

ABSTRACT
Salmonella enterica serovar Typhimurium uses the Salmonella pathogenicity island 1 (SPI1) type III secretion system (T3SS) to induce inflammatory diarrhea and bacterial uptake into intestinal epithelial cells. The expression of hilA, encoding the transcriptional activator of the T3SS structural genes, is directly controlled by three AraC-like regulators, HilD, HilC, and RtsA, each of which can activate hilD, hilC, rtsA, and hilA genes, forming a complex feed-forward regulatory loop. Expression of the SPI1 genes is tightly controlled by numerous regulatory inputs to ensure proper timing in production of the T3SS apparatus. Loss of FadD, an acyl coenzyme A (acyl-CoA) synthetase required for degradation of long-chain fatty acids (LCFAs), was known to decrease hilA expression. We show that free external LCFAs repress expression of hilA independently of FadD and the LCFA degradation pathway. Genetic and biochemical evidence suggests that LCFAs act directly to block primarily HilD activity. Further analyses show that in the absence of FadD, hilA expression is downregulated due to endogenous production of free LCFAs, which are excreted into the culture medium via TolC and then transported back into the bacterial cell via FadL. A fadL mutant is more virulent than the wild-type strain in mouse oral competition assays independently of LCFA degradation, showing that, in the host, dietary LCFAs serve as a signal for proper regulation of SPI1 expression, rather than an energy source.
IMPORTANCE
To cause disease, Salmonella must respond to diverse environmental cues to express its invasion machinery at the appropriate location in the host intestine. We show that host intestinal free long-chain fatty acids (LCFAs) affect Salmonella invasion by reducing expression of the SPI1 type III secretion system, acting primarily via the AraC-like activator HilD. Degradation of LCFAs is not required for this regulation, showing that free LCFAs serve as a cue to proper intestinal localization to invade host epithelial cells and not as a nutrient source.
INTRODUCTION
Bacterial pathogens are challenged with adapting to specific host environments, including spatial detection of particular niches within a given tissue. In the intestine, Salmonella enterica serovar Typhimurium preferentially colonizes the distal ileum. The bacteria induce inflammatory diarrhea and invade nonphagocytic epithelial cells using the type III secretion system (T3SS) encoded on Salmonella pathogenicity island 1 (SPI1; for a review, see reference 1). The T3SS apparatus is a needle-like structure that injects bacterial effector proteins into the host cell cytosol, where they interact with specific host cell proteins to promote actin cytoskeletal rearrangements and activate inflammatory pathways.
The level of SPI1 gene expression is dependent on the level of the SPI1-encoded regulator HilA, which directly activates expression of the SPI1 structural genes (Fig. 1A) (2–4). Expression of hilA is directly controlled by three AraC-like regulators, HilD, HilC, and RtsA (5, 6), each of which is independently capable of inducing expression of the hilD, hilC, and rtsA genes, as well as hilA, forming a complex feed-forward regulatory loop to control SPI1 expression (7). HilD is the dominant regulator of the system, while HilC and RtsA act as amplifiers of the signal (7, 8). The system is tightly controlled by a multitude of external regulatory inputs, which presumably ensure that SPI1 is expressed only at the appropriate time and place within the host (9). These factors are integrated to control the threshold of HilD required for autoactivation (8), which then acts as a switch to turn on SPI1 (10–12).
FIG 1 .
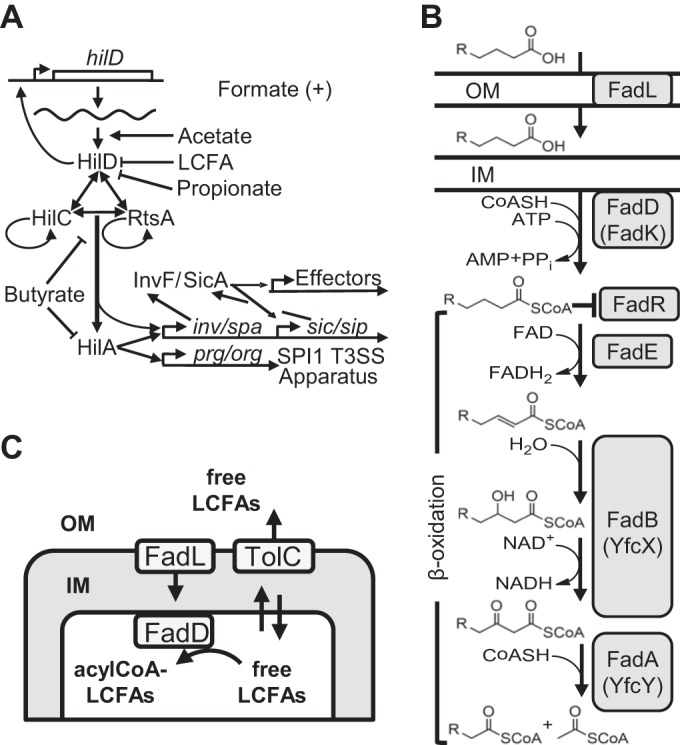
(A) SPI1 regulatory circuit. For clarity, the genes encoding HilC, RtsA, and HilA are not shown (7, 9). (B) Pathway for β-oxidation of fatty acids. Enzymes in the anaerobic pathway are in parentheses (adapted from data in reference 13). OM, outer membrane; IM, inner membrane; FAD, flavin adenine dinucleotide. (C) Schematic representation of the free LCFA import and export in Salmonella.
Transport of external free long-chain fatty acids (LCFAs) across the outer membrane requires the β-barrel protein FadL (13). Subsequently, free LCFAs partition into the inner membrane and can cross the inner membrane bilayer unassisted (14, 15). FadD facilitates unidirectional transport by converting free LCFAs to their acyl coenzyme A (acyl-CoA) form, trapping them in the cytoplasm, where they can enter the β-oxidation cycle (Fig. 1B) (13). Expression of the fad regulon genes is controlled by the transcriptional regulator FadR, which dissociates from regulated promoters upon binding acyl-CoA LCFAs. In the absence of acyl-CoA LCFAs, FadR represses fad (fatty acid degradation) genes and activates fab (fatty acid biosynthesis) genes (13).
Loss of the acyl-CoA synthetase FadD was shown to reduce hilA expression independently of the regulatory protein FadR (16). However, the mechanism of this regulation was not elucidated. Given the potential impact of fatty acids in the intestine, we sought to understand the role of LCFAs, and their utilization, in SPI1 regulation. Here, we show that LCFAs act as a direct signal to control expression of the Salmonella invasion system. This regulation is distinct from the effects of short-chain fatty acids, which also affect SPI1. We propose that Salmonella integrates the concentration information for both long- and short-chain fatty acids to determine the optimal site for intestinal invasion.
RESULTS
Free LCFAs mediate a fast and reversible decrease in hilA transcription.
To study SPI1 regulation by free long-chain fatty acids (LCFAs), we used a single-copy chromosomal hilA-lac transcriptional fusion as an established readout for SPI1 expression (17). Addition of unsaturated (oleate) or saturated (myristate and palmitate) free LCFAs resulted in a concentration-dependent decrease in hilA transcription (Fig. 2), effectively decreasing hilA transcription at submillimolar concentrations. In further experiments, oleate was used as a representative LCFA due to greater solubility. The observed repression of hilA transcription by 0.6 mM oleate was evident at 15 min after oleate addition to the medium, and repression was reversible; transfer to medium without oleate relieved repression within 30 to 45 min (see Fig. S1 in the supplemental material). These results suggested that it was LCFAs per se that caused the effect, rather than metabolic breakdown products or, for example, synthesis of new lipids.
FIG 2 .
