

ABSTRACT
Ebola virus (EBOV) makes extensive and intricate use of host factors in the cellular endosomal/lysosomal pathway to release its genome into the cytoplasm and initiate infection. Following viral internalization into endosomes, host cysteine proteases cleave the EBOV fusion glycoprotein (GP) to unmask the binding site for its intracellular receptor, the cholesterol transporter Niemann-Pick C1 (NPC1). GP-NPC1 interaction is required for viral entry. Despite these and other recent discoveries, late events in EBOV entry following GP-NPC1 binding and culminating in GP-catalyzed fusion between viral and cellular lipid bilayers remain enigmatic. A mechanistic understanding of EBOV membrane fusion has been hampered by the failure of previous efforts to reconstitute fusion in vitro or at the cell surface. This report describes an assay to monitor initial steps directly in EBOV membrane fusion—triggering of GP and virus-cell lipid mixing—by single virions in live cells. Fusogenic triggering of GP occurs predominantly in Rab7-positive (Rab7+) endosomes, absolutely requires interaction between proteolytically primed GP and NPC1, and is blocked by key GP-specific neutralizing antibodies with therapeutic potential. Unexpectedly, cysteine protease inhibitors do not inhibit lipid mixing by virions bearing precleaved GP, even though they completely block cytoplasmic entry by these viruses, as shown previously. These results point to distinct cellular requirements for different steps in EBOV membrane fusion and suggest a model in which host cysteine proteases are dispensable for GP fusion triggering after NPC1 binding but are required for the formation of fusion pores that permit genome delivery.
IMPORTANCE
Ebola virus (EBOV) causes outbreaks of highly lethal disease for which no approved vaccines or treatments exist. Recent work has elucidated key molecular features of the complex EBOV entry process, including stepwise interactions with multiple host factors. However, there is a critical gap in our understanding of events that surround the final membrane fusion step which persists due to the paucity of direct and extensive investigation of EBOV fusion. Here, we report a real-time assay for EBOV glycoprotein fusion triggering and use it to define its cellular location and requirements. We also uncover an unexpected requirement for host proteases at a step after fusion triggering that may reflect their role in formation of fusion pores for genome delivery.
INTRODUCTION
Ebola virus (EBOV) and other members of the family Filoviridae of enveloped, negative-strand RNA viruses are associated with highly lethal disease for which no FDA-approved vaccines or treatments exist. Filovirus particles are characterized by a thread-like morphology, with infectious virions averaging 800 to 1,000 nm in length by 100 nm in diameter (1). The viral genome is encapsidated by several proteins, the nucleoprotein (NP), VP24, VP30, and VP35, to form the ribonucleocapsid, along with the L polymerase (2, 3). The VP40 matrix protein adds structural support through interfaces with the nucleocapsid and the viral envelope (4). Studding the viral membrane are metastable trimeric glycoprotein (GP) spikes composed of disulfide-linked heterodimers, the GP1 receptor-binding subunit, and the GP2 fusion subunit (5).
The filovirus GP2 subunit is categorized as a class I viral fusion protein on the basis of its α-helical major domains or heptad repeat (HR) regions (6). The N-terminal HR of GP2 comprises segments that encircle GP1 along with the internal fusion loop, which packs against an adjacent GP1 monomer (7). For insertion into apposing host membranes, the fusion loop must be extricated from its position in prefusion spikes and presumably extended before GP2 refolds. The postfusion structure of GP2 trimers is marked by six-helix bundle (6HB) formation (8, 9), consistent with other class I viral fusion proteins (10), bringing the host and viral membranes into close proximity to lower the kinetic barriers to fusion. Hemifusion of the outer bilayer leaflets precedes formation of a full fusion pore for the cytoplasmic release of the viral genome.
Like other filoviruses, EBOV is capable of binding a variety of surface molecules, which contributes to its broad cellular tropism. C-type lectins and phosphatidylserine receptors have been implicated in attachment, though their roles in uptake have not been fully defined (11–14), and T-cell immunoglobulin domain-1 may act as a receptor on epithelial cells (15). Internalization of EBOV occurs primarily through a macropinocytosis-like process, and the virus traffics through the endocytic pathway (16–18). Proteolytic cleavage of GP1 by a class of low-pH-activated proteases, cysteine cathepsins, removes the C-terminal glycan cap and mucin domain sequences (19) to reveal a receptor-binding domain. This newly exposed GP1 domain interacts with Niemann-Pick C1 (NPC1), which serves as an obligate intracellular receptor for filoviruses (20, 21). While cathepsin cleavage and subsequent NPC1 binding are indispensable for infection, they are themselves insufficient to mediate release of the viral genome to the cytoplasm (22). A final trigger for fusion of viral and endosomal membranes by GP2 has been postulated but not demonstrated (23, 24), and which precise endosomal compartment actually supports fusion is unknown.
To date, progress in defining the requirements for EBOV fusion triggering has been hampered by the lack of robust assays with true biological relevance. In order to examine GP triggering more fully, we have used a real-time microscopy assay for the detection of GP-mediated lipid mixing within cells. This assay further enables the events of GP triggering and lipid mixing to be uncoupled from full fusion and infection. In this report, we characterize GP-mediated lipid mixing and investigate host factors thought to be involved in the process. Our findings identify essential but distinct roles for cysteine cathepsins and NPC1 in EBOV GP fusion triggering.
RESULTS
Characterization of dequenching virions.
For evaluating EBOV GP triggering under biosafety level 2 (BSL-2) conditions, we generated vesicular stomatitis virus (VSV) pseudotypes, which provide a highly validated surrogate system for recapitulating filovirus entry (25, 26). To facilitate viral tracking in most experiments, virions were engineered to contain a fluorescent protein, monomeric NeonGreen (mNG), fused to the viral phosphoprotein (P) (27, 28). The heavily glycosylated mucin domain (Muc) of EBOV GP is dispensable for entry, and its deletion enhances viral attachment (29, 30). Accordingly, we primarily used VSV pseudotypes bearing mucin domain-deleted GP (GPΔMuc). To validate our viral preparations, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD)-labeled VSV bearing mNG-P and EBOV GPΔMuc was bound to a coverslip and incubated with Alexa Fluor 555-labeled KZ52, a neutralizing antibody that recognizes a conformational epitope at the GP1-GP2 interface (7). On average, over 90% of DiD-labeled particles also possessed NG-P signal, and 72% of total particles exhibited triple-labeling, indicating that the preparations used were of high quality (Fig. 1A).
FIG 1 .

Characterization of dequenching particles. (A) Coverslip-bound VSV pseudotyped with EBOV GPΔMuc displays high incorporation of mNG-P and GP, as indicated by KZ52-AF555 binding. Individual channels are pseudocolored white for greater visibility. DiD labeling was carried out so that the DiD signal remained visible when self-quenched. (B) Dequenching is detected as a sharp increase in DiD signal. An example of hemifusion is seen here. Bar, 10 µm. (C) Intensity trace for the indicated particle. (D) Representative traces for fast/specific (solid line) and slow/nonspecific (dotted line) dequenching. Rates were derived by fitting the traces to a one-phase nonlinear regression model. The plateau for the slow trace begins approximately 28 min after the onset of the signal increase. a.u., arbitrary units.
