

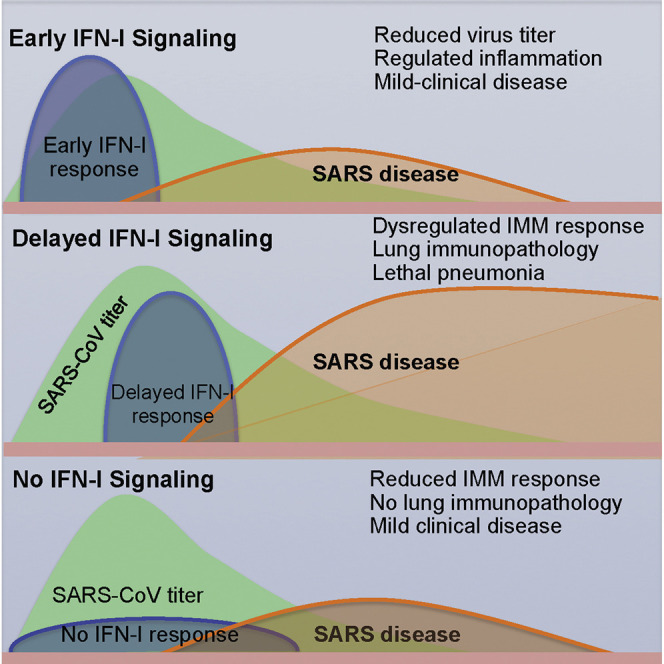
Summary
Highly pathogenic human respiratory coronaviruses cause acute lethal disease characterized by exuberant inflammatory responses and lung damage. However, the factors leading to lung pathology are not well understood. Using mice infected with SARS (severe acute respiratory syndrome)-CoV, we show that robust virus replication accompanied by delayed type I interferon (IFN-I) signaling orchestrates inflammatory responses and lung immunopathology with diminished survival. IFN-I remains detectable until after virus titers peak, but early IFN-I administration ameliorates immunopathology. This delayed IFN-I signaling promotes the accumulation of pathogenic inflammatory monocyte-macrophages (IMMs), resulting in elevated lung cytokine/chemokine levels, vascular leakage, and impaired virus-specific T cell responses. Genetic ablation of the IFN-αβ receptor (IFNAR) or IMM depletion protects mice from lethal infection, without affecting viral load. These results demonstrate that IFN-I and IMM promote lethal SARS-CoV infection and identify IFN-I and IMMs as potential therapeutic targets in patients infected with pathogenic coronavirus and perhaps other respiratory viruses.
Graphical Abstract
Highlights
-
•
SARS-CoV causes a lethal respiratory infection in BALB/c mice
-
•
Robust SARS-CoV replication and delayed IFN-I signaling promote disease
-
•
IFN-I induces influx of pathogenic inflammatory monocytes and vascular leakage
-
•
Disease severity is ameliorated in the absence of IFN signaling
Factors that lead to lethal disease following SARS-CoV infection are not well understood. Channappanavar et al. show that robust virus replication and delayed IFN-I signaling promote severe disease. IFN-I promotes accumulation of pathogenic monocyte-macrophages resulting in lung immunopathology, vascular leakage, and suboptimal T cell responses.
Introduction
Highly pathogenic respiratory coronaviruses (CoV) such as severe acute respiratory syndrome CoV (SARS-CoV) and Middle East respiratory syndrome CoV (MERS-CoV) pose substantial challenges to public health (Peiris et al., 2004, Zumla et al., 2015). During the 2002–2003 epidemic, SARS-CoV infected approximately 8,000 individuals, with a 10% overall mortality (Peiris et al., 2004). More recently, MERS-CoV was identified in 2012 in a patient in the Middle East (Zaki et al., 2012). Since its emergence, MERS-CoV has infected 1,626 patients, resulting in 586 deaths (January 15, 2016, 36.0% mortality) (WHO, 2016). The identification of MERS-CoV and other zoonotic CoVs (Ge et al., 2013, Menachery et al., 2015) makes it likely that these viruses will continue to cross species and cause additional disease outbreaks in human populations.