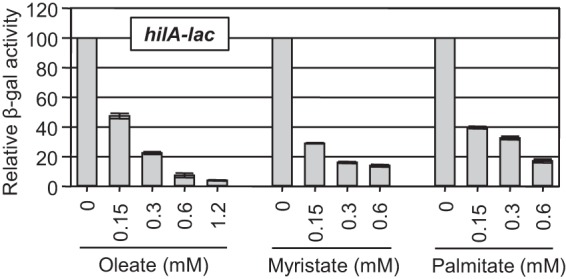
External unsaturated (oleate) and saturated (myristate and palmitate) LCFAs repress hilA transcription in a concentration-dependent manner. Relative β-galactosidase (β-gal) activity in strains containing a hilA-lac transcriptional fusion grown under SPI1-inducing conditions in the presence of indicated concentrations of LCFAs. Relative β-galactosidase activity was calculated as a percentage of the untreated control strain value for each LCFA concentration. The detergent Tergitol NP-40 (2.5%) was present in all samples. β-Galactosidase activity units are defined as (micromoles of ONP formed minute−1) × 106/(OD600 × milliliter of cell suspension) and are reported as mean ± standard deviation, where n is 4. The strain used was JS749.
Decreased hilA expression by external LCFA is independent of FadD, FadK, FadR, and β-oxidation of LCFAs.
A previous study concluded that FadD positively regulates hilA expression in the absence of external LCFAs by an unknown FadR-independent mechanism (16). We wanted to determine the role of FadD in modulation of hilA expression and understand the relationship to the effects of external LCFAs. Loss of FadD caused a 1.6-fold decrease in expression, but addition of oleate still caused a 4-fold decrease in the fadD background, suggesting that external LCFAs act independently of FadD (Fig. 3). Loss of FadK, an acyl-CoA synthetase that functions under anaerobic conditions (13), did not affect the phenotype. Blocking the β-oxidation pathway (fadE fadAB) also had no effect on hilA expression. Moreover, addition of oleate had a slightly greater effect in this background. These results support the concept that the free fatty acids are causing the effect; degradation only lowers the concentration. Loss of the transcriptional regulator FadR caused a slight decrease in hilA transcription in the absence of external oleate, suggesting that FadR does have some regulatory input into SPI1 (Fig. 3). However, repression of hilA by oleate is evident in the absence of FadR. Deletion of both fadD and fadR leads to an apparent additive effect, but oleate still caused a decrease in hilA expression in the double mutant background. Taken together, these data suggest that free LCFAs cause a decrease in hilA expression independently of degradation or the regulatory protein FadR.
FIG 3 .
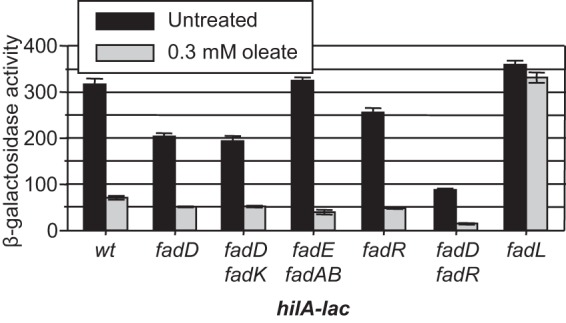
External oleate causes a decrease in hilA expression independent of FadD, FadR, or the β-oxidation pathway; FadL is required for downregulation. Strains containing a hilA-lac fusion and the indicated mutations were grown under SPI1-inducing conditions in the absence or presence of 0.3 mM oleate. Tergitol NP-40 (0.2%) was present in all samples. β-Galactosidase activity is reported as mean ± standard deviation, where n is 4. Strains used were JS749 and JS2057 to -62. wt, wild type.
The LCFA transporter FadL is required for the effects of both external LCFAs and loss of FadD.
The LCFA transporter FadL is required for transport of external LCFAs to the periplasm. Deletion of fadL alone had no effect on hilA expression. However, loss of FadL prevented hilA repression by 0.3 mM external oleate (Fig. 3), showing that free LCFAs must gain access to the periplasm to affect hilA expression. FadL is also required for repression by myristate and palmitate (data not shown). Interestingly, deletion of fadL also suppressed the decrease in hilA transcription caused by loss of FadD. However, the fadL deletion did not suppress the fadR phenotype, as hilA expression was still decreased 1.5-fold in all fadR fadL genetic backgrounds (Fig. 4A). These data suggest that FadD and FadR act independently to control hilA expression. Moreover, the fadD phenotype is dependent on transport of LCFAs across the outer membrane, despite the fact that no external LCFAs were added to the medium. One explanation is that free LCFAs accumulate in the medium in the fadD mutant culture and that FadL is transporting these LCFAs back into the cell, resulting in decreased hilA expression.
FIG 4 .
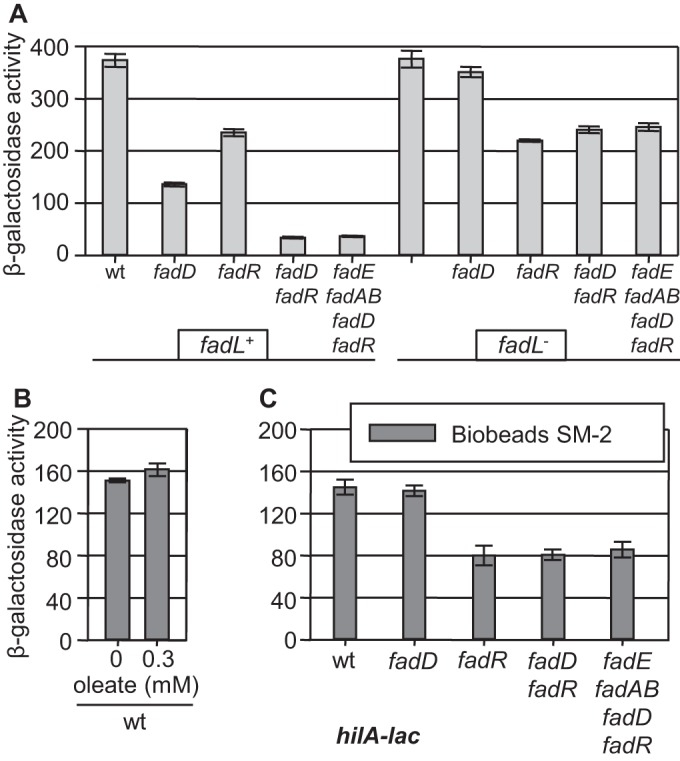
(A) Loss of the FadL transporter suppresses the fadD phenotype. Strains containing a hilA-lac fusion and the indicated mutations were grown under SPI1-inducing conditions. wt, wild type. (B) Addition of SM-2 Bio-Beads relieves hilA repression by external oleate. The wild-type strain containing a hilA-lac fusion was grown under SPI1-inducing conditions with SM-2 Bio-Beads and Tergitol NP-40 (0.2%) with or without 0.3 mM oleate. (C) Addition of SM-2 Bio-Beads relieves the fadD phenotype. Strains containing a hilA-lac fusion and the indicated mutations were grown under SPI1-inducing conditions with SM-2 Bio-Beads. β-Galactosidase activity is reported as mean ± standard deviation, where n is 4. Strains used were JS749, JS2057 to -59, JS2061 to -66, and JS2068.
External absorption of LCFAs relieves the fadD phenotype.