The lipophilic dye DiD spontaneously incorporates into membranes and displays fluorescence self-quenching at high concentrations. Lipid mixing between viral and host bilayers, as in hemifusion or fusion, enables lateral diffusion of the dye accompanied by dequenching or increased fluorescence intensity (31, 32). Virions were labeled so that the fluorescence signal of self-quenched DiD was detectable prior to dequenching. DiD dequenching resulted in a sharp increase in signal, which we define here as at least a doubling of DiD intensity within 20 s, and a net increase in intensity of 2- to 5-fold within 1 min was typically observed (Fig. 1B and C; see also Movie S1 in the supplemental material). In our experiments, increases in DiD signal sorted strictly into one of two groups: very fast or very slow. Fast dequenching, as indicated by a representative trace (Fig. 1D, solid line), occurred irrespective of nucleocapsid release (data not shown) and appears to indicate specifically triggered lipid mixing events. We attribute the significantly lower rate of DiD dequenching (Fig. 1D, dotted line) to nonspecific transfer of the dye to host membranes, possibly through particle degradation. Only fast events constituted “dequenching” in this study. We found that the extent of dequenching varied somewhat among DiD-labeled particles. This may reflect differences in the volumes of endocytic compartments, as well as in the concentration of dye within the viral membrane, as some heterogeneity in self-quenching was noted. Dequenching events occurring at later postinfection time points generally exhibited greater amplitude between the quenched and dequenched states (data not shown).
We initially investigated dequenching kinetics in Vero cells, due to their high permissivity and established use in filovirus infection studies. The time span needed for 50% of particles bearing EBOV GPΔMuc to dequench (t1/2) was 42 min, considerably greater than the t1/2 of 17 min for similar particles bearing VSV G, which is believed to undergo fusion triggering in early endosomes (Fig. 2A) (33, 34). We found that the dequenching kinetics for VSV-EBOV GPΔMuc particles were very similar in U2OS cells (t1/2 = 44 min, Fig. 2B), and, because autofluorescence is much lower than in Vero cells, we used U2OS cells in the bulk of our experiments. Regardless of cell type, virtually all dequenching events occurred within 2 h of the synchronized onset of infection. We also examined the dequenching kinetics of VSV particles bearing full-length EBOV GP. Although the onset and conclusion of dequenching events showed similar timings, the overall rate of dequenching by VSV-EBOV GP (t1/2 = 64 min) was slower than that of VSV-EBOV GPΔMuc, which may reflect differences in internalization or the need for additional proteolytic processing (Fig. 2C). Precleavage of EBOV GPΔMuc by thermolysin, which structurally and functionally mimics the required cleavage of EBOV GP by endosomal/lysosomal cysteine protease cathepsin B (19, 35), did not accelerate dequenching kinetics compared to uncleaved EBOV GPΔMuc.
FIG 2 .

Dequenching kinetics and probabilities. (A) Dequenching kinetics of VSV particles pseudotyped with VSV G or EBOV GPΔMuc in Vero cells. (B) Dequenching kinetics of EBOV GPΔMuc-bearing particles in Vero cells versus U2OS cells. (C) Dequenching kinetics of particles bearing uncleaved or cleaved EBOV GPΔMuc or full-length EBOV GP in U2OS cells. (D) Total percentage of cell-associated particles undergoing dequenching in U2OS and Vero cells. (n > 100 dequenching events.)
Live imaging revealed that the majority of virions did not engage in lipid mixing (Fig. 2D). Of the total number of cell-associated VSV-EBOV GPΔMuc particles, approximately 16% dequenched in Vero cells and 10% in U2OS cells within 2 h. Additionally, 9.1% of particles bearing full-length EBOV GP dequenched in U2OS cells in the same time span. VSV G particles showed similarly low probabilities for lipid mixing in the two cell types. Although we are unable to distinguish definitively between bound and internalized virions, nearly all cell-associated particles had trafficked to perinuclear regions by the end of experiments (data not shown), leading us to believe that uptake levels are high with both EBOV GP and VSV G. The low percentage of particles with actual relevance to infection validates our single-particle approach to elucidating aspects of EBOV entry.
Viral lipid mixing is strictly GP mediated.
In order to confirm that the observed dequenching events indeed represented GP-mediated lipid mixing, we first assessed dequenching in the presence of NH4Cl. Failure to acidify endosomes precludes GP activation and infection (36, 37), as low pH is required for cysteine cathepsin activity (38) and possibly for fusogenic conformational changes in GP (39, 40). We found nearly a complete reduction of VSV-EBOV GPΔMuc dequenching by NH4Cl treatment, which lends support to the observed lipid mixing being specifically GP mediated (Fig. 3A).
FIG 3 .
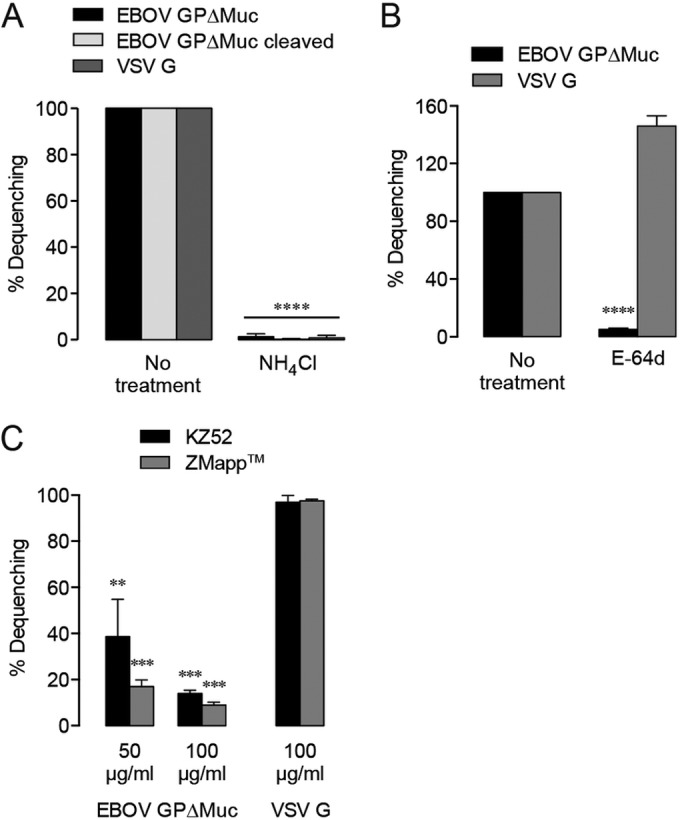
Viral dequenching is glycoprotein mediated. (A) DiD dequenching requires endosomal acidification. (B) Inhibition of cysteine cathepsin activity reduces EBOV GPΔMuc-mediated dequenching. (C) EBOV GP-binding neutralizing antibodies specifically inhibit dequenching. Dequenching is presented here as a percentage of the untreated EBOV GPΔMuc or VSV G control activity. Asterisks indicate that values are significantly lower than those seen with untreated controls.
The need for host cysteine cathepsin cleavage of EBOV GP in infection is well established (19, 35). Pan-cysteine cathepsin inhibitors, such as E-64d, prevent entry by filoviruses, which differ in their preference for cleavage by cathepsin B or L (19, 41). We found that E-64d treatment reduced EBOV GPΔMuc-mediated lipid mixing by more than 90%, while lipid mixing by VSV G-bearing particles was not similarly affected (Fig. 3B), demonstrating that a lack of cysteine cathepsin activity hinders downstream events in lipid mixing for EBOV GP.
As neutralizing antibodies directed against the EBOV GP base, such as KZ52 (7, 42) or c4G7 and c2G4 in the ZMapp antibody cocktail (43–45), have been hypothesized to inhibit fusogenic structural rearrangement, we evaluated their effects on lipid mixing (Fig. 3C). Our results confirm that KZ52 and ZMapp treatments specifically render EBOV GP less able to engage in lipid mixing while leaving viral internalization and trafficking unaltered (see Fig. S1A in the supplemental material). Preincubation of VSV-EBOV GPΔMuc with KZ52 resulted in inhibition of dequenching by 60% at 50 µg/ml and 85% at 100 µg/ml. ZMapp also had a concentration-dependent, if slightly more potent, effect on dequenching, with lipid mixing inhibited by 83% at 50 µg/ml and 91% at 100 µg/ml. Neither antibody treatment was able to abolish dequenching, even though the concentrations used were approximately 30-fold to 40-fold higher than the reported IC90 values for viral neutralization by KZ52, c2G4, and c4G7 individually (42, 45). This supports the idea of the existence of a significant barrier to fusion, with the number of unencumbered GP molecules needed for simple lipid mixing being much lower than the number needed to bring about membrane fusion and genome release. Collectively, our results signify that lipid mixing is the express product of GP triggering, which entails some degree of conformational change by proteolytically primed EBOV GP and which may lead to or encompass full fusion.