Factors responsible for the unusually high pathogenicity of SARS-CoV and MERS-CoV are incompletely understood. High initial virus titers and increased monocyte-macrophage and neutrophil accumulation in the lungs associated with elevated serum pro-inflammatory cytokine (IL-1, IL-6, IL-8, CXCL-10, and MCP-1) levels accompanied extensive lung damage in SARS patients (Franks et al., 2003, Nicholls et al., 2003, Wong et al., 2004). Studies of SARS patients with severe disease revealed diffuse alveolar damage, epithelial necrosis, and fibrin and hyaline deposition, characteristic of acute lung injury (ALI) (Nicholls et al., 2003, Peiris et al., 2004). Additionally, fatal SARS in humans was accompanied by robust and persistent expression of IFN and IFN-stimulated genes (ISGs) (Cameron et al., 2007, Cameron et al., 2008), associated with impaired T cell and antibody responses (Cameron et al., 2007, Cui et al., 2003). Thus, the deleterious clinical manifestations of SARS-CoV could stem from a combination of exuberant innate immune responses and virus-induced direct cytopathic effects. Consistent with a dysregulated innate immune response in patients, infection of human macrophages and dendritic cells is abortive, with elevated expression of several pro-inflammatory cytokines and chemokines but delayed expression of type I interferon (IFN-I), critical for initiation of the anti-viral innate immune response (Law et al., 2005, Spiegel et al., 2006, Yen et al., 2006). IFN-I induction is also delayed after infection of a human airway epithelial cell line (Menachery et al., 2014).
Among animal models employed to study SARS-CoV pathogenesis, infection of mice with a mouse-adapted strain of SARS-CoV (MA15, referred to as SARS-CoV herein) replicates many features of SARS in humans (Roberts et al., 2007). Young mice from many strains (e.g., C57BL/6, 129) support SARS-CoV replication in the lungs but develop mild or subclinical disease, mimicking mild human disease (Frieman et al., 2010, Page et al., 2012, Zhao et al., 2011). In contrast, infection of BALB/c mice with SARS-CoV resulted in rapid virus replication associated with increased monocyte/macrophage accumulation, pulmonary edema, hyaline membrane formation, diffuse alveolar damage, and destruction of the alveolar/airway epithelium (Gralinski and Baric, 2015, Roberts et al., 2007, van den Brand et al., 2014), similar to the disease observed in patients with severe SARS. Infected aged mice of all strains examined also developed severe disease, paralleling the age dependence observed in human SARS (Chen and Subbarao, 2007, van den Brand et al., 2014, Zhao et al., 2011).
Although fatal outcomes due to immunopathological events following SARS-CoV infection have been well established, factors initiating detrimental inflammatory responses are not well understood. Since persistent elevation of IFN-I suggested a pathogenic role in SARS patients, we explored the possibility that IFN-I was critical in the initiation of events that led to lethal lung immunopathology, using BALB/c mice. Previous studies showed that IFN-I, II, and III signaling or combinations thereof were dispensable for protection of young C57BL/6 or 129 mice after SARS-CoV infection (Frieman et al., 2010, Mahlakõiv et al., 2012). However, these studies did not address the role of IFN-I in the context of severe disease. Here, we show that IFN-I signaling is detrimental in highly susceptible SARS-CoV-infected BALB/c mice, in large part by promoting the influx of pathogenic inflammatory monocyte-macrophages (IMMs), and suggest that targeted antagonism of this pathway would improve outcomes in patients with severe coronavirus infections.
Results
Abrogation of IFN-I Signaling Prevents Morbidity and Mortality in SARS-CoV-Infected BALB/c Mice
To examine whether IFN-I signaling contributed to severe SARS in mice, we infected 7- to 9-week-old BALB/c mice and mice lacking expression of IFNαβ receptor (Ifnar −/−) with a lethal dose (3 × 104 PFU) of SARS-CoV and monitored disease severity. BALB/c mice infected with SARS-CoV exhibited lethargy, ruffled fur, hunched posture, and labored breathing as early as day 3 post infection (p.i.), accompanied by progressive weight loss and death of approximately 85% of mice by day 8 p.i. (Figures 1 A–1C). In marked contrast, infected Ifnar −/− mice all survived the infection, exhibiting only moderate (∼15%) weight loss and mild to moderate clinical disease (Figures 1A–1C); even at a higher dosage (105 PFU), all Ifnar −/− mice survived (Figure S1A). Middle-aged BALB/c mice (8–9 months) are highly susceptible to infection with SARS-CoV, but even in these mice survival was increased in the absence of IFN-I signaling (Figure S1B).