SM-2 Bio-Beads (Bio-Rad, Inc.) can be used to absorb LCFAs in culture. As proof of SM-2 Bio-Bead functionality, bacteria were grown with Bio-Beads in the presence of 0.3 mM oleate; under these conditions, the added LCFAs had no effect on hilA transcription (Fig. 4B). Likewise, in the presence of Bio-Beads, loss of FadD had no effect on hilA transcription in the wild-type background (Fig. 4C). A 2-fold decrease in hilA expression was still evident in the presence of Bio-Beads in strains lacking FadR, independent of any other genetic changes. Thus, addition of Bio-Beads specifically relieves hilA repression caused by loss of FadD, suggesting that the fadD phenotype involves free LCFAs in the culture medium. FadR apparently functions independently to control hilA transcription.
Free LCFAs produced by the fadD mutant cells are excreted into the culture medium in a TolC-dependent manner.
Loss of FadD apparently results in an accumulation of free LCFAs in the culture medium (18), which then have to be transported into the cell via the outer membrane transporter FadL to repress hilA expression. This also implies that loss of FadD or addition of the external free LCFAs to the culture medium regulates hilA expression by the same mechanism. Internally produced free LCFAs would have to be subjected to efflux out of the cells and/or be liberated by cell lysis to accumulate in the culture medium. A recent study implicated multidrug efflux pumps that function via the outer membrane channel TolC in the export of free fatty acids in Escherichia coli (19). We introduced a tolC deletion into the transcriptional hilA-lac fusion strains in wild-type, fadD, fadL, or fadD fadL backgrounds. Loss of TolC alone resulted in a slight decrease in hilA transcription (Fig. 5A) as seen previously (20). Simultaneous loss of both FadD and TolC caused a 10-fold decrease in hilA expression, which could no longer be suppressed by the loss of the FadL outer membrane transporter. These data suggest that TolC mediates free LCFA efflux in a fadD mutant. Blocking the efflux of free LCFAs accumulating in the absence of FadD causes an increase in the free LCFA concentration inside the fadD tolC mutant cells, which results in hilA expression being decreased to an even greater level than that in the fadD mutant. Loss of FadL does not suppress loss of FadD in the tolC mutant because the LCFAs are trapped inside the cell. Surprisingly, the addition of Bio-Beads did partially suppress the fadD phenotype in the tolC deletion background (Fig. 5A). However, there was no effect of Bio-Beads in a fadD tolC fadL triple mutant. These data suggest that, consistent with our hypothesis, LCFAs are building up in the fadD tolC fadL mutant and that these LCFAs are not accessible to the culture medium. Free LCFAs are also apparently accumulating in the fadD tolC mutant, as evidenced by the decrease in hilA expression, but in this case, the LCFAs can be absorbed if a “sink” (Bio-Beads) is provided external to the cell. These results imply that LCFAs accumulate and equilibrate across the cytoplasmic membrane in the absence of FadD and that these LCFAs can pass from the periplasm into the external milieu in a FadL-dependent manner, albeit less efficiently than via TolC.
FIG 5 .

Free LCFAs produced by the fadD mutant cells are excreted into the culture medium in a TolC-dependent manner. (A) Strains containing a hilA-lac fusion and the indicated mutations were grown under SPI1-inducing conditions with or without SM-2 Bio-Beads. β-Galactosidase activity is reported as mean ± standard deviation, where n is 4. Strains used were JS749, JS2057, JS2062 and -63, and JS2069 to -72. wt, wild type. (B) The fadL, fadD fadL, and fadD fadL tolC bacterial cultures were grown in defined medium, and LCFA content in 15 ml of filtered supernatant from each culture was determined. Resulting LCFA content was normalized to dry cell weight. For the box plot, the line in the middle of the rectangle is the median (or the second quartile [Q2]). The top whisker denotes the maximum value or the third quartile plus 1.5 times the interquartile range [Q3 + (1.5 × IQR)], whichever is smaller. The bottom whisker denotes either the minimum value or the first quartile minus 1.5 times the interquartile range [Q1 − (1.5 × IQR)], whichever is larger. Strains used were JS2062 and -63 and JS2072.
To further test our model, we determined the free LCFA concentration in the supernatant of fadL, fadL fadD, and fadL fadD tolC mutants via mass spectrometry (MS); the fadL background negated any reuptake of LCFAs. The β-galactosidase activity produced from a hilA-lacZ fusion present in each strain was as expected. Moreover, there was no detectable β-galactosidase activity in any of the supernatants, suggesting that cell lysis was negligible in this experiment (see Fig. S2 in the supplemental material). Figure 5B shows that the concentration of LCFAs present in the supernatant was significantly increased in the fadL fadD mutant compared to the fadL mutant, whereas extracellular LCFAs were significantly reduced in the fadL fadD tolC mutant. These results are perfectly consistent with our model; free LCFAs build up in the fadD mutant and are secreted into the milieu in a TolC-dependent manner.
Decreased hilA expression in response to external free LCFAs is independent of periplasmic stress regulators or the PhoPQ two-component regulatory system.
The data above show that externally added free LCFAs must gain access to either the periplasm or the cytoplasm to reduce hilA expression. The effect of internally produced free LCFAs is enhanced when their efflux across the outer membrane is blocked. However, because there is clear equilibration across the cytoplasmic membrane, it is difficult, based on the above data, to distinguish whether the effect of LCFAs is mediated in the periplasm, the cytoplasmic membrane, or the cytoplasm. It is possible that free LCFAs act in the periplasm to repress hilA, by inducing periplasmic stress or by being sensed by another SPI1 regulator. We tested the role of the periplasmic stress regulators CpxR and RpoE, as well as the PhoPQ two-component system, which was recently shown to sense free unsaturated LCFAs in the periplasm of Salmonella (21). Deleting these systems had no effect on hilA expression in the presence or absence of external LCFAs (see Fig. S3 in the supplemental material). Hence, it seems unlikely that free LCFAs are acting via any periplasmic system to control SPI1 expression.
External LCFAs act primarily via the AraC-like SPI1 regulator HilD.
Vibrio cholerae AraC-like regulator ToxT was recently shown to bind free LCFAs, thus blocking ToxT DNA binding (22–25). Production of the Salmonella SPI1 T3SS is dependent on three AraC-like regulators, HilD, HilC, and RtsA. HilD serves as a major point of integration of regulatory signals into the SPI1 circuit (7–9). We addressed the effect of free LCFAs on the ability of each of these proteins to induce hilA expression by utilizing a Δspi1 ΔrtsA strain (hilD, hilC, rtsA, and hilA genes are deleted). Each of the three AraC-like regulators was then produced individually via a regulated promoter, and activity was monitored using the hilA-lac transcriptional fusion in cultures grown with or without 1.2 mM oleate, the highest feasible concentration. Expression of hilA in the wild-type strain was decreased more than 20-fold in the presence of oleate (Fig. 6A). Expression was dependent on HilD, HilC, and RtsA, as evidenced by the lack of activity in the spi1 rtsA deletion strain. When production of a TetR-controlled HilD protein was induced with anhydrotetracycline, addition of 1.2 mM oleate caused a 7-fold decrease in hilA-lac transcription compared to the untreated control. Thus, the ability of HilD protein alone to activate hilA expression is reduced in the presence of oleate. When only RtsA protein was produced, addition of 1.2 mM oleate resulted in a 1.8-fold reduction in hilA expression (Fig. 6B). HilC was produced from an arabinose-dependent promoter on a plasmid, with some HilC produced even when uninduced. But, whether uninduced or induced, addition of 1.2 mM oleate caused only a slight decrease in hilA-lac transcription (Fig. 6C). These results suggest that free LCFAs affect each of these regulatory proteins independently, with HilD being the most susceptible to inhibition. It should be noted that production of HilC and RtsA is normally dependent on HilD and that HilD is also autoregulatory (7, 8). Thus, this 7-fold effect on HilD seen in this experiment could easily account for the 20-fold effect seen in the wild-type background, despite the limited inhibition of RtsA or HilC activity.