NPC1 is required for fusogenic triggering of EBOV GP.
The endosomal/lysosomal membrane protein NPC1, involved in cholesterol homeostasis, serves as an essential intracellular receptor for filovirus infection, with a cysteine cathepsin-cleaved form of GP directly recognizing the lumenal domain C of NPC1 (20–22, 46). Because the precise role of NPC1 in filovirus entry has not been fully defined, we evaluated lipid mixing in the absence of NPC1 (see Fig. S2 in the supplemental material). Dequenching of VSV-EBOV GPΔMuc particles was inhibited almost entirely in NPC1-deficient U2OS cells, whereas VSV G dequenching was unaffected, indicating that NPC1 expression is vital for EBOV fusion triggering (Fig. 4A).
FIG 4 .
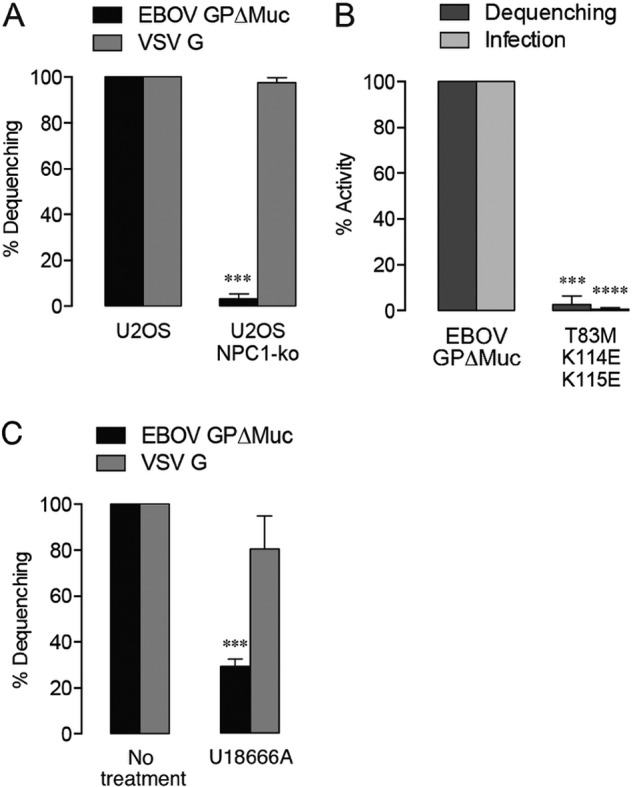
Lipid mixing requires EBOV GP-NPC1 interaction. (A) Lipid mixing by EBOV GPΔMuc-pseudotyped virus is significantly reduced in U2OS cells lacking the NPC1 cholesterol transporter. Asterisks indicate that values are significantly lower than those seen with wt U2OS. (B) A GP mutant unable to bind NPC1 displays impaired lipid mixing activity and infectivity. Asterisks indicate that values are significantly lower than those seen with VSV-EBOV GPΔMuc. (C) Lipid mixing is decreased by addition of the drug U18666A (10 µM), which inhibits cholesterol transport from within endosomes and induces an NPC1 disease phenotype. Asterisks indicate that values are significantly lower than those seen with the untreated control.
Mutations at the putative GP1-NPC1 interface have been previously demonstrated to reduce GP1-NPC1 interaction and greatly impair infectivity (22, 47, 48). Recent work has shown that proteolytic cleavage of GP1 exposes the NPC1-binding site, comprising a basic/polar crest overhanging a deep hydrophobic trough (65). We investigated entry by VSV-EBOV GPΔMuc particles bearing three mutations in the NPC1-binding site (T83M in the trough, K114E/K115E in the crest) that abolish GP1-NPC1 interaction and viral infectivity (65). Lipid mixing was reduced by 97% with this triple mutant (Fig. 4B). The inhibition of lipid mixing by VSV-EBOV GP in NPC1-deficient U2OS cells and by VSV-EBOV GP (T83M/K114E/K115E) in wild-type (wt) U2OS cells was not explained by altered delivery to endosomal/lysosomal compartments, since postinfection colocalization of viral particles and endocytic markers was not significantly reduced (see Fig. S1B in the supplemental material).
NPC1’s functionality as a cholesterol transporter may also have some bearing on EBOV entry, as GP-mediated lipid mixing is selectively inhibited by the cationic amphiphile U18666A (Fig. 4C), which induces an NPC1 disease phenotype marked by cholesterol accumulation within late endosomes and lysosomes (20, 49). GP1-NPC1 binding is not affected by U18666A (20, 24), but short-term U18666A treatments that do not cause substantial cholesterol accumulation (see Fig. S3 in the supplemental material) nevertheless prevent EBOV entry (20), suggesting that this block is due to the drug’s specific effect on NPC1 rather than to the resulting increase in endosomal/lysosomal cholesterol levels. Taken together, our results provide direct evidence that NPC1 facilitates one or more early steps in EBOV membrane fusion.
GP fusion triggering occurs in NPC1-positive (NPC1+), Rab7+ compartments.
Colocalization of dequenching virions with fluorescently labeled endosomal markers was performed in order to elucidate the compartment in which EBOV GP-dependent membrane fusion is triggered (Fig. 5; see also Movies S2 to S5 in the supplemental material). Although a recent report suggested that EBOV fuses exclusively in compartments devoid of NPC1 (50), we found that all dequenching particles in NPC1-mNG-expressing cells colocalized with this marker at the time of dequenching (Fig. 5J; see also Movie S5). Additionally, the high percentage of dequenching particles colocalized with green fluorescent protein (GFP)-Rab7 (98%) indicates the ubiquity of this protein in endosomes supporting GP triggering. Further evidence that EBOV must traffic to a suitably mature compartment for fusion was provided by inhibition of dequenching by the two-pore calcium channel (TPC)-blocking small molecule tetrandrine (Fig. 6). Tetrandrine, which may impair the activity of TPC in endocytic trafficking and heterotypic fusion of vesicles (51), was recently shown to inhibit EBOV infection in vitro and in vivo (50) and decreased GP-mediated lipid mixing in our assay by 95% at 10 µM.
FIG 5 .

(A TO H) Endosomal localization of EBOV GP triggering. Colocalization and associated intensity trace of a dequenching particle with a GFP-Rab5+ (A and B), GFP-Rab7+ (C and D), GPF-LAMP1+ (E and F), or NPC1-mNG+ (G and H) vesicle. Bar, 10 µm. (J) Total percentage of dequenching particles colocalizing with an endocytic marker (n = approximately 50 dequenching events in each set of transduced cells). (K) Time elapsed between colocalization with NPC1 and onset of dequenching. The line indicates the mean value (n = 50 dequenching particles).
FIG 6 .
