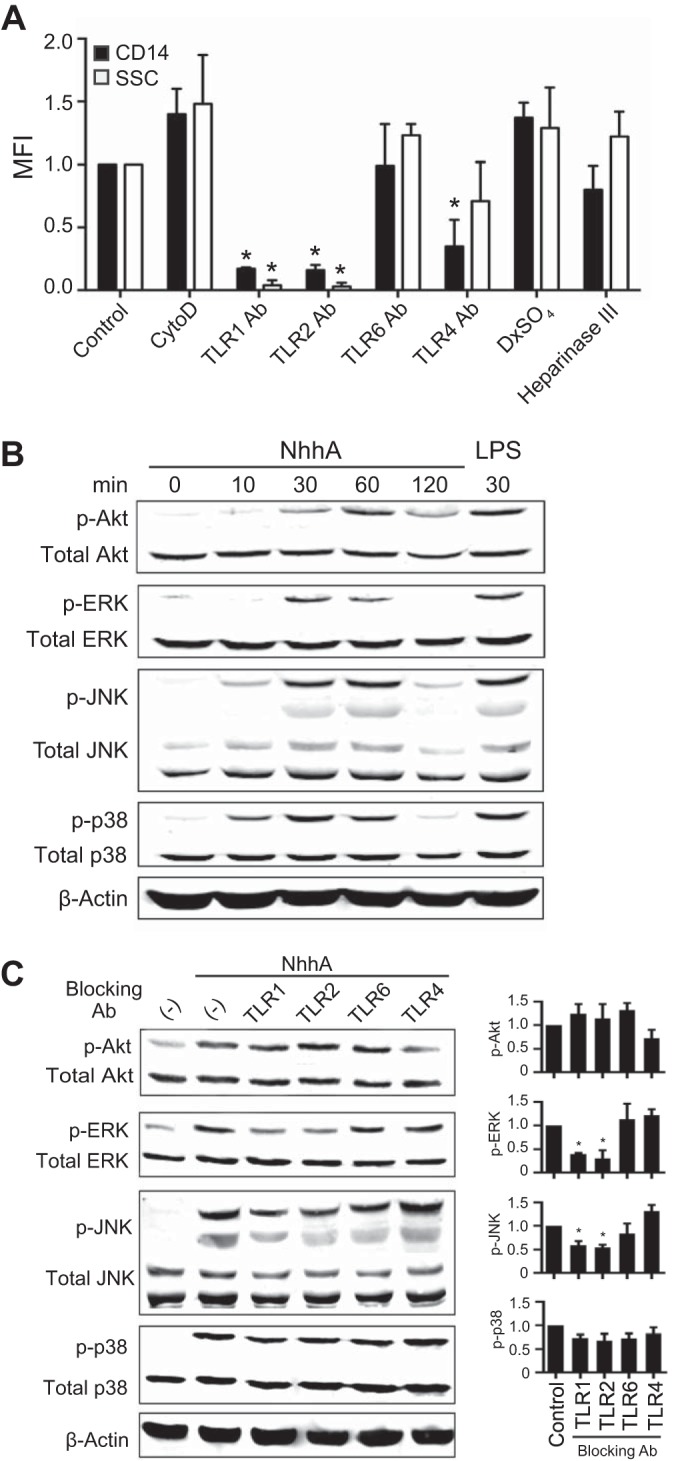
NhhA-triggered Mo differentiation is dependent on TLR1/TLR2 activation. (A) Mo were treated with 50 nM NhhA alone (control) or in the presence of cytochalasin D (1 µM; actin polymerization inhibitor), dextran sulfate (100 µg/ml; scavenger receptor antagonist), heparinase III (5 U/ml), or blocking antibodies (Abs) directed against human TLR1, TLR2, TLR4, and TLR6 (5 µg/ml) for 3 days. Differentiation to Mφ was determined by the flow cytometric analysis of CD14 expression and SSC parameters (shown as median fluorescence intensity [MFI]) and presented as fold change relative to the control. Error bars indicate medians with interquartile ranges of three independent experiments. *, P < 0.05 compared with the control using ANOVA followed by the Bonferroni post hoc test. (B) Mo were treated with 50 nM NhhA for the indicated time points, and whole-cell lysates were analyzed by Western blotting to detect phosphorylation of Akt, ERK, JNK, and p38. β-Actin was stained as the loading control. Mo treated with lipopolysaccharide (LPS; 100 ng/ml) for 30 min were used as the positive control. The data shown are representative of three independent experiments. (C) Mo were treated with 50 nM NhhA alone (control) or cotreated with neutralizing antibodies directed against human TLR1, TLR2, TLR4, and TLR6 (5 µg/ml) for 30 min (ERK, JNK, and p38) to 60 min (Akt). Phosphorylation of the indicated proteins was detected by Western blotting. One representative image is shown in the left panel. Densitometric analysis of phosphoprotein normalized to the respective total protein is shown in the right panel. Data shown are the mean ± standard deviation from three independent experiments and are presented as fold change relative to control. *, P < 0.05 compared with the control using ANOVA followed by the Bonferroni post hoc test.
Recognition by heterodimers of TLR1 and TLR2 activates the nuclear factor NF-κB, mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K)/Akt signaling pathways, which coordinately regulate differentiation- and inflammation-associated gene activities (19). Western blot analysis revealed rapid phosphorylation of Akt, extracellular signal-regulated kinases (ERKs), c-Jun N-terminal protein kinases (JNKs), and p38 in Mo 10 to 60 min after stimulation with NhhA (Fig. 2B). Blocking TLR1 or TLR2 signaling selectively reduced NhhA-triggered ERK and JNK phosphorylation, whereas Akt and p38 activations were not affected (Fig. 2C), indicating that another NhhA-sensing receptor(s) is involved in activating these signaling cascades. We further verified the involvement of the ERK and JNK signaling pathways in NhhA-triggered Mo differentiation by treating the cells with specific inhibitors for ERK (PD98059) and JNK (SP600125), respectively (see Fig. S2 in the supplemental material). Moreover, inhibition of NF-κB, but not activator protein 1 (AP-1) activity, eliminated the differentiating effects of NhhA (see Fig. S2).
Together, these data demonstrate a coordinating role for TLR1/TLR2-activated ERK/JNK MAPK and NF-κB signaling in Mφ differentiation triggered by NhhA.
NhhA-induced Mφ differentiation is dependent on endogenously produced IL-10 and TNF-α.
TLR1/2-mediated NF-κB activation regulates the inducible expression of a broad array of inflammatory mediators that promote Mφ differentiation (19, 22). To identify the factors underlying the differentiating effect of NhhA, we studied the transcriptional profile of a variety of genes coding for cytokines and growth factors in Mo upon NhhA stimulation by quantitative real-time PCR (qPCR) and searched for factors whose production was specifically TLR1/2 or NF-κB pathway dependent. We found that NhhA stimulation had no effect on the transcription of IL-4, IL-13, transforming growth factor β (TGF-β), IFN-γ, IL-15, macrophage colony-stimulating factor (M-CSF), or granulocyte-macrophage CSF (GM-CSF) (Fig. 3A and data not shown). In contrast, the transcription of IL-6, IL-12b, IL-10, tumor necrosis factor alpha (TNF-α), and IL-32 was markedly upregulated by NhhA (Fig. 3A). Furthermore, a blocking assay revealed that the induction of IL-6, IL-12b, IL-10, and TNF-α mRNA was dependent on TLR1/TLR2 or NF-κB signaling (Fig. 3A). To demonstrate the specific effect of the applied blocking antibodies, we treated cells with the isotype-matched control antibody and found that NhhA retained the capacity to induce IL-6 and TNF-α (data not shown). Furthermore, some cells treated with TLR-blocking antibodies were also stimulated with their specific agonists, such as PAM3CSK4, peptidoglycan, and LPS. Under this condition, cytokine production in both Mo and Mφ was specifically blocked (data not shown).
FIG 3 .

NhhA-triggered Mo differentiation is mediated by IL-10 and TNF-α. (A) Mo were stimulated with 50 nM NhhA alone (control) or in the presence of blocking antibodies (Abs) (5 µg/ml) to the indicated human TLRs. Some cells were cotreated with 500 nM Celastrol (an NF-κB inhibitor). RNA was extracted after 3 h, and the mRNA levels of the indicated cytokines were quantified by real-time PCR (qPCR). The data were normalized to the reference gene rpl37A and are presented as fold change relative to that of Mo without NhhA stimulation, which was set to 1. Data shown are the mean ± standard deviation from three independent experiments; *, P < 0.05 compared with the control using ANOVA followed by the Bonferroni post hoc test. (B) Mo were stimulated with 50 nM NhhA alone (control) or in the presence of the blocking agent for the indicated cytokines as described in Materials and Methods. The differentiation state was assessed by the flow cytometric analysis of CD14 expression and SSC parameters (shown as median fluorescence intensity [MFI]). Error bars indicate medians with interquartile ranges of three independent experiments. Data are presented as fold change relative to control; *, P < 0.05 compared with the control using ANOVA followed by the Bonferroni post hoc test.
To identify the mediators that play essential roles in NhhA-triggered Mo differentiation, we stimulated Mo with NhhA in the presence of neutralizing antibodies or inhibitors directed against the indicated cytokines. As shown in Fig. 3B, neutralizing IL-6 or IL-12b had no effects on cell differentiation. In contrast, neutralizing IL-10 or TNF-α diminished NhhA-induced Mo differentiation by 80% or 60%, respectively, as assessed by measuring cell granularity. Furthermore, neutralization of TNF-α reduced the expression of CD14, a coreceptor for detecting LPS, on NhhA-treated cells by nearly 50%. Consistent with the observation that NhhA has no effect on IL-15 and M-CSF transcription (Fig. 3A), blocking IL-15 or M-CSF signaling did not impair NhhA-induced differentiation (Fig. 3B). Moreover, inhibition of IL-32 activation did not show any effect on NhhA-mediated cell differentiation (Fig. 3B). These results provide evidence that NhhA programs Mφ differentiation upon TLR1/TLR2 activation in an IL-10/TNF-α-dependent manner.