Figure 1.

Type I Interferon Signaling Promotes Lung Pathology following Lethal SARS-CoV Infection
(A and B) Percentage of initial weight (A) and survival (B) of mice infected with SARS-CoV.
(C) Clinical scores determined at days 3 and 5 p.i.
(D) SARS-CoV titers in the lungs as determined by plaque assay (ND, not determined).
(E) Gross lung pathology at days 3 and 5 p.i.
(F) Microscopic changes in the lungs of naive and SARS-CoV-challenged mice at day 5 p.i.
(G and H) Weight curves (G) and survival (H) of SARS-CoV infected anti-IFNAR or control (MOPC21) antibody treated mice.
Data are representative of 2–3 independent experiments with 4–5 mice/group. Data in (A), (C), and (D) are represented as ±SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Gross and histopathology results are derived from 4–5 mice/group. See also Figures S1 and S2.
Total lung virus loads were the same in young BALB/c and Ifnar −/− lungs at all days p.i., except for a modest increase at day 3 p.i. in Ifnar −/− mice (Figure 1D). SARS-CoV virus was completely cleared from the lungs of Ifnar −/− mice by day 10 p.i. (Figure 1D). Gross examination of the lungs revealed extensive hyperemia and congestion in BALB/c mice at days 3 and 5 p.i., while those from Ifnar −/− mice appeared nearly normal (Figure 1E). Histopathological examination of lungs showed marked alveolar edema, terminal bronchiolar epithelial sloughing, and thickening of interstitial septa in BALB/c mice at day 5 p.i, while Ifnar −/− lungs showed minimal alveolar edema with increased peribronchial-perivascular immune cell infiltration (Figure 1F). Acute lung injury following SARS is often accompanied by accumulation of protein-rich edema fluid in the perivascular and alveolar spaces (Franks et al., 2003, Gu et al., 2005, Nicholls et al., 2003). To assess vascular leakage, we measured Evan’s blue extravasation in the lungs at day 5 p.i. Lung microvascular leakage was very prominent in SARS-CoV-infected BALB/c mice but was significantly reduced in Ifnar −/− mice (Figure S2).
While these results suggest a critical role for IFN-I signaling in severe disease, developmental abnormalities in the absence of IFNAR signaling could also contribute to improved outcomes. To exclude this possibility, we treated BALB/c mice with either IFN-αβ receptor (IFNAR)-blocking or isotype control antibody prior to and after SARS-CoV infection. Consistent with the disease manifestations in Ifnar −/− mice, anti-IFNAR antibody treatment significantly reduced weight loss and increased survival compared to control antibody-treated mice (Figures 1G and 1H).
To determine the generality of the above results, we infected BALB/c and Ifnar −/− mice with MHV-1 and influenza A virus (IAV, PR8 strain), two RNA viruses that also cause acute respiratory illness in mice. In agreement with previous studies (Cervantes-Barragán et al., 2009, Seo et al., 2011), IFN signaling was critical for protection: all Ifnar −/− mice succumbed to a sub-lethal MHV-1 and PR8 infection, while only ∼10% of MHV-1 and 20% of PR8-infected BALB/c mice, respectively, died (Figures S1C and S1D). These results suggest that there are unique features of the SARS-CoV infection that result in IFN-I signaling being pathogenic rather than protective.