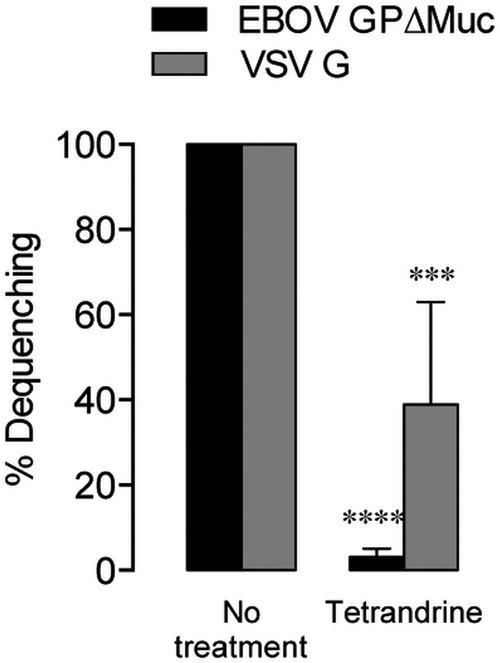
Glycoprotein-mediated lipid mixing is inhibited by the TPC-blocking drug tetrandrine (10 µM). Asterisks indicate that values are lower than those seen with untreated controls.
While considerable lipid mixing also occurred in GFP-LAMP1+ vesicles (75%), we found, surprisingly, that a significant fraction of dequenching VSV-EBOV GPΔMuc particles (15%) colocalized with GFP-Rab5 puncta. Despite their slower rate of lipid mixing events (Fig. 2C), 10% of particles bearing full-length EBOV GP also dequenched in Rab5+ compartments (data not shown). Because Rab7 appears to be a nearly essential component of the EBOV fusion compartment, these Rab5+ vesicles are likely intermediate endosomes, suggesting that GP triggering can occur earlier in the endocytic pathway than commonly believed (20, 50, 52, 53). Our results also imply that some population of endosomes must display Rab5 and NPC1 concurrently, at least in the U2OS cell line used. We detected an average lag of 6 min (ranging from 40 s to more than 20 min) between colocalization of viral and NPC1 signals and dequenching, indicating that the occurrence of GP triggering is not immediate upon delivery to NPC1+ compartments (Fig. 5K).
GP2 fusion loop mutations decouple lipid mixing and infection.
To examine the effect of fusion loop mutation on lipid mixing, we tested two previously published GP2 mutants (Fig. 7) (54, 55). Internalization and late endosomal delivery of VSV particles bearing these mutants did not differ significantly from VSV-EBOV GPΔMuc (see Fig. S1C in the supplemental material), indicating that their defects are specific to aspects of fusion. The GP (F535R) mutant, characterized as having impaired membrane binding and insertion capability (54), exhibited no defect in lipid mixing while being unable to support infection. Another fusion loop mutant, GP (L529A/I544A) (54), showed substantially decreased lipid mixing. The L529A/I544A mutation is believed to prevent formation of the hydrophobic fusion loop tip at low pH, with L529 and I544 creating a scaffold along with F535 (54, 55). While nonviable, the double mutant did effect low-level lipid mixing (Fig. 7), suggesting that even severely disrupted GP fusion loops retain some capacity for membrane insertion in cells.
FIG 7 .
Mutation of the GP2 fusion loop can uncouple EBOV GP-mediated lipid mixing and infection. Asterisks indicate that values are significantly lower than those seen with EBOV GPΔMuc.
Post-NPC1-binding cysteine cathepsin activity is needed for infection but not lipid mixing by EBOV GP.
In order to separate possible requirements for cysteine cathepsins pre- and post-NPC1 binding, we evaluated lipid mixing by GP in various cleavage states in the presence and absence of E-64d. Thermolysin was used to generate particles either fully or partially lacking C-terminal GP1 sequences corresponding to the glycan cap and mucin domains (Fig. 8A). While increased infectivity of particles bearing cleaved GP has been reported (19, 30, 35), we did not observe this phenomenon or enhanced lipid mixing. When particles were bound to cells to the same extent, cleaved and uncleaved GP produced similar levels of lipid mixing and infection (Fig. 8B and C). Unexpectedly, we found that dequenching by particles with cleaved EBOV GP was much less susceptible to E-64d blockage and that the degree of lipid mixing correlated with the extent of GP cleavage (Fig. 8C). Fully cleaved GP exhibited slight hyperfusogenicity that was unaffected in the absence of cathepsin activity. The inability of precleavage to rescue infectivity in E-64d-treated cells (Fig. 8B), however, indicates that a specific requirement for cathepsins in fusion pore formation lies post-NPC1 binding.
FIG 8 .
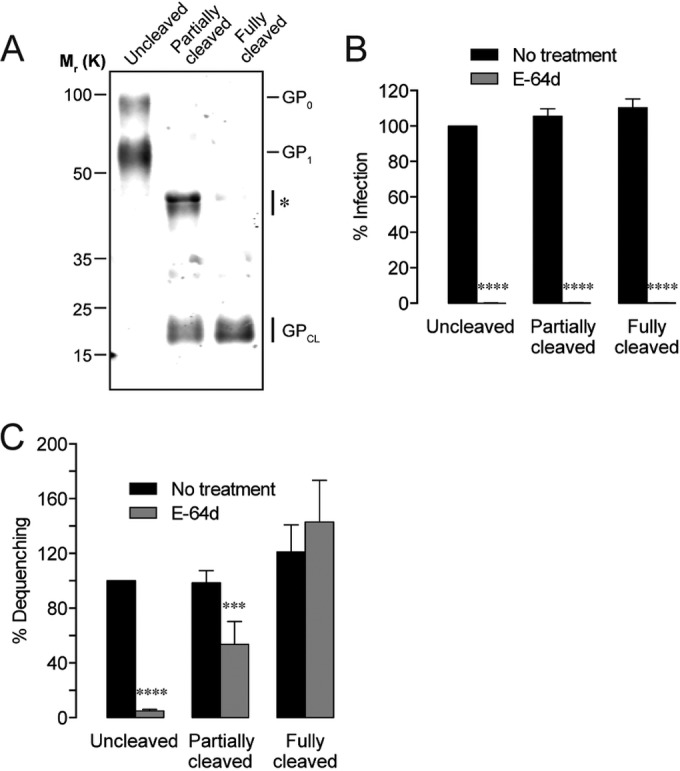
Post-NPC1-binding cathepsin activity is required for infection but not EBOV GP triggering. (A) Immunoblot showing the extent of GP1 cleavage in three EBOV GPΔMuc-pseudotyped virus preparations. The asterisk indicates a cleavage intermediate of GP1. (B) The E-64d cysteine protease inhibitor (25 µM) completely blocks infection. (C) Precleaved EBOV GPΔMuc can induce lipid mixing in the absence of cathepsin activity (25 µM E-64d). Asterisks indicate that values are significantly lower than those seen with the untreated, uncleaved control.
DISCUSSION
Most previous attempts to characterize EBOV membrane fusion have relied on assessing delivery of viral content, such as luciferase or β-lactamase, through which GP triggering is inferred (24, 52, 56). While content delivery assays are useful in characterizing infection on a general scale, they have sizable limitations. For example, they cannot resolve the fates of individual particles or differentiate between hemifusion, incomplete fusion pore expansion, and no GP triggering at all. In order to examine GP fusion triggering directly, we have employed an assay for tracking real-time lipid mixing by single particles that revealed novel and significant aspects of the process.
We found, unsurprisingly, that the majority of cell-associated particles did not advance to lipid mixing (Fig. 2D). The low probability of lipid mixing suggests that the results of previously published experiments utilizing bulk endocytosis may be misleading, with an inadvertent focus on nontriggered virions. This presents a particular obstacle in determining factors involved in EBOV GP fusion triggering or the compartment(s) in which triggering occurs.