NhhA induces CD200R expression on macrophages.
Next, we characterized the phenotype of NhhA-derived Mφ (NhhA-Mφ) by flow cytometry. Expression levels of proinflammatory cytokine-associated parameters, including molecules that support antigen presentation (HLA-DR, CD86, and CD80) or sense bacterial infection (TLR4), were low in NhhA-Mφ (Fig. 4 and data not shown). In contrast, expression of the mannose receptor CD206 and CD200R, a negative regulator of Mφ activation, was significantly upregulated in these cells (Fig. 4).
FIG 4 .
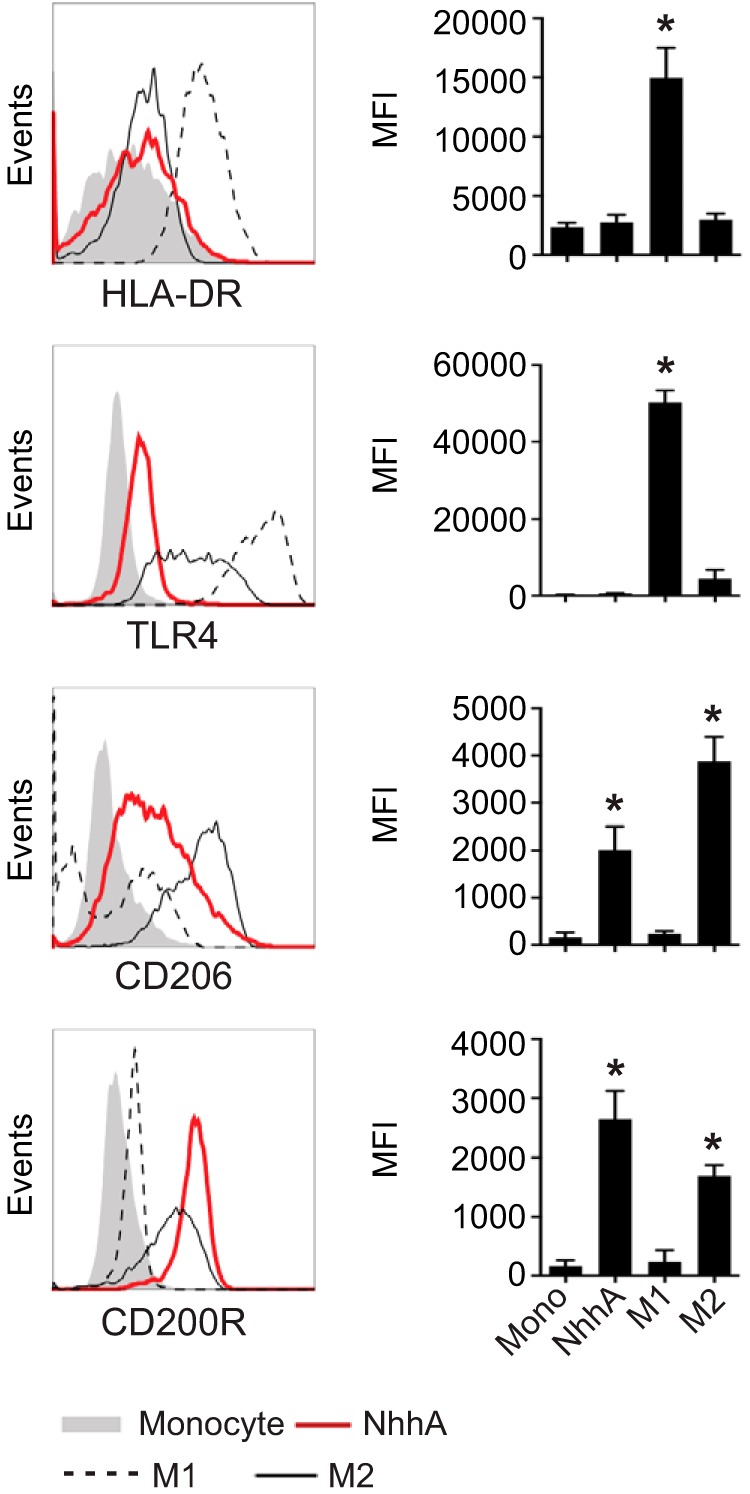
NhhA upregulates surface CD200R expression on Mφ. Mo were treated with 50 nM NhhA for 3 days. M1- or M2-polarized Mφ were generated as described in Materials and Methods. Expression of the indicated surface protein was analyzed by flow cytometry. The left panel shows fluorescence-activated cell sorting plots of one representative staining, and the median fluorescence intensities (MFIs) are shown in the right panel. Error bars indicate medians with interquartile ranges of three independent experiments. *, P < 0.05 compared with the Mo control using ANOVA followed by the Bonferroni post hoc test.
NhhA-Mφ are hyporesponsive to meningococcal stimulation but are competent at eliminating bacteria.
We confirmed the upregulation of CD200R in NhhA-Mφ (Fig. 4). Therefore, we analyzed the profile of inflammatory mediators generated by these cells upon meningococcal stimulation by qPCR (Fig. 5A and data not shown). NhhA-Mφ were inert to meningococcal stimulation, which impeded the induction of proinflammatory cytokines (IL-6, TNF-α, IL-12, IL-23, and IL-1β) compared with Mo. In contrast, transcription levels of anti-inflammatory cytokines (IL-10 and IL-4) were not affected, and a transient upregulation on the first day after NhhA treatment was detected. Moreover, T regulatory cell (Treg)- or Th2-recruiting chemokines (CCL18, CCL17, and CCL22) were consistently upregulated in NhhA-Mφ upon bacterial infection. The trend of cytokine production could be detected in Mφ differentiated by NhhA for up to 5 days, indicating that the observed phenotype is not a consequence of insufficient maturation or differentiation status.
FIG 5 .
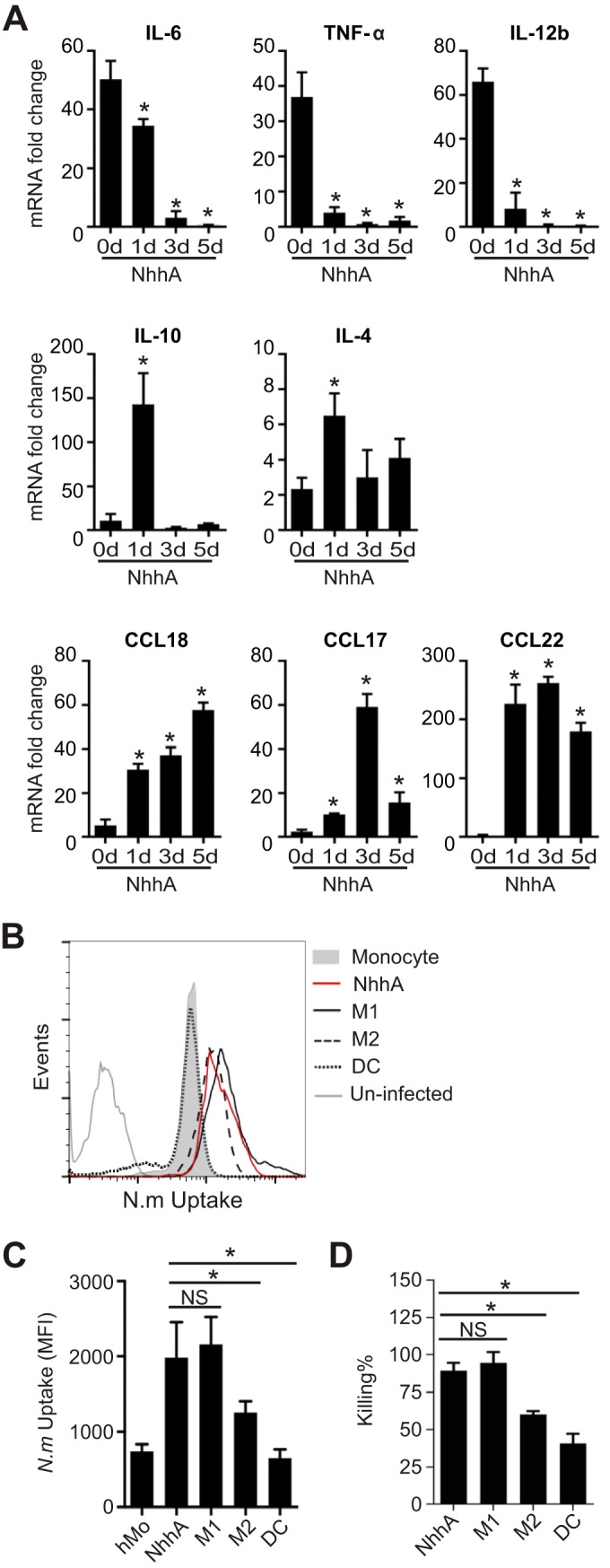
NhhA-Mφ are hyporesponsive to Neisseria meningitidis stimulation but retain the potential to eliminate bacteria. (A) Mo were differentiated with 50 nM NhhA for the indicated time points and subsequently challenged with FAM20 (MOI, 100) for 3 h. The RNA was isolated, and qPCR was performed to estimate the transcription levels of the indicated cytokines or chemokines. Normalized data, shown as the mean ± standard deviation from three independent experiments, are presented as fold change compared with uninfected Mo (day 0). *, P < 0.05 compared with control at day 0 using ANOVA followed by the Bonferroni post hoc test. (B and C) NhhA-Mφ, M1Mφ, M2Mφ, or DCs, generated as described in Materials and Methods, were challenged with fluorescein isothiocyanate (FITC)-labeled N. meningitidis FAM20 (MOI, 100) for 40 min at 37°C. The intracellular fluorescence intensity was determined by flow cytometry, and a representative plot is shown in panel B. Quantitative data for median fluorescence intensity (MFI), shown as medians with interquartile ranges from three independent experiments, are presented in panel C. *, P < 0.05 using ANOVA followed by the Bonferroni post hoc test. (D) NhhA-Mφ, M1Mφ, M2Mφ, or DCs, generated as described for panel B, were challenged with live FAM20 (MOI, 100) for 40 min. After intensive washing and antibiotic treatment to kill extracellular bacteria, survival of intracellular bacteria was determined by a modified gentamicin protection assay, as described in Materials and Methods. The data represent the mean ± standard deviation from three independent experiments. *, P < 0.05 using ANOVA followed by the Bonferroni post hoc test. NS, not significant.