FIG 6 .
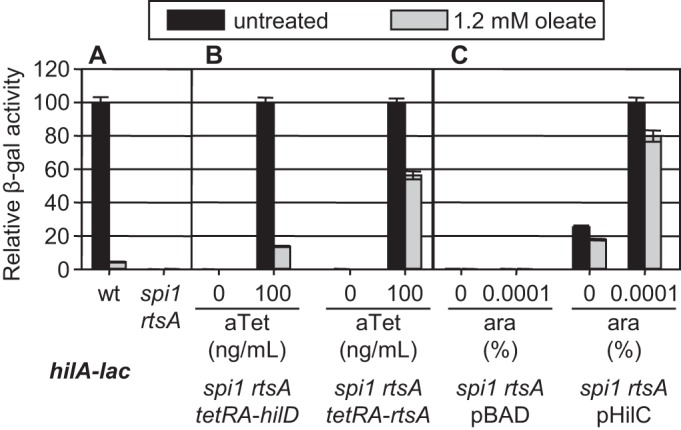
Effect of oleate on HilD, HilC, or RtsA activity. (A) Strains containing a hilA-lac fusion in the wild-type (wt) or spi1 rtsA background grown under SPI1-inducing conditions in the presence or absence of 1.2 mM oleate. Relative β-galactosidase activity is a percentage of untreated wild-type strain value (238.75 units). (B) Strains containing a hilA-lac transcriptional fusion in the spi1 rtsA background with HilD or RtsA proteins produced under a tetracycline-regulated promoter were grown under SPI1-inducing conditions with the indicated anhydrotetracycline (aTet) concentrations in the presence or absence of 1.2 mM oleate. Relative β-galactosidase activity is a percentage of untreated strain value in the presence of 100 ng/ml aTet, inducing production of HilD (127.16 units) or RtsA (34.42 units). (C) Strains containing a hilA-lac transcriptional fusion in the spi1 rtsA background with HilC protein produced under the control of an arabinose-regulated promoter. Strains were grown under SPI1-inducing conditions with the indicated arabinose concentrations in the presence or absence of 1.2 mM oleate. All samples contained 2.5% Tergitol. Relative β-galactosidase activity is a percentage of untreated strain value in the presence of 0.0001% arabinose (243.21 units). β-Galactosidase activity is reported as mean ± standard deviation, where n is 4. Strains used were JS749 and JS2075 to -79.
LCFAs act directly on HilD.
HilD and HilC are homologous, particularly in their DNA binding domains, and thus bind the same sites in the various promoters that they regulate (26). To study DNA binding in vitro, we used Sumo-HilD and Sumo-HilC fusion proteins (27). These fusion proteins specifically bind a hilC promoter fragment at the same relative protein-to-DNA concentrations (see Fig. S4 in the supplemental material). The addition of 10 to 30 µM oleate to these gel shift reactions completely blocked HilD DNA binding but had little effect on the binding of HilC (Fig. 7). These results recapitulate the in vivo data above and suggest that LCFAs directly interact with HilD to affect its DNA binding ability.
FIG 7 .
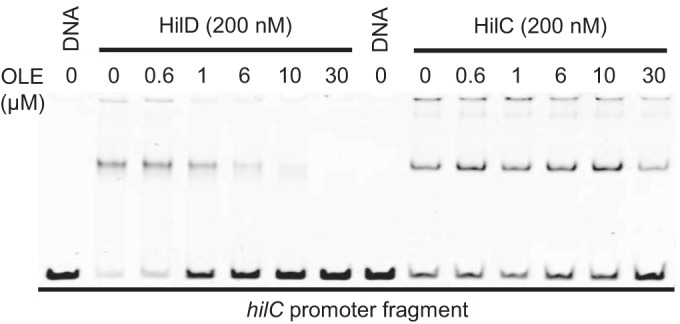
Oleate (OLE) affects HilD DNA binding. HilD- or HilC-Sumo proteins bind to a 210-bp fragment of the hilC promoter. All lanes contain 10 nM DNA and 0.05% Tergitol NP-40 detergent. Oleate was added to some samples at the indicated concentrations.
Free LCFAs repress SPI1 expression in the host in a FadL-dependent manner.
The ability of external free LCFAs to repress SPI1 expression raises the important question of whether this regulation is relevant in the host. Our in vitro data showed that loss of FadL resulted in bacteria being “blind” to external LCFAs, preventing repression of hilA. To address the role of FadL in Salmonella virulence, we used mouse oral and intraperitoneal (i.p.) competition assays to determine the virulence phenotype of a fadL null mutant. In the oral experiments, we recovered bacteria from the distal small intestine, the primary site of Salmonella invasion (28, 29).
Dietary fatty acids could constitute a significant source of free LCFAs in the intestine. Accordingly, we compared the outcomes of the infection in mice where food was withheld for 4 h before oral inoculation with those in mice that were actively feeding (8664 Teklad rodent diet; 6.4% fat). The competitive index (CI) data in Table 1 show that loss of FadL conferred a small (1.4-fold) but statistically significant increase in virulence compared to the wild-type strain when food was withheld. In contrast, when mice were fed, loss of FadL resulted in a 3-fold increase in competitiveness. This effect was evident only upon oral infection, since loss of FadL had no effect when bacteria were inoculated intraperitoneally, suggesting that degradation of LCFAs is irrelevant; SPI1 is also dispensable when bacteria are administered i.p. (7).
TABLE 1 .
Dietary free LCFAs repress SPI1 expression in the host in a FadL-dependent manner
| Infection type | Strain Aa | Strain Ba | No. of mice | CIb | Pc |
|---|---|---|---|---|---|
| Oral infectione | fadL (no food) | wt (no food) | 7 | 1.39 | 0.026 |
| fadL | wt | 10 | 2.99 | 0.025 | |
| fadE fadAB fadD fadR | wt | 9 | 0.05 | 0.034 | |
| fadE fadAB fadD fadR Δspi1 | Δspi1 | 9 | 1.11 | NS | |
| fadE fadAB fadD fadR fadL | fadE fadAB fadD fadR | 10 | 13.78 | <0.0005 | |
| i.p. infectiond | fadL | wt | 5 | 0.71 | NS |
| fadE fadAB fadD fadR fadL | fadE fadAB fadD fadR | 5 | 0.94 | NS |
The strains used were JS135, JS749, JS2066 to -68, JS2080, and JS2081. wt, wild type.
The competitive index (CI) was calculated as described in Materials and Methods.
The Student t test was used to compare the CIs to the inocula. NS, not significant.
Bacteria were recovered from the spleen in the case of intraperitoneal (i.p.) competition assays.
Bacteria were recovered from the distal portion of the small intestine in oral competition assays. Mice were actively feeding before oral inoculation in all oral infection experiments except as specified otherwise (no food).