The interaction between cysteine cathepsin-primed GP1 and NPC1 has been previously shown to be vital for filovirus infection (22); however, the precise role of NPC1 in this process was unknown. Here, we used an NPC1-deficient cell line and a GP mutant unable to bind NPC1 to prove that the GP-NPC1 interaction, and not just virus-NPC1 colocalization, is indispensable for EBOV fusion triggering and lipid mixing (Fig. 3 and 4). We found that the appearance of NPC1 in virus-bearing endosomes always preceded GP triggering, though the time span between the two events varied (Fig. 5K), as also reported by Simmons and colleagues through a live-cell imaging assay with GP-pseudotyped retroviruses while the current article was in preparation (53). Mingo et al. proposed that delivery to NPC1+ vesicles represented a key rate-limiting step for EBOV fusion (52), but we found a lag ranging from 40 s to more than 20 min between virus-NPC1 colocalization and dequenching. It is unknown at present whether NPC1 binding directly promotes fusogenic conformational changes in GP or primes it for interaction with additional host factors. Therefore, while the lag may represent the span of time needed for the virus to bind NPC1 as its lumenal domains appear in the compartment, it may also signify a need for one or more host factors to act on GP prior to or following GP1-NPC1 interaction.
We provide direct evidence that EBOV GP fusion triggering occurs in NPC1+/Rab7+ vesicles, which may, in fact, encompass a range of endosomal compartments. Previously thought to undergo triggering only quite late in the endocytic pathway (50, 53), EBOV GP could mediate lipid mixing in intermediate endosomes, as indicated by their Rab5 signal, in our live assays. Because the presence of NPC1 is an absolute requirement for dequenching in our assay, we infer that some population of Rab5+ vesicles must contain NPC1 in U2OS cells. Such colocalization is not impossible, as immunofluorescence and fractionation studies have shown that NPC1 localizes primarily, but not entirely, to late endosomes, lysosomes, and the trans-Golgi network (49); therefore, NPC1 levels in some intermediate endosomes may be sufficient to support GP triggering. However, it is unclear whether viral genome release from relatively immature endosomes can occur following GP fusion triggering. Indeed, Simmons et al. found no evidence for GP-mediated retroviral content release from Rab5+ compartments (53), raising the possibility that virions must traffic further in the endocytic pathway to encounter host factors at levels necessary for complete fusion and genome release.
Previous work has revealed that a conformational epitope at the GP1-GP2 interface in the GP base is targeted by multiple neutralizing antibodies, including two of three components of the ZMapp therapeutic cocktail (7, 44, 45), leading to the hypothesis that this epitope is an “Achilles heel” for filoviruses. It was proposed on structural grounds that these base-binding antibodies potently neutralize by blocking fusogenic rearrangement and membrane insertion by GP2. Here, we provide the first direct evidence that neutralizing antibodies targeting the EBOV GP base inhibit viral fusion triggering (Fig. 3C). Interestingly, higher concentrations of neutralizing antibody were required to inhibit lipid mixing to the same extent as infection, but this may typify fusion by enveloped viruses. A recent study by Otterstrom et al. (57) examined the levels of antibody binding needed to inhibit influenza virus hemifusion. Using a pair of potent, broadly effective neutralizing antibodies, whose inhibitory profiles against influenza virus strains are very similar to those of KZ52 and ZMapp against filoviruses (58, 59), the authors showed that only high concentrations of these stem-binding antibodies were able to block the majority of hemifusion events. Although their assay (viral particles bound to a supported bilayer) differed from ours, their data strongly corroborate our results, with approximately 50 to 150 µg/ml of either anti-influenza virus antibody needed to reduce in vitro hemifusion by ~90%. The requirement for antibody concentrations beyond relevance to infection indicates a far lower threshold for lipid mixing than for full fusion and content release.
Further highlighting the distinction between EBOV lipid mixing and fusion, we found that GP mutations that abrogated infectivity did not necessarily extend to lipid mixing (Fig. 7). Although the F535R mutation was shown to reduce the binding of GP2 ectodomains with liposomes (54), we noted that lipid mixing in live cells was unaffected. Additionally, a peptide corresponding to the fusion loop of the L529A/I544A GP mutant was unable to insert into model membranes or promote lipid mixing in a cell-free system (38). While we likewise found that the L529A/I544A double mutant could not support infection, lipid mixing activity within cells was significantly reduced but not completely inhibited, indicating that the amino acid sequence may behave differently in native GP and in an isolated peptide, much less in an in vitro system. Shallow insertion of the fusion loop may be sufficient to yield lipid mixing in cells, even while failing to interact with the target membrane extensively enough for progression to full fusion pore formation.
Entry by virions bearing precleaved EBOV GP remains sensitive to inhibitors of host cysteine cathepsin activity, suggesting the existence of an additional protease-dependent step in filovirus membrane fusion (19, 35, 60). Surprisingly, however, our experiments revealed that lipid mixing by cleaved GP was entirely unaffected by inhibition of host cysteine cathepsin activity, even as infectivity was ablated (as reported previously) (Fig. 8). The latter finding is in agreement with that of Simmons et al., who found no evidence of content release by retroviral particles bearing cleaved GP in E-64d-treated cells; however, they were unable to examine lipid mixing consistently in their assay (53).
Our findings suggest that, following GP-NPC1 binding, cysteine cathepsins are dispensable for GP fusion triggering and lipid mixing but play a critical role in the formation and/or expansion of fusion pores that allow cytoplasmic release of viral genomes. We advance two mechanistic scenarios to account for our observations. First, the fusion trigger (comprising NPC1 and possibly an additional signal, e.g., acid pH, but not cysteine cathepsins) releases GP from its prefusion conformation, allowing formation of a membrane-bound prehairpin intermediate and driving hemifusion of viral and cellular membranes. A final cysteine cathepsin cleavage event then enables GP refolding to a 6HB, which mediates fusion pore formation and genome release. A similar two-step mechanism, with sequential roles for receptor binding and low pH, has been proposed for fusion by the avian sarcoma/leukosis retrovirus (61). Alternatively, it is conceivable that proteolytic cleavage of NPC1-bound GP is indeed the fusion trigger but that GP fusion triggering and 6HB formation can nevertheless occur inefficiently in the absence of cysteine cathepsin activity. Rearrangement of a few GP trimers may permit lipid mixing but not energetically costly fusion pore formation and expansion.
While this initial study focused primarily on the GP triggering and lipid mixing steps of EBOV entry, further work is needed to elucidate outstanding issues, such as the precise mechanism that explains why post-NPC1-binding cathepsin activity is required for infection but not lipid mixing. Future live-cell studies will examine the dynamics of fusion pore expansion and viral genome release and provide a more comprehensive understanding of the EBOV entry process.
MATERIALS AND METHODS
Cells.
U2OS human osteosarcoma cells were cultured in McCoy’s 5a medium (Gibco), and Vero grivet monkey kidney cells and human embryonic kidney 293T cells were cultured in Dulbecco’s modified Eagle medium (Gibco). All culture media were supplemented with 10% fetal bovine serum (FBS) and with 100 U/ml penicillin and 100 µg/ml streptomycin.