The intrinsic action of NhhA on the functional polarization of Mφ was confirmed by infecting Mo with WT or ΔNhhA meningococci. As shown in Fig. S3 in the supplemental material, WT bacteria containing NhhA triggered the transcription of IL-10 and, to a greater extent, inducible nitric oxide synthase (iNOS), whereas ΔNhhA bacteria more efficiently induced IL-12b.
We further studied the potential of NhhA-Mφ to take up and eliminate bacteria by flow cytometry and a gentamicin protection assay, respectively. We found that NhhA-Mφ could engulf and kill bacteria at a level comparable to that observed for M1Mφ, which was significantly higher than that of M2Mφ and DCs (Fig. 5B to D). Taken together, these data indicate that NhhA-Mφ resist N. meningitidis stimulation but exhibit an augmented capacity to eliminate bacteria.
NhhA modulates phenotype polarization of macrophages in vivo.
To elucidate the in vivo effect of NhhA on Mφ polarization and its impact on host responses to meningococcal infection, a CD46+/+ transgenic mouse model of meningococcal infection was applied. The immune responses observed in this mouse model may be of particular interest with respect to human pathogens because CD46, a ubiquitously expressed type I transmembrane glycoprotein in humans, is not only a regulator of complement activation but is also engaged in cytokine production (23). We challenged mice intraperitoneally (i.p.) with NhhA (50 nM) for 3 days and found that this treatment did not affect the total number of peritoneal cells (Fig. 6A) or peritoneal Mφ (Fig. 6B) compared with control mice. Upon meningococcal infection (i.p. with 108 CFU of FAM20 per mouse), the number of peritoneal cells was dramatically increased in NhhA-treated mice, indicating a rapid influx of immune cells (Fig. 6A). The increase in the number of peritoneal cells may be primarily attributed to infiltrating Mφ (F4/80hi CD11bhi Ly6Cint Ly6G− [Fig. 6B]). In contrast, the numbers of peritoneal neutrophils (Ly6G+ F4/80− CD11b+ Ly6Cint), DCs (CD205+ CD19− CD3−), T cells (CD3+ CD19−), or B cells (CD19+ CD3−) were not markedly different (see Fig. S4 in the supplemental material). Although we could not rule out the possibility that NhhA might polarize the functional phenotype of other immune cells, our data do support the role of NhhA in targeting Mo-Mφ differentiation in vivo.
FIG 6 .
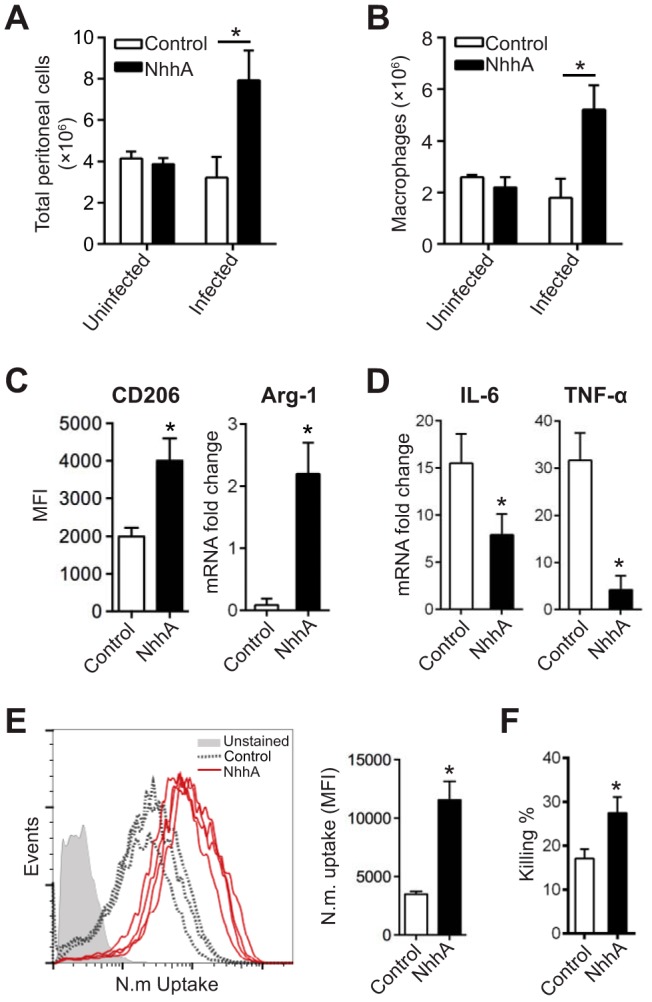
NhhA modulates phenotype polarization of macrophages in vivo. CD46+/+ mice were challenged i.p. with 50 nM native or heat-inactivated NhhA (control) for 3 days prior to infection with FAM20 (108 CFU/mouse). At 12 h postinfection, peritoneal cells were collected from both the bacterium-infected and uninfected mice. In some experiments (C to F), peritoneal cells were cultured in cell culture plates for 2 h, and attached Mφ were isolated for functional analysis. (A) The total number of peritoneal cells was quantified by trypan blue staining and presented as the mean ± standard deviation (n = 6) (*, P < 0.05 using paired Student’s t test). (B) The Mφ subset was quantified by flow cytometry by gating for F4/80hi CD11bhi Ly6Cint Ly6G− cells. Data presented are mean ± standard deviation (n = 6) (*, P < 0.05 using paired Student’s t test). (C and D) Effects of NhhA on Mφ polarization. (C) Surface expression of CD206 was quantified by flow cytometry and shown as median fluorescence intensity (MFI) with interquartile ranges (n = 6). Arg-1 mRNA was analyzed by qPCR. Error bars indicate mean ± standard deviation (n = 6). (D) TNF-α and IL-6 mRNAs were quantified by qPCR. All data from qPCR were normalized to the reference gene gapdh and are presented as fold change relative to uninfected samples. Data shown are the mean ± standard deviation (n = 6) (*, P < 0.05 using paired Student’s t test). (E and F) NhhA treatment enhances phagocytosis and bacterial elimination by peritoneal Mφ. (E) Peritoneal Mφ collected from the control (n = 3) or NhhA-treated (n = 4) mice were challenged with FITC-labeled N. meningitidis FAM20 (MOI, 100) for 40 min at 37°C. The number of intracellular bacteria was quantified by flow cytometry. The plots of bacterial uptake are presented in the left panel, and quantified data, presented as median fluorescence intensity (MFI), are shown in the right panel. Error bars indicate medians with interquartile ranges. *, P < 0.05 using paired Student’s t test. (F) Some peritoneal Mφ were treated with live FAM20 (MOI, 100) for 40 min, and bacterial elimination was determined as described in Fig. 5D. Data are presented as the mean ± standard deviation (n = 3 or 4). *, P < 0.05 compared with the control group using paired Student’s t test. Data shown in panels A to F are representative of at least three independent experiments.
Peritoneal Mφ were isolated for phenotypic and functional analyses. In contrast to cells collected from control mice, Mφ from NhhA-treated mice expressed significantly higher levels of CD206 and Arg-1 mRNA (Fig. 6C, P < 0.05), indicating an M2 Mφ-like phenotype. Indeed, these cells exhibited a decreased potential to generate proinflammatory cytokines such as IL-6 and TNF-α (Fig. 6D) upon meningococcal stimulation. Furthermore, they displayed a marked capacity to engulf and kill the internalized bacteria (Fig. 6E and F), which is consistent with our in vitro findings.
The intrinsic effect of NhhA on immunomodulation of Mφ in vivo was further tested by infecting CD46+/+ mice i.p. with WT or ΔNhhA meningococcal bacteria (108 CFU/mouse, for 12 h), and peritoneal cells were analyzed by flow cytometry. The absence of NhhA in meningococci did not affect the Mo (F4/80med CD11bmed) population in the peritoneal cavity (23.6% ± 1.8% versus 25.4% ± 4.7%, P = 0.59). However, a significantly larger amount of Mφ (F4/80hi CD11bhi) was detected in the peritoneal cavity of mice challenged with the WT strain compared with the ΔNhhA bacteria (30.6% ± 4.0% versus 16.7% ± 3.3%, P = 0.005) (Fig. 7A). Real-time PCR and enzyme-linked immunosorbent assay (ELISA) studies revealed that peritoneal Mφ in response to ΔNhhA bacteria produced more proinflammatory cytokine IL-12b but smaller amounts of anti-inflammatory cytokine IL-10 (Fig. 7B). The bactericidal mediator iNOS was diminished in the ΔNhhA bacterium-treated cells. Consistently, lower nitrite levels in the peritoneal cavity were detected in the ΔNhhA bacterium-treated mice than in the mice stimulated with the WT meningococci (Fig. 7C). These results were in line with our in vitro observations (see Fig. S3 in the supplemental material) and clearly demonstrated the intrinsic role of NhhA in modulating Mφ differentiation in vivo.
FIG 7 .