To prove that the fadL virulence phenotype was not dependent on the ability of Salmonella to degrade free LCFAs in vivo, we competed the mutant lacking FadE, FadAB, FadD, and FadR (β-oxidation−) against the wild-type strain (β-oxidation+). Loss of β-oxidation resulted in a 20-fold attenuation in virulence compared to the wild-type strain in oral mouse infection (Table 1). If this virulence defect was due to SPI1 downregulation, then loss of β-oxidation should have no effect on virulence in the spi1 deletion background. Indeed, the spi1 mutant lacking fadE, fadAB, fadD, and fadR genes was not attenuated in oral mouse competition assays compared to the spi1 mutant strain, consistent with the model that LCFAs are acting as a signal rather than a nutritional source. We then asked whether loss of FadL still had an effect on virulence in the absence of β-oxidation. The β-oxidation− mutant lacking FadL was 14-fold more virulent than the isogenic β-oxidation− mutant in the oral competition assay (Table 1), effectively suppressing the virulence defect conferred by loss of β-oxidation; the fadL mutation had no effect on virulence when bacteria were administered i.p. Taken together, our in vitro and in vivo results suggest that dietary free LCFAs have an impact on Salmonella virulence by downregulating SPI1 genes in the host intestine. Indeed, the virulence defect caused by loss of the β-oxidation pathway is explained completely by the effect on SPI1 expression. Moreover, additional deletion of fadL suppresses this defect. Thus, Salmonella is using dietary free LCFAs as an environmental cue in the host intestine, not as a significant energy source.
DISCUSSION
Here, we show that free long-chain fatty acids (LCFAs) act as an environmental signal in the intestine that dictates appropriate expression of the SPI1 T3SS during oral infection. Submillimolar concentrations of extracellular free LCFAs added to the growth medium are sufficient for significant downregulation of hilA, encoding the primary regulator of the SPI1 structural genes. Both saturated and unsaturated free LCFAs are effective. The simplest model that explains our data is that free LCFAs above some threshold concentration in the cytoplasm bind directly to HilD protein to block its ability to bind DNA and activate transcription, effectively blocking expression of hilD, hilC, rtsA, and hilA. LCFAs have a lesser effect on the activity of the homologous SPI1 regulators HilC and RtsA. This is, perhaps, not surprising given that the three proteins share only 10% identity in their N-terminal domains, the most likely site of LCFA binding. This mechanism of SPI1 regulation by free LCFAs in Salmonella is similar to that of ToxT in V. cholerae (22–25). Indeed, it has been suggested that the activities of other AraC-like regulatory proteins, including VirF from Yersinia enterocolitica and Rns from enterotoxigenic E. coli, are negatively affected by binding to unsaturated fatty acids (30). Interestingly, the SphR protein in Pseudomonas aeruginosa is activated by direct binding to sphingosine (31). Additional studies, ideally including crystal structures of the SPI1 AraC-like regulators, will be required to understand the binding of free LCFA to HilD, enabling direct comparison to the structural models of the ToxT-LCFA binding (23).
Our results suggest that free LCFAs can traverse/equilibrate across the cytoplasmic membrane in the absence of FadD enzymatic activity, which normally would convert the LCFAs to acyl-CoA derivatives, thus trapping them in the cytoplasm. This is in agreement with both experimental (14) and computational (15) studies showing that LCFAs in protonated form can cross a phospholipid bilayer unassisted, where the absorption and flip-flop stages are fast (milliseconds) and desorption from the opposite membrane leaflet is slower (seconds). Accordingly, loss of FadD causes a buildup of free LCFAs that reach some equilibrium across the inner membrane. They are then exported out of the cell by TolC and are imported back into the periplasm by FadL. The result is some concentration equilibrium between external and cytoplasmic LCFAs. Loss of either TolC or FadL disrupts this equilibrium, altering the steady-state concentration of free LCFAs in the cytoplasm.
Loss of FadR also causes a decrease in SPI1 expression. Thus, FadR is a positive regulator of the SPI1 system. However, the mechanism of FadR-mediated regulation is independent of that caused by free LCFAs. This is evidenced by the fact that loss of FadL or the addition of SM-2 Bio-Beads suppresses the effects of LCFAs or loss of FadD but does not affect the phenotype conferred by loss of FadR. More experiments are required to determine how this transcriptional regulator feeds into the SPI1 regulatory circuit.
The distribution of the environmental signals encountered inside the host is likely critical for Salmonella infection. Both dietary sources and bile secretions in the proximal small intestine would likely account for the LCFAs encountered by Salmonella in the host intestinal tract (22). Medium-chain FAs (C6 to C10) are also known to both decrease hilA expression and inhibit Salmonella growth at relatively high concentrations (32), but the mechanism behind this regulation is unknown. Short-chain FAs, specifically, acetate, formate, propionate, and butyrate, have also been shown to significantly affect SPI1 gene expression. However, these compounds act by very different mechanisms. Acetate activates translation of the hilD mRNA via the SirA/Csr system (9, 33, 34). Formate also activates the SPI1 system, but the mechanism is unknown (35). In contrast, butyrate downregulates the SPI1 system and does so independently of HilD (9, 36). Propionate also negatively affects SPI1 expression. Although propionate does act via HilD protein, it does not do so directly. Rather, production of propionyl-CoA is required (37). Our results expand an overall model put forward by Altier and colleagues whereby Salmonella cells sense the relative location in the gastrointestinal tract (33, 35, 37, 38). We propose that the luminal concentration of LCFAs should decrease as they are absorbed along the length of the small intestine. Thus, at the distal ileum, the concentration of LCFAs would be low (39, 40). In contrast, the concentrations of acetate and formate are high in the distal ileum (38), signaling the optimal location for induction of the SPI1 T3SS and invasion of the host epithelia. In the cecum, past the site of invasion, increased concentrations of propionate and butyrate would result in repression of SPI1 genes (37). It is also interesting that Salmonella is incapable of using butyrate as a carbon source (13, 41), and our results suggest that utilizing LCFAs is irrelevant during infection. Thus, these compounds in particular are acting primarily as signals for proper intestinal location.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All Salmonella strains used in this study (see Table S1 in the supplemental material) are isogenic derivatives of Salmonella enterica serovar Typhimurium 14028 (American Type Culture Collection) and were constructed using P22 HT105/1 int-201 (P22)-mediated transduction (42). Deletion of various genes and concomitant insertion of an antibiotic resistance cassette were carried out using lambda Red-mediated recombination (43, 44) as described previously (45). The endpoints of each deletion/insertion are indicated in Table S1 in the supplemental material. The appropriate insertion of the antibiotic resistance marker was confirmed by PCR analysis. In each case, the constructs resulting from this procedure were moved into a clean wild-type background (14028) by P22 transduction. Each mutant strain was reconstructed at least once to ensure that the phenotype was the result of the designated mutation(s). In some cases, antibiotic resistance cassettes were removed using the temperature-sensitive plasmid pCP20 carrying the FLP recombinase (46).
Luria-Bertani (LB) medium containing 10 g tryptone, 5 g yeast extract, and 5 g or 10 g NaCl per liter was used for growth of bacteria in aeration. Superoptimal broth with catabolite repression (SOC) was used for the recovery of transformants (42). Bacterial strains were grown at 37°C, except for the strains containing temperature-sensitive plasmids pCP20 and pKD46 (43, 46), which were grown at 30°C. Antibiotics were used at the following concentrations: 100 µg/ml ampicillin (Ap), 20 µg/ml chloramphenicol (Cm) (but 10 µg/ml Cm was used for strains containing a ΔtolC::Cm deletion), 50 µg/ml kanamycin (Kn), 25 µg/ml tetracycline (Tet), and 50 µg/ml apramycin. Enzymes were purchased from Invitrogen or New England Biolabs and used according to the manufacturer’s recommendations. Primers were purchased from IDT Inc.
To induce the SPI1 system (SPI1-inducing conditions), bacteria were initially inoculated into LB medium (0.5% NaCl), grown overnight, and then subcultured 1/100 in 1.5 ml LB medium with 1% NaCl (high-salt LB [HSLB] medium) in 13- by 100-mm tubes and grown for 4.5 to 6 h on a roller drum. In all experiments requiring added LCFAs, the indicated concentration of Tergitol NP-40 was included in all subcultures. The LCFAs were initially solubilized in 90% ethanol for oleate (or in 70 to 75% ethanol and 3 to 7% Tergitol NP-40 for saturated LCFAs), and the pH was adjusted to 6.8 to 7.0. Equivalent ethanol/detergent was also added to the no-LCFA controls.