Generation of U2OS NPC1-ko cells was carried out by CRISPR/Cas9-mediated genome editing (62) using previously described methods (26). Briefly, a CRISPR guide RNA (gRNA) cloning vector (Addgene plasmid 41824) encoding an NPC1-specific gRNA that targets nucleotides 768 to 790 (5′ GGCCTTGTCATTACTTGAGGGGG 3′ on the complementary strand) of human NPC1 mRNA was cotransfected along with a plasmid encoding human-codon-optimized endonuclease Cas9 (Addgene plasmid 41815), a red fluorescent protein expression plasmid (to monitor transfection efficiency), and pMX-IRES-Bla (conferring blasticidin resistance to transfected cells). Following selection of transfected cells by blasticidin treatment, genomic DNA flanking the gRNA target site was amplified by PCR using forward (5′ TCATAAACACACCAAACTTGGAATC 3′) and reverse (5′ TCCTGCGGCAGAGGTTTTC 3′) primers and tested for indels by Surveyor assay as previously described (26). Once genome editing was confirmed at the population level, multiple single-cell clones were isolated for NPC1 gene sequencing. A clonal cell line, NPC1-#6, was found to contain a deletion of 13 nucleotides in both of its NPC1 alleles. The deletion in NPC1-#6 cells is predicted to cause a frame shift at amino acid position 167 that leads to production of a truncated protein of 215 amino acids, in comparison to the 1,278 amino acids of wild-type NPC1. We confirmed the absence of any remaining wild-type NPC1 alleles by reverse transcription-PCR (RT-PCR) using NPC1-F (5′ AGGCCCCCTCAAGTAATGAC 3′, specific for the deleted sequence in the NPC1-knockout cell line) and npc1-R (5′ GCCCAAAGTGCTGGTCAAAG 3′) primers. Amplification of the site-1 protease (S1P) gene using S1P-F (5′ GATGTGCTCTGGCAGATGGG 3′) and S1P-R (5′ TTTCACGCCAGAACCCCGC 3′) primers was used as a positive control for RT-PCR.
Viruses.
Pseudotyped virions were generated by infecting 293T cells expressing viral glycoproteins with VSV lacking its G gene, as previously described (19, 26, 28). GP cDNA derived from EBOV/H.sap/COD/76/Yam-Mayinga (EBOV “Mayinga” isolate) was used to generate VSV-EBOV GP. EBOV GPΔMuc lacks GP residues 309 to 489 (29). All EBOV GP mutants were generated in the GPΔMuc background. Infection supernatants were collected upon observation of significant cytopathy (1 to 4 days postinfection). Clarified supernatants were centrifuged at 4°C and 70,000 × g using an SW28 rotor to pellet the virus. Viral pellets were resuspended in phosphate-buffered saline (PBS) and further purified through a 10% sucrose cushion at 4°C and 107,000 × g using an SW41 rotor. Pellets were then resuspended in PBS at an approximate viral protein concentration of 1 mg/ml, and aliquots were stored at −80°C until use. Viral infectivity was assessed as previously described (19).
Cleavage of EBOV-GP was performed by incubating the virus with 250 µg/ml thermolysin (Sigma) for 1 h for partially cleaved GP and 500 µg/ml thermolysin for 2 h for fully cleaved GP at 37°C after the first ultracentrifugation step. Protease activity was quenched by addition of 1 mM phosphoramidon (Peptides International) and incubation on ice for 20 min prior to the second ultracentrifugation step through a sucrose cushion. The extent of cleavage was confirmed by immunoblotting with an anti-GP1 antibody.
Viral membranes were labeled with self-quenching concentrations of DiD (Life Technologies). Virus (50 µg) was incubated with 250 µM DiD for 90 min with gentle agitation in the dark at room temperature. Labeled virus was passed through a NAP-5 column (GE Healthcare) to remove excess dye and large aggregates and was used for imaging within 48 h. DiD labeling reduced viral infectivity by less than 10% (data not shown).
Antibodies and inhibitors.
ZMapp was a gift from John Dye (USAMRIID). For production of KZ52, various heavy- and light-chain domains of KZ52 were chemically synthesized as codon-optimized sequences for expression in human cells by Epoch Life Sciences and cloned into pMAZ-IgH (containing the expression cassette of human γ1 chain constant domains) and pMAZ-IgL (containing the expression cassette of human κ light-chain constant domains) vectors (63), respectively, using a Gibson assembly kit (New England Biolabs). Sanger sequencing confirmed the identity of the cloned vectors. Antibody production and purification were carried out as described earlier (64), with modifications. Briefly, suspension-adapted HEK293-Freestyle cells–grown in suspension cultures in Freestyle F17 expression media (Thermo Scientific)–were cotransfected with pMAZ-IgH-KZ52 and pMAZ-IgL-KZ52 (1:1 ratio) vectors using polyethyleneimine. At 6 to 7 days posttransfection, the supernatant was collected and incubated overnight at 4°C with protein A agarose resin (GenScript Corporation) in the presence of cOmplete Mini EDTA-free protease inhibitor cocktail (Roche Life Sciences). After the resin was washed with PBS (pH 8.0), bound IgG was eluted with 100 mM glycine (pH 3.0) into 1 M Tris-HCl (pH 8.0). The eluate was concentrated and buffer exchanged with PBS by use of an Amicon centrifugal filter concentrator (Millipore) with a molecular weight cutoff of 30,000 Da. Purified antibody was stored at −20°C. Protein concentration and yield were determined by UV absorbance, and purity was determined by SDS-PAGE. For labeling coverslip-bound virus, KZ52 was conjugated with Zenon Alexa Fluor 555 (Life Technologies) according to the manufacturer’s instructions.
Stock solutions of drugs were prepared in dimethyl sulfoxide (DMSO) or water and stored as frozen aliquots until use. Cells were incubated with ammonium chloride (Sigma) for 15 min or with E-64d (Peptides International) and tetrandrine (Sigma) for 1 h prior to experiments performed at the indicated concentrations. No preincubation with U18666A (Calbiochem) was performed. Inhibitors were maintained at the same concentrations during spinoculation and imaging.
Expression of fluorescent endosome markers.
For expression of fluorescently labeled endocytic pathway markers, cells were transduced with CellLight vectors (Life Technologies) for early endosomes (GFP-Rab5), late endosomes (GFP-Rab7), or lysosomes (GFP-LAMP1) 18 h prior to imaging according to the manufacturer’s instructions. A retroviral packaging vector bearing NPC1-mNG was also generated, and cells were transduced 18 h in advance of imaging.
Live-cell imaging.
Live imaging was performed on a DeltaVision widefield epifluorescence microscope (GE Healthcare) at 37°C. The microscope was equipped with a heated environmental chamber, an electron-multiplying charge-coupled device (EMCCD) camera (Photometrics), a 40×/1.35 numerical-aperture (NA) oil immersion objective, and a DAPI (4′,6-diamidino-2-phenylindole)/fluorescein isothiocyanate (FITC)/tetramethyl rhodamine isocyanate (TRITC)/Cy5 filter set.
Cells were seeded on fibronectin-coated 35-mm-diameter coverslip dishes (MatTek) 24 h in advance of experiments and were grown to approximately 70% confluence. Dishes were placed on ice for several minutes before addition of virus. Virus in imaging buffer (140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, 5 mM glucose, 2 µg/ml Hoechst stain, and 2% FBS, pH 7.4) was spinoculated onto cells by 20 min of centrifugation at 1,500 × g at 4°C. Inoculum volumes were adjusted so that 200 to 400 cell-associated particles were visible per field. Unbound particles were removed by four washes with cold PBS, and 500 µl cold imaging buffer was added to the dish. Samples were mounted on the microscope and quickly focused, and then 1.5 ml warm imaging buffer was added to start the experiment (t = 0). Images were acquired every 4 s over the duration of the experiments using a single Z-section, as the cells were very flat. Excluding the initial kinetics studies, all experiments were limited to 2 h.
Data analysis.
Image analysis and single-particle tracking were performed using Volocity software (PerkinElmer). Excluding minor adjustments in brightness and contrast, image files were not manipulated. For single-particle dequenching measurements, puncta were thresholded by initial intensity and size. Puncta falling outside the range of 0.125 to 1.0 µm2 expected of discrete VSV particles were excluded from analysis.
For colocalization of dequenching particles with cellular markers, a virion was considered colocalized only if the GFP or mNG punctum exceeded the cellular background signal by 30% or more and if the GFP or mNG and DiD puncta cotrafficked (greater than 80% overlap of DiD signal with GFP or mNG signal) for at least 20 s before and after a dequenching event. The majority of colocalized dequenching events far exceeded these criteria.