Intrinsic effect of NhhA on Mφ differentiation and polarization in vivo. CD46+/+ mice (n = 6) were challenged i.p. with 108 wild-type (WT) or NhhA-deficient (ΔNhhA) FAM20 bacteria for 12 h, and the total peritoneal cells and peritoneal fluids were isolated for further analysis. (A) Representative fluorescence-activated cell sorting plots showing percentage of Mo (F4/80med CD11bmed) and Mφ (F4/80hi CD11bhi) in isolated peritoneal cells. (B) Cytokine response in peritoneal Mφ. The left panel shows transcriptional levels of IL-12b and IL-10 in peritoneal Mφ as determined by qPCR; the right panel shows protein levels of IL-10 in peritoneal fluids as determined by ELISA. (C) iNOS response in peritoneal Mφ. The left panel shows transcriptional levels of iNOS in peritoneal Mφ as determined by qPCR; the right panel shows the amounts of nitrite determined in peritoneal fluids. qPCR data in panels B and C were normalized and expressed as fold change compared with the WT bacterium-stimulated group. Bars represent the mean ± standard deviation (*, P < 0.05 [paired Student’s t test]).
NhhA-Mφ prevent bacterial dissemination in an experimental meningococcal infection model.
The data reported above suggested that NhhA-programmed Mφ might dampen the immune response to meningococcal stimulation. To test this hypothesis, CD46+/+ transgenic mice pretreated with NhhA were challenged with FAM20 (i.p.), and the development of septic shock was monitored and compared with that of control mice. As shown in Fig. 8A, NhhA pretreatment resulted in complete survival compared with control mice (100% versus 20%) 3 days postinfection. Bacteremia levels were significantly lower in the NhhA-treated mice than in the control group even as early as 2 h postinfection, and no bacteria could be detected 24 h postinfection (Fig. 8B). Bacterial survival levels in whole blood collected from control and NhhA-pretreated mice were comparable, indicating that bacterial elimination occurred mainly in the peritoneal cavity (see Fig. S5A in the supplemental material). Furthermore, NhhA treatment alleviated the host proinflammatory immune responses, as indicated by significantly decreased levels of IL-6 and TNF-α, but comparable levels of IL-10 in both the blood (Fig. 8C) and peritoneal cavity (see Fig. S5B), compared with the control mice.
FIG 8 .
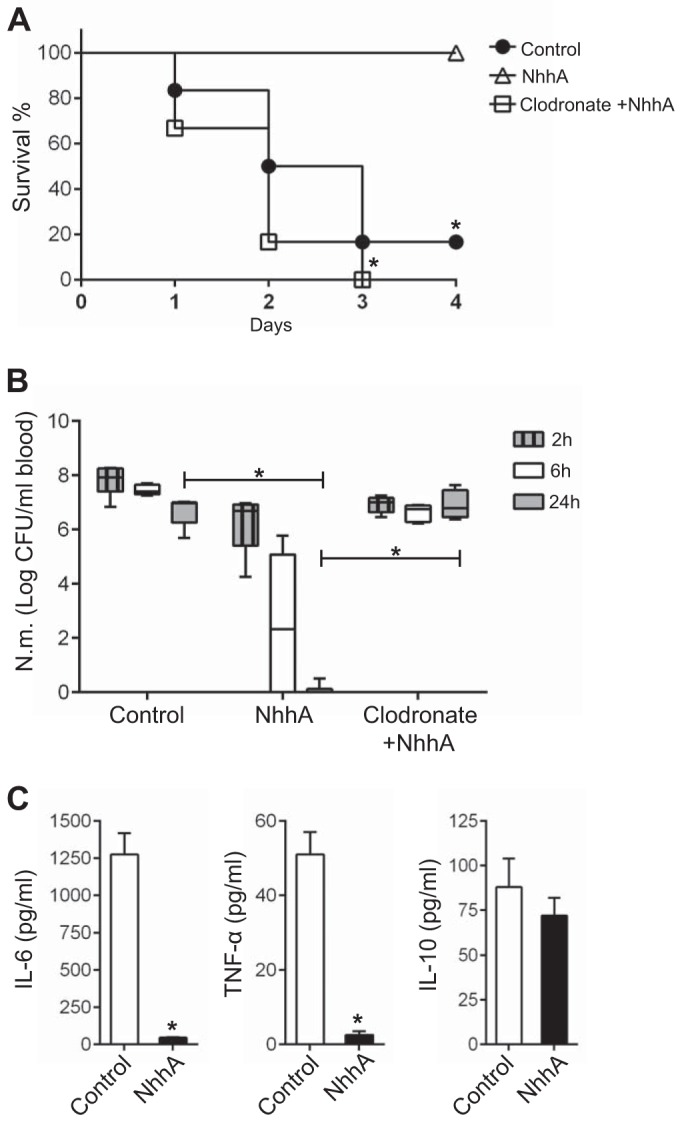
NhhA-Mφ prevent bacterial dissemination in vivo. CD46+/+ mice (n = 6 to 8) were treated i.p. with 50 nM heat-inactivated (control) or native NhhA for 3 days prior to being challenged i.p. with FAM20 (108 CFU). To deplete Mo/Mφ, some mice were treated with clodronate-containing liposomes (200 µl i.p.) 4 days prior to NhhA treatment, and depletion was confirmed by monitoring of stained blood smears. (A) Survival of mice at indicated time points postinfection. *, P < 0.05 versus the control group using the Mann-Whitney U test. (B) Bacterial levels in the blood at the indicated postinfection time points. The detection limit is 500 CFU/ml blood. Box plots are divided into upper quartiles and lower quartiles by medians. Error bars indicate the minimum and maximum of data values. *, P < 0.05 using paired Student’s t test. (C) Levels of IL-6, TNF-α, and IL-10 in the serum at 24 h postinfection were determined by ELISA. The data represent the mean ± standard deviation. *, P < 0.05 compared with the control group using paired Student’s t test. The results in panels A to C represent data pooled from three experiments.
To confirm the role of NhhA-modulated Mφ polarization in the host immune response to meningococcal infection, we performed the in vivo experiments under Mo/Mφ-deficient conditions generated by pretreating mice with clodronate-containing liposomes. We found that the NhhA-triggered protection was abolished, as assessed by survival curves (Fig. 8A) and bacteremia levels (Fig. 8B). These data demonstrate that NhhA targets the functional programming of Mφ to dampen proinflammatory responses and occlude bacterial dissemination in vivo.
NhhA potentiates nasopharyngeal persistence of N. meningitidis.
To determine whether NhhA modulates mucosal defense against N. meningitidis at the nasopharynx, a natural niche of meningococci, we pretreated CD46+/+ transgenic mice with NhhA (50 nM) before challenging them with meningococci through an intranasal (i.n.) route. Bacterial burden and cytokine levels in the nasal lavage fluid collected from mice at 12 h postinfection were analyzed. Mice pretreated with NhhA harbored more bacteria at the nasopharynx than did control mice (Fig. 9A). Surprisingly, we observed a sharp reduction in IL-6 levels in these mice compared with those observed in the control group. On the other hand, IL-10 levels were comparable between the two groups (Fig. 9B). The enhanced levels of proinflammatory cytokines may contribute to tissue injury. Therefore, we hypothesized that the nasopharyngeal mucosa of mice pretreated with NhhA would display a lower inflammatory response upon meningococcal infection, whereas the control mice would display a greater inflammatory response, although the bacterial level was diminished in the control mice. Our hypothesis was confirmed by histological studies: the nasopharyngeal mucosa of NhhA-treated mice showed less leukocyte infiltration and enhanced mucosal integrity features that were not observed in the control mice (Fig. 9C). We tried different antibodies to stain Mφ in the nasopharyngeal mucosa of mice; however, we could not obtain satisfactory images. The agents used for decalcification and embedding during tissue preparation may influence the immunohistochemical detection of specific markers.
FIG 9 .
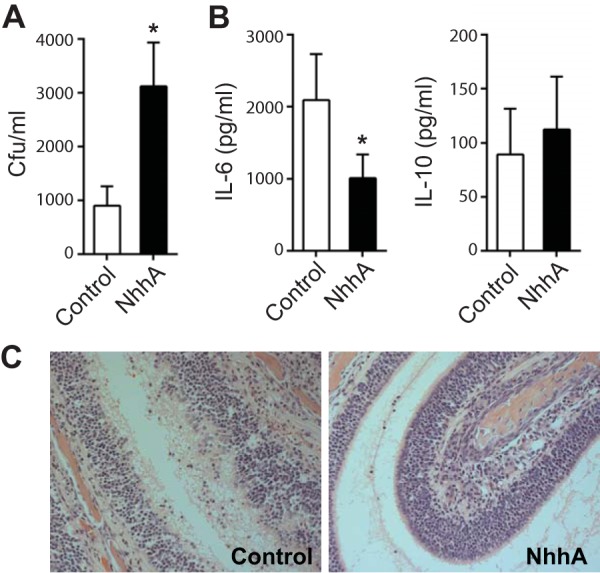
NhhA potentiates the commensal persistence of N. meningitidis. CD46+/+ mice (n = 6 to 8) were pretreated with 50 nM heat-inactivated (control) or native NhhA for 3 days prior to intranasal bacterial challenge (108 CFU of FAM20 per mouse). Nasal lavage fluid and head tissue were collected 12 h after infection. (A) Bacterial burden in nasal lavage fluid. (B) Levels of IL-6 and IL-10 in the nasal fluid as determined by ELISA. Bars in panels A and B represent the mean ± standard deviation of data pooled from three experiments. *, P < 0.05 compared to control group using paired Student’s t test. (C) H&E-stained thin section of the nasal mucosa of both control and NhhA-pretreated mice. Original magnification of images, ×100. Representative images of three independent experiments are presented.
DISCUSSION
Two long-standing mysteries are why many healthy individuals are asymptomatic carriers of N. meningitidis and how meningococci colonize nasopharyngeal mucosa without activating the host innate immune response. Here, we provide evidence that the highly conserved meningococcal surface protein NhhA has a profound effect on modulating the host immune response through programming Mφ polarization toward an immune homeostatic phenotype, named NhhA-Mφ. These Mφ have an enhanced capacity to clear invading bacteria. More importantly, they are hyporesponsive to meningococcal stimulation, likely through CD200R-triggered anti-inflammatory signaling in immune cells (24). Together, all of these influences may favor bacterial commensal colonization at the nasopharyngeal mucosa.