External absorption of LCFAs by SM-2 Bio-Beads.
For experiments with the SM-2 Bio-Beads (Bio-Rad Inc.), 0.25 g of beads (for 1.5 ml of medium) was aliquoted into 13- by 100-mm tubes, washed with 70% ethanol, and then extensively washed 3 times with sterile phosphate-buffered saline (PBS) and 3 times with the HSLB medium. Bacterial strains grown in LB medium overnight were subcultured 1/100 in 1.5 ml HSLB medium containing SM-2 Bio-Beads and grown for 4.5 to 6 h on a roller drum.
β-Galactosidase assays.
β-Galactosidase assays were performed using a microtiter plate assay as previously described (47) on strains grown under the indicated conditions. β-Galactosidase activity units are defined as (micromoles of o-nitrophenol [ONP] formed per minute) × 106/(optical density at 600 nm [OD600] × milliliter of cell suspension) and are reported as means ± standard deviations, where n is 4.
Analysis of LCFA content in culture supernatant.
For the determination of LCFA content in the supernatants of the fadL, fadD fadL, and fadD fadL tolC mutants, bacteria were initially inoculated into morpholinepropanesulfonic acid (MOPS) EZ rich defined medium (Teknova Inc.), grown overnight, and then subcultured 1/100 in 25 ml modified MOPS EZ medium (1× MOPS rich buffer, 1× ACGU solution, 0.04 mM K2HPO4, 1× Supplement EZ, 0.2% glucose) in 125-ml flasks and grown for 7 h with aeration at 200 rpm. Triplicate cultures were grown for each mutant background. Fifteen milliliters of each resulting bacterial culture was centrifuged for 5 min at 7,000 rpm, the supernatants were collected and filtered through 0.45-µm polyethersulfone (PES) filters (Foxx Life Sciences Inc.), and the bacterial pellets were collected for normalization to dry cell weight. All glassware used in the experiment was washed with acetone prior to use to minimize LCFA contamination of the initial growth medium.
Fatty acids were extracted with 2 ml of chloroform, dried under an N2 stream, and converted into their methyl esters with 50 µl of 2.0 M (trimethylsilyl)diazomethane dissolved in hexanes (Sigma, St. Louis, MO, USA) and 30 µl of methanol for 15 min at room temperature. Twenty microliters of internal standard (1 mg ⋅ ml−1 nonadecanoic acid) was added to each sample prior to derivatization. One microliter of each sample was injected in split mode (7:1) into a gas chromatography-mass spectrometry (GC-MS) system consisting of an Agilent 7890 gas chromatograph, an Agilent 5975 mass selective detector (MSD), and an Agilent 7683B autosampler (Agilent Inc., Palo Alto, CA). Separation was performed on a ZB-5MS (60 m by 0.32-mm inside diameter [i.d.], 0.25-µm film thickness) capillary column (Phenomenex, Torrance, CA). The inlet and MSD interface temperatures were 250°C, and the ion source temperature was adjusted to 230°C. The helium carrier gas was kept at a constant flow rate of 2 ml ⋅ min−1. The temperature program was 3 min of isothermal heating at 120°C, followed by an oven temperature increase of 5°C ⋅ min−1 to 260°C and a final 5 min at 310°C. The mass spectrometer was operated in positive electron impact (EI) mode at 69.9-eV ionization energy at an m/z 50 to 800 scan range. The instrument variability was 5%, which is within the standard acceptance limit.
The spectra of all chromatogram peaks were compared with NIST08 electron impact mass spectrum libraries (National Institute of Standards and Technology [NIST], MD, USA), W8N08 (Palisade Corporation, NY, USA), and a custom-built database (460 unique metabolites). All known artificial peaks were identified and removed. To allow comparison between samples, all data were normalized to the internal standard in each chromatogram and the cell dry weight (DW). The spectra of all chromatogram peaks were evaluated using the AMDIS 2.71 (NIST, MD, USA) program.
Metabolite concentrations are reported as “(analyte concentration relative to nonadecanoic acid) per gram dry weight” (relative concentration), i.e., as target compound peak area divided by the internal standard (IS) peak area (IS concentrations are the same in all samples): Ni = Xi × XIS−1 × g dry weight−1. Only those LCFAs not present in growth medium were plotted (LCFAs found in the initial growth medium were subtracted from the data). Box plots were generated in R v.3.1.2 (http://www.r-project.org) or in XLStat 2015v 2.02 programs. For the box plot, the line in the middle of the rectangle is the median (or the second quartile [Q2]). The top whisker denotes the maximum value or the third quartile plus 1.5 times the interquartile range [Q3 + (1.5 × IQR)], whichever is smaller. The bottom whisker denotes either the minimum value or the first quartile minus 1.5 times the interquartile range [Q1 − (1.5 × IQR)], whichever is larger.
Purification of HilD and HilC proteins.
HilD and HilC were purified with N-terminal His6-Sumo fusion tags as described previously (27) and stored at 4°C in 20% glycerol. Dilutions of proteins were made using protein dilution buffer (50 mM phosphate buffer, pH 7.4, 20 mM NaCl, 10% glycerol).
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed using the SYBR green EMSA kit (Thermo Fisher Inc.) according to the manufacturer’s instructions. A 10 nM concentration of a double-stranded 210-bp DNA fragment of the hilC promoter region (corresponding to −162 to +48) or a 258-bp nonspecific DNA fragment was incubated for 20 min at room temperature (RT) with indicated amounts of purified HilD or HilC protein in a binding buffer (20 mM HEPES, pH 7.3, 20 mM KCl, 1% glycerol, 1 mM dithiothreitol [DTT], 0.04 mM EDTA, 0.05% Tergitol NP-40) and 0.5× Novex Hi-Density Tris-borate-EDTA (TBE) sample buffer. Oleate (or 90% ethanol at pH 6.8 to 7.0 as a control) was added to samples as indicated. The samples were separated on a 6% DNA retardation gel using 0.5× TBE running buffer according to the manufacturer’s instructions (Invitrogen, Inc.).
Virulence assays.
Bacteria were initially inoculated into LB medium (0.5% NaCl), grown overnight, and then subcultured 1/35 in 4 ml LB medium without NaCl in 50-ml flasks and grown for 4 h with aeration at 200 rpm. BALB/c mice (Harlan) (6 to 8 weeks old) were inoculated either orally or intraperitoneally (i.p.) with 0.2 ml of a bacterial suspension. For oral infections, the bacteria were washed and suspended at 5 × 108 or 5 × 109 cells per ml in sterile 0.1 M sodium phosphate buffer, pH 8.0. For intraperitoneal infections, the cells were diluted to 5 × 103 cells per ml in sterile PBS. For oral infections, mice were sacrificed by CO2 asphyxiation at 2 to 2.5 days after inoculation and the ileal small intestines were harvested. For i.p. infections, the mice were sacrificed by CO2 asphyxiation between 4 and 5 days after inoculation and spleens were harvested. These organs were homogenized, and serial dilutions were plated on the appropriate medium to determine the number of CFU per organ. The relative percentage of each strain recovered was determined by replica plating to the appropriate antibiotic-containing medium. In all competition assays, the inoculum consisted of a 1:1 mix of two bacterial strains. The actual CFU and relative percentage represented by each strain were determined by direct plating of the inoculum. The competitive index (CI) was calculated as (percentage of strain A recovered/percentage of strain B recovered)/(percentage of strain A inoculated/percentage of strain B inoculated). The Student t test was used to determine whether the output ratio was significantly different from the input ratio. All animal work was reviewed and approved by the University of Illinois Institutional Animal Care and Use Committee (IACUC) and performed under protocol no. 10050.