Mean measurements (± standard deviations [SD]) were derived from three separate experiments, unless otherwise indicated. Statistical significance was established by Student’s t test or by one-way analysis of variance (ANOVA) with a post hoc Dunnett’s test (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Viral internalization. (A) Percentage of cell-associated KZ52- or ZMapp-treated (100 µg/ml) EBOV-GPΔMuc-bearing particles colocalized with NPC1 at 45 min after synchronized infection in U2OS cells. (B) Percentage of cell-associated EBOV GPΔMuc-bearing particles colocalized with LAMP1 at 45 min after synchronized infection in U2OS and U2OS NPC1-ko cells. (B) Percentage of cell-associated particles bearing mutant EBOV GPΔMuc colocalized with NPC1 at 45 min after synchronized infection in U2OS cells. Download
Characterization of U2OS NPC1-ko cells. (A) The wt NPC gene sequence is aligned with the sequences of both alleles in the NPC1-ko (NPC1-#6) cell clone. The gRNA target sequence is in red. (B) RT-PCR results for wt and NPC1-ko (NPC1-#6) cells determined using gRNA target site-specific primers. Human site-1 protease (S1P)-specific primers were used as a control. (C) Infectivity of wt and NPC1-ko U2OS cells by VSV pseudotyped with EBOV GP. Infectivity was assessed 12 to 16 h postinfection as described previously (19). Download
Cholesterol sequestration in U2OS cells as depicted by filipin staining. (A to E) Cells were left untreated (A) or incubated with U18666A (10 µM) for 1 h (B), 2 h (C), 6 h (D), or 24 h (E). (F) U2OS NPC1-ko cells are shown as a positive control. Download
Dequenching of a DiD-labeled particle in a U2OS cell. Here, the VSV particles bear mNG-P and EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in a Rab5+ compartment. U2OS cells expressing Rab5-GFP were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in a Rab7+ compartment. U2OS cells expressing Rab7-GFP were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in a LAMP1+ compartment. U2OS cells expressing LAMP1-GFP were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in an NPC1+ compartment. U2OS cells expressing NPC1-mNG were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
ACKNOWLEDGMENTS
We thank Cecelia Harold and Tanwee Alkutkar for technical support, Esther Ndungo for sharing unpublished data on a GP mutant, Jonathan Lai for help with engineering of the antibody KZ52, and the Analytical Imaging Facility at the Albert Einstein College of Medicine for assistance.
This work was supported by funding from NIH/NIAID (R01 AI088027 to K.C.). The funding agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Spence JS, Krause TB, Mittler E, Jangra RK, Chandran K. 2016. Direct visualization of Ebola virus fusion triggering in the endocytic pathway. mBio 7(1):e01857-15. doi:10.1128/mBio.01857-15.
REFERENCES
- 1.Geisbert TW, Jahrling PB. 1995. Differentiation of filoviruses by electron microscopy. Virus Res 39:129–150. doi: 10.1016/0168-1702(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 2.Huang Y, Xu L, Sun Y, Nabel GJ. 2002. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and post-translational modification of nucleoprotein. Mol Cell 10:307–316. doi: 10.1016/S1097-2765(02)00588-9. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe S, Noda T, Kawaoka Y. 2006. Functional mapping of the nucleoprotein of Ebola virus. J Virol 80:3743–3751. doi: 10.1128/JVI.80.8.3743-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Booth TF, Rabb MJ, Beniac DR. 2013. How do filovirus filaments bend without breaking? Trends Microbiol 21:583–593. doi: 10.1016/j.tim.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez A, Yang ZY, Xu L, Nabel GJ, Crews T, Peters CJ. 1998. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J Virol 72:6442–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallaher WR. 1996. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell 85:477–478. doi: 10.1016/S0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 7.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weissenhorn W, Calder LJ, Wharton SA, Skehel JJ, Wiley DC. 1998. The central feature of the membrane fusion subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc Natl Acad Sci U S A 95:6032–6036. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malashkevich VN, Schneider BJ, McNally ML, Milhollen MA, Pang JX, Kim PS. 1999. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-Å resolution. Proc Natl Acad Sci U S A 96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez CP, Lasala F, Carrillo J, Muñiz O, Corbí AL, Delgado R. 2002. C-type lectins dc-sign and l-sign mediate cellular entry by Ebola virus in cis and in trans. J Virol 76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simmons G, Reeves JD, Grogan CC, Vandenberghe LH, Baribaud F, Whitbeck JC, Burke E, Buchmeier MJ, Soilleux EJ, Riley JL, Doms RW, Bates P, Pöhlmann S. 2003. Dc-sign and dc-signr bind Ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305:115–123. doi: 10.1006/viro.2002.1730. [DOI] [PubMed] [Google Scholar]
- 13.Matsuno K, Nakayama E, Noyori O, Marzi A, Ebihara H, Irimura T, Feldmann H, Takada A. 2010. C-type lectins do not act as functional receptors for filovirus entry into cells. Biochem Biophys Res Commun 403:144–148. doi: 10.1016/j.bbrc.2010.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol 80:10109–10116. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, Meyerholz DK, Rennert P, Mullins RF, Brindley M, Sandersfeld LM, Quinn K, Weller M, McCray PB Jr, Chiorini J, Maury W. 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc Natl Acad Sci U S A 108:8426–8431. doi: 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog 6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. 2010. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. 2011. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 419:72–82. doi: 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. 2012. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J 31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bale S, Liu T, Li S, Wang Y, Abelson D, Fusco M, Woods VL Jr, Saphire EO. 2011. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl Trop Dis 5:e1395. doi: 10.1371/journal.pntd.0001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. 2013. Multiple cationic amphiphiles induce a Niemann-pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A 94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinfelter LM, Jangra RK, Jae LT, Herbert AS, Mittler E, Stiles KM, Wirchnianski AS, Kielian M, Brummelkamp TR, Dye JM, Chandran K. 2015. Haploid genetic screen reveals a profound and direct dependence on cholesterol for hantavirus membrane fusion. mBio 6:e00801-15. doi: 10.1128/mBio.00801-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J. 2013. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular virus entirely from cDNA clones. Proc Natl Acad Sci U S A 92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeffers SA, Sanders DA, Sanchez A. 2002. Covalent modifications of the Ebola virus glycoprotein. J Virol 76:12463–12472. doi: 10.1128/JVI.76.24.12463-12472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaletsky RL, Simmons G, Bates P. 2007. Proteolysis of the Ebola virus glycoproteins enhances virus binding and infectivity. J Virol 81:13378–13384. doi: 10.1128/JVI.01170-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stegmann T, Morselt HW, Scholma J, Wilschut J. 1987. Fusion of influenza virus in an intracellular acidic compartment measured by fluorescence dequenching. Biochim Biophys Acta 904:165–170. doi: 10.1016/0005-2736(87)90100-3. [DOI] [PubMed] [Google Scholar]
- 32.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. 2003. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A 100:9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sieczkarski SB, Whittaker GR. 2003. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 34.Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. 2009. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 83:440–453. doi: 10.1128/JVI.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol 80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wool-Lewis RJ, Bates P. 1998. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol 72:3155–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez A. 2007. Analysis of filovirus entry into vero E6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J Infect Dis 196(Suppl 2):S364–S371. doi: 10.1086/520597. [DOI] [PubMed] [Google Scholar]