The ability of NhhA to modulate Mφ polarization was further confirmed in vivo using a murine system. We used both the peritoneal and intranasal routes to study the effect of NhhA-mediated immune modulation on meningococcal intravascular dissemination and colonization, respectively. When the mice were challenged peritoneally, NhhA pretreatment induced significantly decreased levels of not only proinflammatory cytokines but also invading bacteria in the blood, which were associated with enhanced survival compared with that observed for the control group. When an intranasal route of challenge was applied, the NhhA-induced hyporesponsiveness to meningococcal stimulation was also detected in the nasopharyngeal mucosa, which was associated with enhanced bacterial colonization. Thus, the conserved meningococcal surface protein NhhA plays a key role in inducing host immune homeostasis at the mucosal surface and contributes bacterial asymptomatic colonization. As a corroborative example, previous studies by Krysko et al. have shown that chronic rhinosinusitis characterized with M2 polarization of Mφ is associated with enhanced colonization by Staphylococcus aureus in the nasal mucosa (25). A recent study demonstrated that meningococcal LOS, via TLR4, augments CD200 expression on Mφ (26). Meningococcal infection at the nasopharyngeal mucosa is a dynamic process, including adhesion and invasion of bacterial cells (15), tissue damage (27), and shedding of bacterial outer membrane vesicles (28), as well as influx of immune cells to the infection site (15, 29). It is therefore reasonable to speculate that N. meningitidis may interact with and modulate the functional polarization of Mφ or even other immune cells through distinct mechanisms and skew the host mucosal immune response, thereby favoring asymptomatic colonization of the host by the bacterium.
A novel finding of this study is that meningococci block the differentiation of Mo into DCs; a similar effect has been observed for a range of chronic-infection-associated pathogens such as M. tuberculosis (30) and Salmonella Typhimurium (30, 31). Inhibition of Mo-DC differentiation and the generation of anti-inflammatory Mφ may decrease the chances of meningococcal dissemination, which could in turn diminish the overactivation of local inflammatory responses and hence facilitate bacterial containment at the nasopharyngeal mucosa. Our finding that NhhA-Mφ exhibit high potential to eliminate the bacteria corroborates this idea. NhhA expression is conserved in nearly all analyzed meningococcal isolates (13), suggesting that the functions of NhhA are likely comparable throughout the species. NhhA exhibits a certain sequence variation between different species; further studies are necessary to determine whether NhhA has a strain-specific role.
NhhA-Mφ exhibited efficient phagocytosis coupled with attenuated induction of proinflammatory cytokines upon meningococcal stimulation, a character that is in sharp contrast to the M2Mφ triggered by the Th2 cytokine IL-4 (32). Our results revealed that NhhA, in contrast to IL-4, induces rapid activation of the Akt/PI3K signaling pathway that is associated with macropinocytic uptake by Mφ (33), suggesting that differential signaling activation by NhhA may be the underlying mechanism. A recent study by Frodermann et al. (34) further revealed the role of the activation of the PI3K pathway in inducing the IL-10 response in Mφ upon TLR2 activation by staphylococcal peptidoglycan. We also observed that NhhA-stimulated Mφ induced IL-10 expression in a PI3K-dependent manner (data not shown). However, the NhhA-triggered Akt/PI3K activation was dispensable for Mo differentiation and was uncoupled from TLR1/TLR2 signaling, suggesting that an additional receptor(s) is involved in sensing NhhA and activating this signaling pathway.
Although NhhA-triggered monocyte differentiation is dependent on the activation of TLR1/TLR2 signaling, the results of this study suggest a unique role for endogenously produced IL-10 and TNF-α in Mo differentiation and Mφ polarization upon NhhA stimulation. This role is mechanistically different from that of the TLR1/2 agonist PAM3CSK4, in which endogenously produced IL-15 and GM-CSF initiate Mo-Mφ and Mo-DC differentiation, respectively (19). Previous studies have shown that TNF-α triggers Mo differentiation by impeding the internalization of the M-CSF receptor (35), whereas IL-10 augments monocyte differentiation (36) and Fc receptor-mediated phagocytosis (37). A recent study by Nguyen et al. (38) demonstrated the in vivo role of IL-10 in Mφ functional polarization; peritoneal administration of IL-10 induced Mφ polarization toward a low-level major histocompatibility complex class II (MHC-IIlo) phenotype. These cells were defective in antigen presentation, but they exhibited extensive phagocytosis capacity (38), which is in line with our observations. Indeed, both commensal microbiota and pathogens, such as group B Streptococcus (39), Staphylococcus aureus (34), and Bordetella bronchiseptica (40), have been shown to be able to modulate IL-10 production in immune cells, which benefits bacterial colonization. Enhancing epithelial homeostasis (41, 42) and/or inducing a Treg response (43, 44) might be the underlying mechanism. Nevertheless, it would be worthwhile to further elucidate the role of IL-10 and IL-10-producing cells in the context of meningococcal colonization or even bacterial survival.
IL-32 can trigger monocyte differentiation into either CD14+ Mφ-like (45) or CD1b+ DC-like cells (46); therefore, we investigated the possible role of IL-32 in NhhA-induced Mo differentiation. Although transcription of il-32 was highly induced, an in vitro blocking assay revealed that NhhA-triggered Mo-Mφ differentiation was independent of IL-32. Nevertheless, we could not exclude the possibility that IL-32 might be involved in polarizing the Mφ phenotype because it has been shown that IL-32 triggers Mφ differentiation with both the M1 and M2 phenotypes (47). Moreover, IL-10, generated upon NhhA stimulation, may block the formation of DC-like cells (22).
Understanding the role of protein components in the programming of antigen-presenting cells is important considering that many of these factors are potential vaccine candidates against meningococcal disease. NhhA-triggered Mφ exhibit a profound capacity to produce mediators that play key roles in priming and polarizing adaptive immune responses, such as CCL17, CCL18, and CCL22. The antigenic character of NhhA has been demonstrated in a mouse model (14), and our findings further indicate its adjuvant activity. A recent study has demonstrated that targeting the reprogramming of antigen-presenting cells to induce immune tolerance is a persistence strategy for H. pylori (48). Thus, the identification of surface receptors and signaling networks exploited by NhhA may provide a foundation for understanding vaccine-induced protective immunity against N. meningitidis. It is conceivable that the mucosal hyporeaction induced by NhhA-Mφ can also be harnessed to prevent various mucosal inflammatory disorders.
In conclusion, our data provide novel insights into the complex actions of bacterial components in modulating the host immune responses. We reveal the molecular mechanisms that N. meningitidis might employ to avoid activating the host’s inflammatory response, thereby generating an immune homeostatic microenvironment compatible with commensal persistence of the bacterium.
MATERIALS AND METHODS
Bacteria.
The N. meningitidis serogroup C strain FAM20 and the NhhA-deletion mutant (ΔNhhA) strain generated previously (5) were cultured on gonococcal (GC) agar (Acumedia, MI) supplemented with Kellogg’s solution. To prepare fluorescein isothiocyanate (FITC)-labeled bacteria, FAM20 cells were harvested and incubated in FITC (0.1 mg/ml)-NaHCO3 (0.1 M) buffer (pH 9.0) for 60 min at 25°C followed by intensive washing with phosphate-buffered saline (PBS).
Recombinant NhhA proteins.
The full-length passenger region of NhhA (NhhA-FL, amino acids [aa] 51 to 510) was generated in a previous study (5). The N-terminal (NhhA-N, aa 50 to 164), C-terminal (NhhA-C, aa 380 to 510), and middle (NhhA-M, aa 164 to 380) regions of the NhhA passenger domain were amplified with the primer pairs listed in Table S2 in the supplemental material. After purification and digestion, the PCR products were cloned into a pET-21a expression vector. Expressed proteins were purified under native conditions (5).
NhhA was routinely pretreated with the LPS antagonist polymyxin B (PMB), over the course of the entire study, although we observed that PMB omission did not affect the differentiating effect of the protein.
Monocyte isolation and differentiation.
Human CD14+ primary Mo were isolated from freshly prepared buffy coat (Karolinska Institutet Biobank, Stockholm, Sweden) using a pluriBead cell separation kit (pluriSelect GmbH, Leipzig, Germany). Mo were cultured in RPMI 1640 medium containing 10% fetal calf serum (FCS) in six-well culture plates at a density of 1 × 106/ml and exposed to NhhA, as indicated, to induce differentiation. Control cells were primed with M-CSF (50 ng/ml) for 5 days. A subset of these cells was then polarized for 24 h by either IFN-γ (10 ng/ml) or LPS (20 ng/ml; Sigma-Aldrich, St. Louis, MO) to M1Mφ or IL-4 (20 ng/ml) to M2Mφ. To generate DCs, Mo were cultured in GM-CSF (50 ng/ml) and IL-4 (20 ng/ml) for 5 days followed by LPS (20 ng/ml) for 2 days to enhance maturation. M-CSF and GM-CSF were purchased from ProSpec-Tany TechnoGene Ltd. (Israel). IL-4, IFN-γ, and IL-13 were purchased from ImmunoTools (Germany).
In experiments including inhibitors, cytochalasin D (1 µM), PD98095 (10 µM), SP600125 (10 µM), Celastrol (500 nM), or SR11302 (1 µM) was added to the cell culture 30 min before NhhA stimulation. All inhibitors were purchased from InvivoGen (San Diego, CA) and tested for cytotoxicity using trypan blue and propidium iodide (PI) exclusion assays. Only inhibitor concentrations that ensured viability above 90% were used; control cells were treated with appropriate vehicles for the inhibitors.
Neutralization assays.