SUPPLEMENTAL MATERIAL
Experimental procedures for results presented in supplemental figures. Download
Addition of oleate results in fast and reversible repression of hilA transcription. (A) β-Galactosidase activity in a strain containing a hilA-lac transcriptional fusion after initial growth in the absence of oleate, with 0.6 mM oleate added at t = 0 (see Materials and Methods), and further growth as indicated. (B) β-Galactosidase activity in a strain containing a hilA-lac transcriptional fusion grown in the presence of 0.6 mM oleate and then washed twice with fresh medium (t = 0) and grown further as indicated. β-Galactosidase activity units are defined as (micromoles of ONP formed minute−1) × 106/(OD600 × milliliter of cell suspension) and are reported as mean ± standard deviation, where n is 4. The strain used was JS749. Download
Accumulation of free LCFAs in the culture medium is not due to cell lysis. Average relative β-galactosidase activity determined in cells or supernatant from 3 replicate cultures for each strain submitted for metabolomics analysis. Strains used were JS749, JS2062, JS2063, and JS2072. Download
External free LCFAs repress hilA independently of periplasmic stress regulators or the PhoPQ two-component regulatory system. (A) β-Galactosidase activity in strains containing a hilA-lac transcriptional fusion and the indicated mutations grown under SPI1-inducing conditions in the absence or presence of 0.3 mM oleate, 0.3 mM myristate, or 0.3 mM palmitate. (B) β-Galactosidase activity in strains containing a hilA-lac transcriptional fusion in the wild type with a phoPQ background grown under SPI1-inducing conditions in the absence or presence of 0.3 mM oleate, 0.3 mM myristate, or 0.3 mM palmitate. β-Galactosidase activity is reported as mean ± standard deviation, where n is 4. Strains used were JS749, JS1069, JS2073, and JS2074. Download
HilD and HilC proteins bind a 210-bp fragment of the hilC promoter. All lanes contain 10 nM DNA and 0.05% Tergitol NP-40 detergent. A 258-bp nonspecific DNA fragment was used as a control. Download
Strains and plasmids used in this study.
ACKNOWLEDGMENTS
We thank John Cronan for helpful discussions and suggestions, Alexander Ulanov of the Carver Metabolomics Center at UIUC for help with the GC-MS analysis, Marc Erhardt for providing the pHilD-SUMO plasmid and protocols, Cari Vanderpool for valuable comments on the manuscript, and Taras Pogorelov and members of the Slauch lab for helpful discussions.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Golubeva YA, Ellermeier JR, Chubiz JEC, Slauch JM. 2016. Intestinal long-chain fatty acids act as a direct signal to modulate expression of the Salmonella pathogenicity island 1 type III secretion system. mBio 7(1):e02170-15. doi:10.1128/mBio.02170-15.
REFERENCES
- 1.Galán JE. 2001. Salmonella interactions with host cells: type III secretion at work. Annu Rev Cell Dev Biol 17:53–86. doi: 10.1146/annurev.cellbio.17.1.53. [DOI] [PubMed] [Google Scholar]
- 2.Bajaj V, Hwang C, Lee CA. 1995. HilA is a novel OmpR/ToxR family member that activates the expression of Salmonella typhimurium invasion genes. Mol Microbiol 18:715–727. doi: 10.1111/j.1365-2958.1995.mmi_18040715.x. [DOI] [PubMed] [Google Scholar]
- 3.Eichelberg K, Galán JE. 1999. Differential regulation of Salmonella typhimurium type III secreted proteins by pathogenicity island 1 (SPI-1)-encoded transcriptional activators InvF and HilA. Infect Immun 67:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lostroh CP, Lee CA. 2001. The HilA box and sequences outside it determine the magnitude of HilA-dependent activation of P(prgH) from Salmonella pathogenicity island 1. J Bacteriol 183:4876–4885. doi: 10.1128/JB.183.16.4876-4885.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mills DM, Bajaj V, Lee CA. 1995. A 40-kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Mol Microbiol 15:749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 6.Ellermeier CD, Slauch JM. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J Bacteriol 185:5096–5108. doi: 10.1128/JB.185.17.5096-5108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellermeier CD, Ellermeier JR, Slauch JM. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol 57:691–705. doi: 10.1111/j.1365-2958.2005.04737.x. [DOI] [PubMed] [Google Scholar]
- 8.Saini S, Ellermeier JR, Slauch JM, Rao CV. 2010. The role of coupled positive feedback in the expression of the SPI1 type three secretion system in Salmonella. PLoS Pathog 6:e1001025. doi: 10.1371/journal.ppat.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. 2012. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190:79–90. doi: 10.1534/genetics.111.132779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song M, Kim HJ, Kim EY, Shin M, Lee HC, Hong Y, Rhee JH, Yoon H, Ryu S, Lim S, Choy HE. 2004. ppGpp-dependent stationary phase induction of genes on Salmonella pathogenicity island 1. J Biol Chem 279:34183–34190. doi: 10.1074/jbc.M313491200. [DOI] [PubMed] [Google Scholar]
- 11.Passerat J, Got P, Dukan S, Monfort P. 2009. Respective roles of culturable and viable-but-nonculturable cells in the heterogeneity of Salmonella enterica serovar Typhimurium invasiveness. Appl Environ Microbiol 75:5179–5185. doi: 10.1128/AEM.00334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailly-Bechet M, Benecke A, Hardt WD, Lanza V, Sturm A, Zecchina R. 2011. An externally modulated, noise-driven switch for the regulation of SPI1 in Salmonella enterica serovar Typhimurium. J Math Biol 63:637–662. doi: 10.1007/s00285-010-0385-1. [DOI] [PubMed] [Google Scholar]
- 13.Clark D, Cronan J. 7 October 2005. Two-carbon compounds and fatty acids as carbon sources. EcoSal Plus 2005 doi: 10.1128/ecosalplus.3.4.4. [DOI] [PubMed] [Google Scholar]
- 14.Simard JR, Pillai BK, Hamilton JA. 2008. Fatty acid flip-flop in a model membrane is faster than desorption into the aqueous phase. Biochemistry 47:9081–9089. doi: 10.1021/bi800697q. [DOI] [PubMed] [Google Scholar]
- 15.Wei C, Pohorille A. 2014. Flip-flop of oleic acid in a phospholipid membrane: rate and mechanism. J Phys Chem B 118:12919–12926. doi: 10.1021/jp508163e. [DOI] [PubMed] [Google Scholar]
- 16.Lucas RL, Lostroh CP, DiRusso CC, Spector MP, Wanner BL, Lee CA. 2000. Multiple factors independently regulate hilA and invasion gene expression in Salmonella enterica serovar Typhimurium. J Bacteriol 182:1872–1882. doi: 10.1128/JB.182.7.1872-1882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin D, Rao CV, Slauch JM. 2008. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol 190:87–97. doi: 10.1128/JB.01323-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pech-Canul Á, Nogales J, Miranda-Molina A, Álvarez L, Geiger O, Soto MJ, López-Lara IM. 2011. FadD is required for utilization of endogenous fatty acids released from membrane lipids. J Bacteriol 193:6295–6304. doi: 10.1128/JB.05450-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lennen RM, Politz MG, Kruziki MA, Pfleger BF. 2013. Identification of transport proteins involved in free fatty acid efflux in Escherichia coli. J Bacteriol 195:135–144. doi: 10.1128/JB.01477-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Virlogeux-Payant I, Baucheron S, Pelet J, Trotereau J, Bottreau E, Velge P, Cloeckaert A. 2008. TolC, but not AcrB, is involved in the invasiveness of multidrug-resistant Salmonella enterica serovar Typhimurium by increasing type III secretion system-1 expression. Int J Med Microbiol 298:561–569. doi: 10.1016/j.ijmm.