- 38.Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. 2012. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824:68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregory SM, Harada E, Liang B, Delos SE, White JM, Tamm LK. 2011. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc Natl Acad Sci U S A 108:11211–11216. doi: 10.1073/pnas.1104760108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrison JS, Higgins CD, Chandran K, Lai JR. 2011. Designed protein mimics of the Ebola virus glycoprotein GP2 α-helical bundle: stability and pH effects. Protein Sci 20:1587–1596. doi: 10.1002/pro.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misasi J, Chandran K, Yang JY, Considine B, Filone CM, Côté M, Sullivan N, Fabozzi G, Hensley L, Cunningham J. 2012. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J Virol 86:3284–3293. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maruyama T, Rodriguez LL, Jahrling PB, Sanchez A, Khan AS, Nichol ST, Peters CJ, Parren PW, Burton DR. 1999. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J Virol 73:6024–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu X, Alimonti JB, Melito PL, Fernando L, Ströher U, Jones SM. 2011. Characterization of Zaire ebolavirus glycoprotein-specific monoclonal antibodies. Clin Immunol 141:218–227. doi: 10.1016/j.clim.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Murin CD, Fusco ML, Bornholdt ZA, Qiu X, Olinger GG, Zeitlin L, Kobinger GP, Ward AB, Saphire EO. 2014. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc Natl Acad Sci U S A 111:17182–17187. doi: 10.1073/pnas.1414164111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Audet J, Wong G, Wang H, Gao G, Kobinger GF, Qiu X. 2014. Molecular characterization of the monoclonal antibodies composing ZMAb: a protective cocktail against Ebola virus. Sci Rep 4:6881. doi: 10.1038/srep06881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krishnan A, Miller EH, Herbert AS, Ng M, Ndungo E, Whelan SP, Dye JM, Chandran K. 2012. Niemann-pick C1 (NPC1)/NPC1-like chimeras define sequences critical for NPC1’s function as a filovirus entry receptor. Viruses 4:2471–2484. doi: 10.3390/v4112471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manicassamy B, Wang J, Jiang H, Rong L. 2005. Comprehensive analysis of Ebola virus GP1 in viral entry. J Virol 79:4793–4805. doi: 10.1128/JVI.79.8.4793-4805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dube D, Brecher MB, Delos SE, Rose SC, Park EW, Schornberg KL, Kuhn JH, White JM. 2009. The primed Ebolavirus glycoprotein (19-kilodalton GP1,2): sequence and residues critical for host cell binding. J Virol 83:2883–2891. doi: 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higgins ME, Davies JP, Chen FW, Ioannou YA. 1999. Niemann-pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab 68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- 50.Sakurai Y, Kolokoltsov AA, Chen CC, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl-Schott C, Biel M, Davey RA. 2015. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347:995–998. doi: 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruas M, Rietdorf K, Arredouani A, Davis LC, Lloyd-Evans E, Koegel H, Funnell TM, Morgan AJ, Ward JA, Watanabe K, Cheng X, Churchill GC, Zhu MX, Platt FM, Wessel GM, Parrington J, Galione A. 2010. Purified TPC isoforms form NAADP receptors with distinct roles in Ca2+ signaling ad endolysosomal trafficking. Curr Biol 20:703–709. doi: 10.1016/j.cub.2010.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mingo RM, Simmons JA, Shoemaker CJ, Nelson EA, Schornberg KL, D’Souza RS, Casanova JE, White JM. 2015. Ebola virus and severe acute respiratory syndrome coronavirus display late cell entry kinetics: evidence that transport to NPC1+ endolysosomes is a rate-defining step. J Virol 89:2931–2943. doi: 10.1128/JVI.03398-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simmons JA, D’Souza RS, Ruas M, Galione A, Casanova JE, White JM. 2015. The ebolavirus glycoprotein directs fusion through NPC1+ endolysosomes. J Virol 90:605–610. doi: 10.1128/JVI.01828-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brecher M, Schornberg KL, Delos SE, Fusco ML, Saphire EO, White JM. 2012. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J Virol 86:364–372. doi: 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregory SM, Larsson P, Nelson EA, Kasson PM, White JM, Tamm LK. 2014. Ebolavirus entry requires a compact hydrophobic fist at the tip of the fusion loop. J Virol 88:6636–6649. doi: 10.1128/JVI.00396-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. 2008. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog 4:e1000141. doi: 10.1371/journal.ppat.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Otterstrom JJ, Brandenburg B, Koldijk MH, Juraszek J, Tang C, Mashaghi S, Kwaks T, Goudsmit J, Vogels R, Friesen RH, van Oijen AM. 2014. Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level. Proc Natl Acad Sci U S A 111:E5143—E5148. doi: 10.1073/pnas.1411755111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Throsby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, Guan Y, Cinatl J, ter Meulen J, Lasters I, Carsetti R, Peiris M, de Kruif J, Goudsmit J. 2008. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ekiert DC, Friesen RHE, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJWM, Brandenburg B, Vogels R, Brakenhoff JPJ, Kompier R, Koldijk MH, Cornelissen LAHM, Poon LLM, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. 2011. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333:843–850. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. 2010. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alters its protease dependence during cell entry. J Virol 84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JA. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103:679–689. doi: 10.1016/S0092-8674(00)00170-7. [DOI] [PubMed] [Google Scholar]
- 62.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mazor Y, Barnea I, Keydar I, Benhar I. 2007. Antibody internalization studied using a novel IgG binding toxin fusion. J Immunol Methods 321:41–59. doi: 10.1016/j.jim.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Karauzum H, Chen G, Abaandou L, Mahmoudieh M, Boroun AR, Shulenin S, Devi VS, Stavale E, Warfield KL, Zeitlin L, Roy CJ, Sidhu SS, Aman MJ. 2012. Synthetic human monoclonal antibodies toward staphylococcal enterotoxin B (SEB) protective against toxic shock syndrome. J Biol Chem 287:25203–25215. doi: 10.1074/jbc.M112.364075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bornholdt ZA, et al. mBio, in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Viral internalization. (A) Percentage of cell-associated KZ52- or ZMapp-treated (100 µg/ml) EBOV-GPΔMuc-bearing particles colocalized with NPC1 at 45 min after synchronized infection in U2OS cells. (B) Percentage of cell-associated EBOV GPΔMuc-bearing particles colocalized with LAMP1 at 45 min after synchronized infection in U2OS and U2OS NPC1-ko cells. (B) Percentage of cell-associated particles bearing mutant EBOV GPΔMuc colocalized with NPC1 at 45 min after synchronized infection in U2OS cells. Download
Characterization of U2OS NPC1-ko cells. (A) The wt NPC gene sequence is aligned with the sequences of both alleles in the NPC1-ko (NPC1-#6) cell clone. The gRNA target sequence is in red. (B) RT-PCR results for wt and NPC1-ko (NPC1-#6) cells determined using gRNA target site-specific primers. Human site-1 protease (S1P)-specific primers were used as a control. (C) Infectivity of wt and NPC1-ko U2OS cells by VSV pseudotyped with EBOV GP. Infectivity was assessed 12 to 16 h postinfection as described previously (19). Download
Cholesterol sequestration in U2OS cells as depicted by filipin staining. (A to E) Cells were left untreated (A) or incubated with U18666A (10 µM) for 1 h (B), 2 h (C), 6 h (D), or 24 h (E). (F) U2OS NPC1-ko cells are shown as a positive control. Download
Dequenching of a DiD-labeled particle in a U2OS cell. Here, the VSV particles bear mNG-P and EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in a Rab5+ compartment. U2OS cells expressing Rab5-GFP were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in a Rab7+ compartment. U2OS cells expressing Rab7-GFP were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in a LAMP1+ compartment. U2OS cells expressing LAMP1-GFP were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download
Lipid mixing in an NPC1+ compartment. U2OS cells expressing NPC1-mNG were infected with DiD-labeled particles bearing EBOV GPΔMuc. The white arrow indicates the particle of interest. The red arrow marks the onset of dequenching. Bar, 10 µm. Download