To inhibit the function of surface receptors, cells were pretreated with dextran sulfate (100 µg/ml), heparinase III (5 U/ml), or neutralizing antibodies (5 µg/ml) directed against human TLR1, TLR2, TLR4, and TLR6 (InvivoGen) for 30 min. To suppress the biological activity of the cytokines, 10 µg/ml of antibody-neutralizing IL-6, IL-12p40, or TNF-α (BioLegend, San Diego, CA) or IL-15 (R&D Systems, Minneapolis, MN) was added. Generation of IL-32 was blocked by treating cells with α-1 antitrypsin (0.5 mg/ml; Sigma-Aldrich) (49). M-CSF receptor (M-CSFR) was blocked using GW2580 (10 μM; Abcam, Cambridge, United Kingdom) to stop M-CSF signaling. Control cells were treated with an isotype-matched antibody or an appropriate vehicle.
Mouse model of infection and sampling.
Experiments were performed with gender-matched 6- to 8-week-old hCD46Ge (CD46+/+) transgenic mice (50). For the i.p. infection model, each mouse was pretreated i.p. once with 50 nM native or heat-inactivated (30 min at 95°C) NhhA (0.5 µg NhhA per mouse with 0.2 ml of peritoneal fluid volume) for 1 to 3 days and then challenged with FAM20 (108 CFU). Preliminary experiments showed that NhhA treatments for 1 day and for 3 days yielded comparable results. Data from the 3-day treatment were presented to match conditions used in in vitro studies. In some experiments, mice were infected i.p. with wild-type (WT) or NhhA-deficient (ΔNhhA) FAM20 meningococcal strains (108 CFU) for 12 h. For the intranasal (i.n.) infection model, each mouse was pretreated intranasally twice with 50 nM native or heat-inactivated NhhA (0.03 µg NhhA in 10 µl of instillation volume) prior to bacterial challenge on day 3. Pretreatment with NhhA did not induce proinflammatory responses, as assessed by measuring the levels of IL-6 and TNF-α in either the blood or peritoneal fluid. In some experiments, mice were treated with clodronate-containing liposomes (200 µl i.p.) 4 days before the procedure to delete Mo/Mφ, and successful depletion (>95%) was confirmed by monitoring blood smears stained with Wright-Giemsa solution (Sigma-Aldrich).
The health status of the mice was closely monitored for 7 days, and blood samples (5 µl) were collected from the tail vein at indicated time points to determine bacterial loads. Blood samples collected from the orbital sinus were used for the enzyme-linked immunosorbent assay (ELISA). The peritoneal cells were collected and incubated in complete Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal calf serum (FCS) in cell culture plates for 2 h at 37°C. After removal of the floating cells, the attached Mφ (>90% purity, as estimated by an F4/80+ assay) were used for further analysis. Sections of paraffin-embedded nasal mucosa were stained with hematoxylin and eosin (H&E) solution. Animal care and experiments were handled in accordance with the guidelines issued by the Swedish Ethical Committee on Animal Experiments.
Flow cytometry.
Cells were stained with the indicated antibodies according to manufacturers’ protocols and analyzed on a BD LSR Fortessa cell analyzer. The data were analyzed using the FlowJo software program (Tree Star, San Carlos, CA). The phenotype of the human cells was determined using antibodies detecting membrane CD14, CD1a, CD80, CD83, CD86, HLA-DR, CD206, or TLR4 conjugated with Horizon V450, FITC, phycoerythrin (PE), PE-Cy7, or allophycocyanin (APC) (BD Biosciences, Erembodegem, Belgium) and FITC-conjugated CD200R (AbD Serotec, Raleigh, NC). The mouse peritoneal cells were analyzed using antibodies to Ly6G, Ly6C, CD206, and CD11b (BD Pharmingen); F4/80 (AbD Serotec); and CD3, CD19, and CD205 (MyBioSource, San Diego, CA) conjugated with PE, FITC, APC, or APC-H7. Dead or apoptotic cells generated during detachment were excluded by dual staining with FITC-conjugated annexin V in each experiment.
Bacterial phagocytosis and intracellular killing.
Cells were grown in six-well plates and infected with FITC-labeled FAM20 (multiplicity of infection [MOI], 100) for 40 min at 37°C or 4°C to determine the number of total cell-associated bacteria or adherent bacteria, respectively. After washing, the cells were harvested and fixed with 4% paraformaldehyde, and FITC fluorescence was quantified via flow cytometric analysis. The number of intracellular bacteria was determined by subtracting the fluorescence of adherent bacteria from that of the total cell-associated bacteria.
The killing ability of cells was determined using a gentamicin protection assay. Briefly, cells were infected with FAM20 (MOI, 100) for 40 min, followed by incubation in gentamicin (200 µg/ml) for 30 min. Cells were lysed with 1% saponin, and “total internalized bacteria” were quantified after plating appropriate dilutions of the lysates. Following gentamicin treatment, some cells were incubated with fresh medium for an additional 1 h, and the intracellular “surviving bacteria” were quantified as described above. The proportion of intracellular killed bacteria was calculated using the following formula: 100 × (total internalized bacteria − surviving bacteria)/total internalized bacteria. All samples were tested in triplicate, and the experiments were repeated at least three times.
Measurement of cytokines and nitrite.
Levels of human IL-6, IL-10, IL-12, and TNF-α were quantified using ELISA kits from BioLegend, Levels of mouse IL-6, TNF-α, and IL-10 were determined using ELISA kits from BioSite (Täby, Sweden). Nitrite levels were measured using a Griess assay (Promega). All analyses were performed according to the manufacturer’s instructions.
qRT-PCR.
Total cellular RNA was isolated with an RNeasy minikit (Qiagen, Valencia, CA) and transcribed to cDNA using a RevertAid H Minus First-Strand cDNA synthesis kit (Fermentas, Glen Burnie, MD). Real-time reverse transcription-PCR (qRT-PCR) was performed on a LightCycler 480 real-time PCR system (Roche Diagnostics, Mannheim, Germany) using SYBR Green for real-time monitoring of the PCR. The primers used are listed in Table S1 in the supplemental material. The threshold cycle (CT) values of the target genes were normalized to the CT value of the reference gene (mouse glyceraldehyde 3-phosphate dehydrogenase [GAPDH] or human ribosomal protein L37A [RPL37A]) and expressed as fold change compared with the control sample.
Immunoblotting.
To quantify protein phosphorylation, the cell lysates were subjected to 10% sodium dodecyl sulfate (SDS)-PAGE, and immunoblotting was performed with mouse antibodies to ERK1/2 (pT202/pY204), JNK (pT183/pY185), and p38 (pT180/pY182) (MAPK activation sampler kit; BD Biosciences) or rabbit anti-phospho-Akt (Cell Signaling Technology, Danvers, MA). β-Actin was used as a control protein. Donkey anti-rabbit immunoglobulin G (IgG) or donkey anti-mouse IgG (1:15,000; Li-Cor Biosciences) labeled with IRDye 800CW or IRDye 680RD was used as the secondary antibody. Signals were detected using an Odyssey infrared imaging system (Li-Cor Biosciences), and densitometric analysis was performed using the ImageJ software program (http://imagej.nih.gov/ij).
Statistics.
Differences between the groups were analyzed by analysis of variance (ANOVA) followed by the Bonferroni post hoc test. The Mann-Whitney U test was applied for the murine survival assay. The other animal experiments were analyzed by paired Student’s t test. P values of less than 0.05 were considered statistically significant. Statistical analysis was performed using the Prism 5.0 software (GraphPad, La Jolla, CA).
SUPPLEMENTAL MATERIAL
The differentiating effect of NhhA requires the full passenger domain. Recombinant proteins corresponding to the full-length (NhhA-FL, amino acids [aa] 51 to 510, 50 kDa), middle (NhhA-M, aa 164 to 380, 24 kDa), N-terminal (NhhA-N, aa 51 to 164, 13 kDa), and C-terminal (NhhA-C, aa 380 to 510, 14 kDa) regions of the NhhA passenger domain were generated and purified under native conditions. (A) Mo were treated with recombinant NhhA proteins (50 nM) corresponding to the full-length (NhhA-FL) or truncated passenger domains for 3 days. After extensive washing, the cells were stained for NhhA as described in Materials and Methods. Representative micrographs are shown in the upper panel. Original magnification, ×400. Some cells were stained with a Horizon V450-labeled anti-CD14 antibody and analyzed by flow cytometry. One representative SSC/CD14 plot of three independent experiments is shown in the lower panel. (B) Proteins (50 nM each) were separated by native polyacrylamide gel electrophoresis (PAGE) and detected using polyclonal rabbit anti-NhhA antibodies (1:1,000). The size in kilodaltons of the protein ladder is indicated. Download
ERK, JNK, and NF-κB signaling pathways are involved in NhhA-triggered Mo differentiation. Mo were treated with 50 nM NhhA alone (control) or cotreated with the indicated inhibitors for ERK (PD98095, 10 µM), JNK (SP600125, 10 µM), NF-κB (Celastrol, 500 nM), or activator protein 1 (AP-1) (SR11302, 1 µM) for 3 days. Differentiation to Mφ was determined by the flow cytometric analysis of CD14 expression and SSC parameters. Medians with interquartile ranges of median fluorescence intensity (MFI) from three independent experiments are presented as fold change relative to control. *, P < 0.05 compared with control group using ANOVA followed by the Bonferroni post hoc test. Download
Intrinsic effect of NhhA on Mφ polarization. Mo were stimulated with the FAM20 WT or NhhA-deficient mutant (ΔNhhA) strains at an MOI of 100 for 3 h, mRNA was isolated, and qPCR analysis was performed for IL-12b, IL-10, and inducible nitric oxide synthase (iNOS). Normalized data, expressed as fold change compared with uninfected cells, are presented as the mean ± standard deviation (SD) from three independent experiments. *, P < 0.05 compared with WT-stimulated cells using ANOVA followed by the Bonferroni post hoc test. Download
Peritoneal cell populations. CD46+/+ mice were challenged i.p. with 50 nM native or heat-inactivated NhhA (control) for 3 days prior to infection with FAM20 (108 CFU/mouse, i.p.). At 12 h postinfection, peritoneal cells were collected from both uninfected (upper panel) and bacterium-infected (lower panel) mice. Cell populations were analyzed by flow cytometry. Mφ (F4/80hi CD11bhi Ly6Cint Ly6G−), neutrophils (Ly6G+ F4/80− CD11b+ Ly6Cint), DCs (CD205+ CD19− CD3−), T cells (CD3+ CD19−), and B cells (CD19+ CD3−) were gated. Data are presented as the mean ± standard deviation (SD) (n = 6) and are representative of at least three independent experiments. *, P < 0.05 compared with the control group using paired Student’s t test. Download
NhhA treatment alleviates the host proinflammatory responses. CD46+/+ mice (n = 6 to 8) were treated i.p. with 50 nM heat-inactivated (control) or native NhhA for 3 days. (A) Whole blood was collected from mice and mixed with FAM20 (MOI, 100) for 3 h. Bacterial growth was determined at indicated time points. Data shown are the mean ± standard deviation (SD) (n = 6). (B) Mice were challenged i.p. with 108 CFU FAM20. Levels of IL-6, TNF-α, and IL-10 in the peritoneal cavity at 24 h postinfection were estimated by ELISA. Data represent the mean ± standard deviation, and they are pooled from three independent experiments. *, P < 0.05 compared with the control group using paired Student’s t test. Download
Primers used in real-time PCR.