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Viarengo G, Sciara MI, Salazar MO, Kieffer PM, Furlán RL, García Véscovi E. 2013. Unsaturated long chain free fatty acids are input signals of the Salmonella enterica PhoP/PhoQ regulatory system. J Biol Chem 288:22346–22358. doi: 10.1074/jbc.M113.472829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun 75:1946–1953. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, Kull FJ. 2010. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc Natl Acad Sci U S A 107:2860–2865. doi: 10.1073/pnas.0915021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Childers BM, Cao X, Weber GG, Demeler B, Hart PJ, Klose KE. 2011. N-terminal residues of the Vibrio cholerae virulence regulatory protein ToxT involved in dimerization and modulation by fatty acids. J Biol Chem 286:28644–28655. doi: 10.1074/jbc.M111.258780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plecha SC, Withey JH. 2015. Mechanism for inhibition of Vibrio cholerae ToxT activity by the unsaturated fatty acid components of bile. J Bacteriol 197:1716–1725. doi: 10.1128/JB.02409-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olekhnovich IN, Kadner RJ. 2002. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J Bacteriol 184:4148–4160. doi: 10.1128/JB.184.15.4148-4160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singer HM, Kühne C, Deditius JA, Hughes KT, Erhardt M. 2014. The Salmonella Spi1 virulence regulatory protein HilD directly activates transcription of the flagellar master operon flhDC. J Bacteriol 196:1448–1457. doi: 10.1128/JB.01438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter PB, Collins FM. 1974. The route of enteric infection in normal mice. J Exp Med 139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones BD, Ghori N, Falkow S. 1994. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med 180:15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Day J, Kovacikova G, Taylor RK, Kull FJ. 2014. Unsaturated fatty acid regulation of AraC/XylS transcription factors. Biophys J 106(Suppl 1):497a. doi: 10.1016/j.bpj.2013.11.2779.24507590 [DOI] [Google Scholar]
- 31.LaBauve AE, Wargo MJ. 2014. Detection of host-derived sphingosine by Pseudomonas aeruginosa is important for survival in the murine lung. PLoS Pathog 10:e1003889. doi: 10.1371/journal.ppat.1003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Immerseel F, De Buck J, Boyen F, Bohez L, Pasmans F, Volf J, Sevcik M, Rychlik I, Haesebrouck F, Ducatelle R. 2004. Medium-chain fatty acids decrease colonization and invasion through hilA suppression shortly after infection of chickens with Salmonella enterica serovar Enteritidis. Appl Environ Microbiol 70:3582–3587. doi: 10.1128/AEM.70.6.3582-3587.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawhon SD, Maurer R, Suyemoto M, Altier C. 2002. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol 46:1451–1464. doi: 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- 34.Martínez LC, Yakhnin H, Camacho MI, Georgellis D, Babitzke P, Puente JL, Bustamante VH. 2011. Integration of a complex regulatory cascade involving the SirA/BarA and Csr global regulatory systems that controls expression of the Salmonella SPI-1 and SPI-2 virulence regulons through HilD. Mol Microbiol 80:1637–1656. doi: 10.1111/j.1365-2958.2011.07674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y, Suyemoto M, Garner CD, Cicconi KM, Altier C. 2008. Formate acts as a diffusible signal to induce Salmonella invasion. J Bacteriol 190:4233–4241. doi: 10.1128/JB.00205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gantois I, Ducatelle R, Pasmans F, Haesebrouck F, Hautefort I, Thompson A, Hinton JC, Van Immerseel F. 2006. Butyrate specifically down-regulates Salmonella pathogenicity island 1 gene expression. Appl Environ Microbiol 72:946–949. doi: 10.1128/AEM.72.1.946-949.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hung CC, Garner CD, Slauch JM, Dwyer ZW, Lawhon SD, Frye JG, McClelland M, Ahmer BM, Altier C. 2013. The intestinal fatty acid propionate inhibits Salmonella invasion through the post-translational control of HilD. Mol Microbiol 87:1045–1060. doi: 10.1111/mmi.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garner CD, Antonopoulos DA, Wagner B, Duhamel GE, Keresztes I, Ross DA, Young VB, Altier C. 2009. Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica serovar Typhimurium murine model of infection. Infect Immun 77:2691–2702. doi: 10.1128/IAI.01570-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl A, Hirsch DJ, Gimeno RE, Punreddy S, Ge P, Watson N, Patel S, Kotler M, Raimondi A, Tartaglia LA, Lodish HF. 1999. Identification of the major intestinal fatty acid transport protein. Mol Cell 4:299–308. doi: 10.1016/S1097-2765(00)80332-9. [DOI] [PubMed] [Google Scholar]
- 40.Carey MC, Small DM, Bliss CM. 1983. Lipid digestion and absorption. Annu Rev Physiol 45:651–677. doi: 10.1146/annurev.ph.45.030183.003251. [DOI] [PubMed] [Google Scholar]
- 41.Gutnick D, Calvo JM, Klopotowski T, Ames BN. 1969. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J Bacteriol 100:215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maloy SR, Stewart VJ, Taylor RK. 1996. Genetic analysis of pathogenic bacteria: a laboratory manual. Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- 43.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. doi: 10.1016/S0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- 46.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 47.Slauch JM, Silhavy TJ. 1991. Cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J Bacteriol 173:4039–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures for results presented in supplemental figures. Download
Addition of oleate results in fast and reversible repression of hilA transcription. (A) β-Galactosidase activity in a strain containing a hilA-lac transcriptional fusion after initial growth in the absence of oleate, with 0.6 mM oleate added at t = 0 (see Materials and Methods), and further growth as indicated. (B) β-Galactosidase activity in a strain containing a hilA-lac transcriptional fusion grown in the presence of 0.6 mM oleate and then washed twice with fresh medium (t = 0) and grown further as indicated. β-Galactosidase activity units are defined as (micromoles of ONP formed minute−1) × 106/(OD600 × milliliter of cell suspension) and are reported as mean ± standard deviation, where n is 4. The strain used was JS749. Download
Accumulation of free LCFAs in the culture medium is not due to cell lysis. Average relative β-galactosidase activity determined in cells or supernatant from 3 replicate cultures for each strain submitted for metabolomics analysis. Strains used were JS749, JS2062, JS2063, and JS2072. Download
External free LCFAs repress hilA independently of periplasmic stress regulators or the PhoPQ two-component regulatory system. (A) β-Galactosidase activity in strains containing a hilA-lac transcriptional fusion and the indicated mutations grown under SPI1-inducing conditions in the absence or presence of 0.3 mM oleate, 0.3 mM myristate, or 0.3 mM palmitate. (B) β-Galactosidase activity in strains containing a hilA-lac transcriptional fusion in the wild type with a phoPQ background grown under SPI1-inducing conditions in the absence or presence of 0.3 mM oleate, 0.3 mM myristate, or 0.3 mM palmitate. β-Galactosidase activity is reported as mean ± standard deviation, where n is 4. Strains used were JS749, JS1069, JS2073, and JS2074. Download
HilD and HilC proteins bind a 210-bp fragment of the hilC promoter. All lanes contain 10 nM DNA and 0.05% Tergitol NP-40 detergent. A 258-bp nonspecific DNA fragment was used as a control. Download
Strains and plasmids used in this study.