PCR primers used for NhhA cloning.
ACKNOWLEDGMENTS
This work was supported by the Swedish Research Council (grant no. 2008-2572, 2008–3367), the Swedish Society of Medicine (SLS-327941), Clas Groschinskys Minnesfond (M12212), the China Scholarship Council (CSC), and Stockholm University.
We thank Xia Shen at the Department of Medical Epidemiology and Biostatistics of Karolinska Institutet for statistical consulting.
Footnotes
Citation Wang X, Sjölinder M, Gao Y, Wan Y, Sjölinder H. 2016. Immune homeostatic macrophages programmed by the bacterial surface protein NhhA potentiate nasopharyngeal carriage of Neisseria meningitidis. mBio 7(1):e01670-15. doi:10.1128/mBio.01670-15.
REFERENCES
- 1.Rosenstein NE, Perkins BA, Stephens DS, Popovic T, Hughes JM. 2001. Meningococcal disease. N Engl J Med 344:1378–1388. doi: 10.1056/NEJM200105033441807. [DOI] [PubMed] [Google Scholar]
- 2.Fontanals D, Van Esso D, Pons I, Pineda V, Sanfeliu I, Mariscal D, Vázquez JA, Coll P, Prats G. 1996. Asymptomatic carriage of Neisseria meningitidis in a randomly sampled population. Serogroup, serotype and subtype distribution and associated risk factors. Clin Microbiol Infect 2:145–146. doi: 10.1111/j.1469-0691.1996.tb00222.x. [DOI] [PubMed] [Google Scholar]
- 3.Andersen J, Berthelsen L, Bech Jensen B, Lind I. 1998. Dynamics of the meningococcal carrier state and characteristics of the carrier strains: a longitudinal study within three cohorts of military recruits. Epidemiol Infect 121:85–94. doi: 10.1017/S0950268898008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sprong T, Stikkelbroeck N, van der Ley P, Steeghs L, van Alphen L, Klein N, Netea MG, van der Meer JW, van Deuren M. 2001. Contributions of Neisseria meningitidis LPS and non-LPS to proinflammatory cytokine response. J Leukoc Biol 70:283–288. [PubMed] [Google Scholar]
- 5.Sjölinder M, Altenbacher G, Wang X, Gao Y, Hansson C, Sjölinder H. 2012. The meningococcal adhesin NhhA provokes proinflammatory responses in macrophages via Toll-like receptor 4-dependent and -independent pathways. Infect Immun 80:4027–4033. doi: 10.1128/IAI.00456-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cecchini P, Tavano R, de Laureto P, Franzoso S, Mazzon C, Montanari P, Papini E. 2011. The soluble recombinant Neisseria meningitidis adhesin NadAΔ351–405 stimulates human monocytes by binding to extracellular Hsp90. PLoS One 6:e25089. doi: 10.1371/journal.pone.0025089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.León B, López-Bravo M, Ardavín C. 2007. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26:519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 8.Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, Mukundan L, Chawla A, Monack DM. 2013. Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe 14:171–182. doi: 10.1016/j.chom.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redente EF, Higgins DM, Dwyer-Nield LD, Orme IM, Gonzalez-Juarrero M, Malkinson AM. 2010. Differential polarization of alveolar macrophages and bone marrow-derived monocytes following chemically and pathogen-induced chronic lung inflammation. J Leukoc Biol 88:159–168. doi: 10.1189/jlb.0609378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis ND, Asim M, Barry DP, Singh K, de Sablet T, Boucher J-L, Gobert AP, Chaturvedi R, Wilson KT. 2010. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J Immunol 184:2572–2582. doi: 10.4049/jimmunol.0902436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zughaier SM. 2011. Neisseria meningitidis capsular polysaccharides induce inflammatory responses via TLR2 and TLR4-MD-2. J Leukoc Biol 89:469–480. doi: 10.1189/jlb.0610369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peak IR, Srikhanta Y, Dieckelmann M, Moxon ER, Jennings MP. 2000. Identification and characterisation of a novel conserved outer membrane protein from Neisseria meningitidis. FEMS Immunol Med Microbiol 28:329–334. doi: 10.1111/j.1574-695X.2000.tb01494.x. [DOI] [PubMed] [Google Scholar]
- 14.Peak IR, Srikhanta YN, Weynants VE, Feron C, Poolman JT, Jennings MP. 2013. Evaluation of truncated NhhA protein as a candidate meningococcal vaccine antigen. PLoS One 8:e72003. doi: 10.1371/journal.pone.0072003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sjölinder H, Eriksson J, Maudsdotter L, Aro H, Jonsson AB. 2008. Meningococcal outer membrane protein NhhA is essential for colonization and disease by preventing phagocytosis and complement attack. Infect Immun 76:5412–5420. doi: 10.1128/IAI.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffiths NJ, Hill DJ, Borodina E, Sessions RB, Devos NI, Feron CM, Poolman JT, Virji M. 2011. Meningococcal surface fibril (Msf) binds to activated vitronectin and inhibits the terminal complement pathway to increase serum resistance. Mol Microbiol 82:1129–1149. doi: 10.1111/j.1365-2958.2011.07876.x. [DOI] [PubMed] [Google Scholar]
- 17.Sjölinder M, Altenbacher G, Hagner M, Sun W, Schedin-Weiss S, Sjölinder H. 2012. Meningococcal outer membrane protein NhhA triggers apoptosis in macrophages. PLoS One 7:e29586. doi: 10.1371/journal.pone.0029586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scarselli M, Rappuoli R, Scarlato V. 2001. A common conserved amino acid motif module shared by bacterial and intercellular adhesins: bacterial adherence mimicking cell-cell recognition? Microbiology 147:250–252. doi: 10.1099/00221287-147-2-250. [DOI] [PubMed] [Google Scholar]
- 19.Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, Sieling PA, Liu Y-J, Rea TH, Bloom BR, Modlin RL. 2005. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med 11:653–660. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peiser L, Makepeace K, Plüddemann A, Savino S, Wright JC, Pizza M, Rappuoli R, Moxon ER, Gordon S. 2006. Identification of Neisseria meningitidis nonlipopolysaccharide ligands for class A macrophage scavenger receptor by using a novel assay. Infect Immun 74:5191–5199. doi: 10.1128/IAI.00124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scarselli M, Serruto D, Montanari P, Capecchi B, Adu-Bobie J, Veggi D, Rappuoli R, Pizza M, Aricò B. 2006. Neisseria meningitidis NhhA is a multifunctional trimeric autotransporter adhesin. Mol Microbiol 61:631–644. doi: 10.1111/j.1365-2958.2006.05261.x. [DOI] [PubMed] [Google Scholar]
- 22.Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, Mantovani A. 1998. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur J Immunol 28:359–369. doi:. [DOI] [PubMed] [Google Scholar]
- 23.Cardone J, Le Friec G, Vantourout P, Roberts A, Fuchs A, Jackson I, Suddason T, Lord G, Atkinson JP, Cope A, Hayday A, Kemper C. 2010. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol 11:862–871. doi: 10.1038/ni.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenmalm MC, Cherwinski H, Bowman EP, Phillips JH, Sedgwick JD. 2006. Regulation of myeloid cell function through the CD200 receptor. J Immunol 176:191–199. doi: 10.4049/jimmunol.176.1.191. [DOI] [PubMed] [Google Scholar]
- 25.Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, Claeys S, Hammad H, Brusselle GG, Vandenabeele P, Krysko DV, Bachert C. 2011. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy 66:396–403. doi: 10.1111/j.1398-9995.2010.02498.x. [DOI] [PubMed] [Google Scholar]
- 26.Mukhopadhyay S, Plüddemann A, Hoe JC, Williams KJ, Varin A, Makepeace K, Aknin M-L, Bowdish DM, Smale ST, Barclay AN, Gordon S. 2010. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host Microbe 8:236–247. doi: 10.1016/j.chom.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Sjölinder H, Jonsson A-B. 2010. Olfactory nerve—a novel invasion route of Neisseria meningitidis to reach the meninges. PLoS One 5:e14034. doi: 10.1371/journal.pone.0014034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devoe IW, Gilchrist JE. 1973. Release of endotoxin in the form of cell wall blebs during in vitro growth of Neisseria meningitidis. J Exp Med 138:1156–1167. doi: 10.1084/jem.138.5.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juliusson S, Bachert C, Klementsson H, Karlsson G, Pipkorn U. 1991. Macrophages on the nasal mucosal surface in provoked and naturally occurring allergic rhinitis. Acta Otolaryngol 111:946–953. doi: 10.3109/00016489109138435. [DOI] [PubMed] [Google Scholar]
- 30.Castaño D, Barrera LF, Rojas M. 2011. Mycobacterium tuberculosis alters the differentiation of monocytes into macrophages in vitro. Cell Immunol 268:60–67. doi: 10.1016/j.cellimm.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Rydström A, Wick MJ. 2010. Salmonella inhibits monocyte differentiation into CD11chiMHC-IIhi cells in a MyD88-dependent fashion. J Leukoc Biol 87:823–832. doi: 10.1189/jlb.0909615. [DOI] [PubMed] [Google Scholar]
- 32.Varin A, Mukhopadhyay S, Herbein G, Gordon S. 2010. Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood 115:353–362. doi: 10.1182/blood-2009-08-236711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.BoseDasgupta S, Pieters J. 2014. Inflammatory stimuli reprogram macrophage phagocytosis to macropinocytosis for the rapid elimination of pathogens. PLoS Pathog 10:e1003879. doi: 10.1371/journal.ppat.1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. 2011. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis 204:253–262. doi: 10.1093/infdis/jir276. [DOI] [PubMed] [Google Scholar]
- 35.Chomarat P, Dantin C, Bennett L, Banchereau J, Palucka AK. 2003. TNF skews monocyte differentiation from macrophages to dendritic cells. J Immunol 171:2262–2269. doi: 10.4049/jimmunol.171.5.2262. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto S, Yamada M, Motoyoshi K, Akagawa KS. 1997. Enhancement of macrophage colony-stimulating factor–induced growth and differentiation of human monocytes by interleukin-10. Blood 89:315–321. [PubMed] [Google Scholar]
- 37.Capsoni F, Minonzio F, Ongari AM, Carbonelli V, Galli A, Zanussi C. 1995. IL-10 up-regulates human monocyte phagocytosis in the presence of IL-4 and IFN-gamma. J Leukoc Biol 58:351–358. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen HH, Tran BT, Muller W, Jack RS. 2012. IL-10 acts as a developmental switch guiding monocyte differentiation to macrophages during a murine peritoneal infection. J Immunol 189:3112–3120. doi: 10.4049/jimmunol.1200360. [DOI] [PubMed] [Google Scholar]
- 39.Bebien M, Hensler ME, Davanture S, Hsu L-C, Karin M, Park JM, Alexopoulou L, Liu GY, Nizet V, Lawrence T. 2012. The pore-forming toxin β hemolysin/cytolysin triggers p38 MAPK-dependent IL-10 production in macrophages and inhibits innate immunity. PLoS Pathog 8:e1002812. doi: 10.1371/journal.ppat.1002812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagamatsu K, Kuwae A, Konaka T, Nagai S, Yoshida S, Eguchi M, Watanabe M, Mimuro H, Koyasu S, Abe A. 2009. Bordetella evades the host immune system by inducing IL-10 through a type III effector, BopN. J Exp Med 206:3073–3088. doi: 10.1084/jem.20090494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. 2011. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 42.Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. 2006. IL-10 gene-deficient mice lack TGF-beta/Smad-mediated TLR2 degradation and fail to inhibit proinflammatory gene expression in intestinal epithelial cells under conditions of chronic inflammation. Ann N Y Acad Sci 1072:389–394. doi: 10.1196/annals.1326.023. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Q, Leong SC, McNamara PS, Mubarak A, Malley R, Finn A. 2011. Characterisation of regulatory T cells in nasal associated lymphoid tissue in children: relationships with pneumococcal colonization. PLoS Pathog 7:e1002175. doi: 10.1371/journal.ppat.1002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang X-L, Zhang G-L, Yang T, Yang B-H, Wang L-J, Wang Q-H, Luo Z-X, Liu E-M, Fu Z. 2015. Association of pneumococcal carriage and expression of Foxp3+ regulatory T cells and Th17 cells in the adenoids of children. Respiration 90:25–32. doi: 10.1159/000381724. [DOI] [PubMed] [Google Scholar]
- 45.Netea MG, Lewis EC, Azam T, Joosten LA, Jaekal J, Bae SY, Dinarello CA, Kim SH. 2008. Interleukin-32 induces the differentiation of monocytes into macrophage-like cells. Proc Natl Acad Sci U S A 105:3515–3520. doi: 10.1073/pnas.0712381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schenk M, Krutzik SR, Sieling PA, Lee DJ, Teles RM, Ochoa MT, Komisopoulou E, Sarno EN, Rea TH, Graeber TG, Kim S, Cheng G, Modlin RL. 2012. NOD2 triggers an interleukin-32-dependent human dendritic cell program in leprosy. Nat Med 18:555–563. doi: 10.1038/nm.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osman A, Bhuyan F, Hashimoto M, Nasser H, Maekawa T, Suzu S. 2014. M-CSF inhibits anti–HIV-1 activity of IL-32, but they enhance M2-like phenotypes of macrophages. J Immunol 192:5083–5089. doi: 10.4049/jimmunol.1302732. [DOI] [PubMed] [Google Scholar]
- 48.Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, Gerhard M, Taube C, Müller A. 2013. Helicobacter pylori γ-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A 110:3047–3052. doi: 10.1073/pnas.1211248110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcondes AM, Li X, Tabellini L, Bartenstein M, Kabacka J, Sale GE, Hansen JA, Dinarello CA, Deeg HJ. 2011. Inhibition of IL-32 activation by α-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood 118:5031–5039. doi: 10.1182/blood-2011-07-365247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johansson L, Rytkonen A, Bergman P, Albiger B, Källström H, Hökfelt T, Agerberth B, Cattaneo R, Jonsson AB. 2003. CD46 in meningococcal disease. Science 301:373–375. doi: 10.1126/science.1086476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The differentiating effect of NhhA requires the full passenger domain. Recombinant proteins corresponding to the full-length (NhhA-FL, amino acids [aa] 51 to 510, 50 kDa), middle (NhhA-M, aa 164 to 380, 24 kDa), N-terminal (NhhA-N, aa 51 to 164, 13 kDa), and C-terminal (NhhA-C, aa 380 to 510, 14 kDa) regions of the NhhA passenger domain were generated and purified under native conditions. (A) Mo were treated with recombinant NhhA proteins (50 nM) corresponding to the full-length (NhhA-FL) or truncated passenger domains for 3 days. After extensive washing, the cells were stained for NhhA as described in Materials and Methods. Representative micrographs are shown in the upper panel. Original magnification, ×400. Some cells were stained with a Horizon V450-labeled anti-CD14 antibody and analyzed by flow cytometry. One representative SSC/CD14 plot of three independent experiments is shown in the lower panel. (B) Proteins (50 nM each) were separated by native polyacrylamide gel electrophoresis (PAGE) and detected using polyclonal rabbit anti-NhhA antibodies (1:1,000). The size in kilodaltons of the protein ladder is indicated. Download
ERK, JNK, and NF-κB signaling pathways are involved in NhhA-triggered Mo differentiation. Mo were treated with 50 nM NhhA alone (control) or cotreated with the indicated inhibitors for ERK (PD98095, 10 µM), JNK (SP600125, 10 µM), NF-κB (Celastrol, 500 nM), or activator protein 1 (AP-1) (SR11302, 1 µM) for 3 days. Differentiation to Mφ was determined by the flow cytometric analysis of CD14 expression and SSC parameters. Medians with interquartile ranges of median fluorescence intensity (MFI) from three independent experiments are presented as fold change relative to control. *, P < 0.05 compared with control group using ANOVA followed by the Bonferroni post hoc test. Download
Intrinsic effect of NhhA on Mφ polarization. Mo were stimulated with the FAM20 WT or NhhA-deficient mutant (ΔNhhA) strains at an MOI of 100 for 3 h, mRNA was isolated, and qPCR analysis was performed for IL-12b, IL-10, and inducible nitric oxide synthase (iNOS). Normalized data, expressed as fold change compared with uninfected cells, are presented as the mean ± standard deviation (SD) from three independent experiments. *, P < 0.05 compared with WT-stimulated cells using ANOVA followed by the Bonferroni post hoc test. Download
Peritoneal cell populations. CD46+/+ mice were challenged i.p. with 50 nM native or heat-inactivated NhhA (control) for 3 days prior to infection with FAM20 (108 CFU/mouse, i.p.). At 12 h postinfection, peritoneal cells were collected from both uninfected (upper panel) and bacterium-infected (lower panel) mice. Cell populations were analyzed by flow cytometry. Mφ (F4/80hi CD11bhi Ly6Cint Ly6G−), neutrophils (Ly6G+ F4/80− CD11b+ Ly6Cint), DCs (CD205+ CD19− CD3−), T cells (CD3+ CD19−), and B cells (CD19+ CD3−) were gated. Data are presented as the mean ± standard deviation (SD) (n = 6) and are representative of at least three independent experiments. *, P < 0.05 compared with the control group using paired Student’s t test. Download
NhhA treatment alleviates the host proinflammatory responses. CD46+/+ mice (n = 6 to 8) were treated i.p. with 50 nM heat-inactivated (control) or native NhhA for 3 days. (A) Whole blood was collected from mice and mixed with FAM20 (MOI, 100) for 3 h. Bacterial growth was determined at indicated time points. Data shown are the mean ± standard deviation (SD) (n = 6). (B) Mice were challenged i.p. with 108 CFU FAM20. Levels of IL-6, TNF-α, and IL-10 in the peritoneal cavity at 24 h postinfection were estimated by ELISA. Data represent the mean ± standard deviation, and they are pooled from three independent experiments. *, P < 0.05 compared with the control group using paired Student’s t test. Download
Primers used in real-time PCR.
PCR primers used for NhhA cloning.