Rapid SARS-CoV Replication and Delayed IFN-I Expression in Lungs
The results from previous reports (Frieman et al., 2010, Roberts et al., 2007, Zhao et al., 2009) and those in Figure 1D demonstrate rapid kinetics of SARS-CoV replication in the lungs; as shown in Figure 2 A, virus titers are nearly maximal by 16 hr p.i. Immunohistochemical (IHC) examination of SARS-CoV-infected lungs revealed virus replication in the lung airways and parenchyma with viral antigen staining detected in type II pneumocytes at 16 hr p.i. (Figure 2B). By 24 and 48 hr p.i., we detected viral antigen distributed throughout the lung airways and in parenchyma with intense antigen distribution in type II and to a lesser extent in type I pneumocytes (Figure 2B), similar to findings in SARS-CoV-infected human autopsy samples (Gu et al., 2005, Nicholls et al., 2003). The distribution and number of virus antigen-containing cells were the same in BALB/c and Ifnar −/− mice, consistent with the virus titers shown in Figure 2A.
Figure 2.

Characterization of Virus Replication and IFN-I Production in SARS-CoV-Infected Lungs
(A) Lung virus titers determined at early times after SARS-CoV infection.
(B) Immunohistochemical examination of SARS-CoV N protein at different times p.i.
(C) BALF cytokine/chemokine levels from BALB/c and Ifnar−/− mice at different times p.i.
(D) Weight curves and survival after treatment with IFN-β (2,000 U, i.n., single dose at 6, 12, or 24 hr p.i.).
(E) Lung viral loads at days 1 and 3 p.i. in IFN-β (2,000 U, 6 hr p.i.) and PBS-treated mice.
(F) Lung cells harvested from SARS-CoV-infected mice (24 hr p.i.) were MACS sorted into Siglec-H positive and negative and Siglec-F positive and negative cells. Sorted cells were analyzed for IFN-α4 and IFN-β mRNA transcript levels.
(G) Immunohistochemical examination for IFN-β expression in the lung airway (top panel) and lung parenchyma (bottom panel) at different times p.i. (C).
Data in (A) and (C)–(E) are derived from two independent experiments, 4–5 mice/group/experiment. Data in (A) and (C)–(F) are represented as ±SEM. ∗∗p < 0.01, ∗∗∗p < 0.001.
IFN-I is critical for initiation of a protective immune response, but IFN-I was not detected in the broncho-alveolar lavage fluid (BALF) of BALB/c mice until 24 hr p.i. Similarly, other cytokines and chemokines involved in the innate response also showed delayed expression in the BALF (Figure 2C). In contrast, Ifnar −/− mice had significantly reduced levels of cytokines and chemokines in the BALF compared to BALB/c mice (Figure 2C). Since this delayed inflammatory mediator expression might contribute to a subsequent excessive innate immune response, we next examined whether IFN-I delivered prior to the peak of virus replication would reverse this phenotype and enhance survival. For this purpose, we administered recombinant IFN-β (2000 U) intranasally at various times after infection. In agreement with previous results (Kumaki et al., 2011), IFN-I delivered 6 hr p.i., prior to the peak of virus replication, but not at 24 hr p.i., completely protected mice from weight loss and clinical disease. IFN-β delivery at 12 hr p.i. resulted in an intermediate level of protection (Figure 2D). Treatment with IFN-β at 6 hr p.i. moderately reduced virus titers in the lungs at day 1 p.i (Figure 2E).
We next investigated the cellular source of IFN-I in SARS-CoV-infected lungs. Since plasmacytoid dendritic cells (pDCs) and alveolar macrophages (AMs) are known to be important sources of IFN-I in RNA virus infections (Killip et al., 2015), we initially purified pDCs (Siglec-H+) and AMs (Siglec-F+) using magnetic beads and measured IFN-β and IFNα4 mRNA levels by qRT-PCR. While Siglec-H is expressed on pDCs and macrophage subsets in lymphoid tissues, it is primarily expressed on pDCs in the lungs (Swiecki et al., 2014; data not shown). pDCs from infected mice expressed significantly higher levels of IFN-β and IFNα4 mRNA compared to Siglec-H− cells (Figure 2F). Consistent with these results, human pDCs are also robust IFN producers after SARS-CoV infection in vitro (Cervantes-Barragan et al., 2007). AMs also expressed IFN-I, but not to a greater extent than non-AMs. We also stained tissue sections for IFN-β to assess IFN-I in other pulmonary cells and detected expression in airway cells and interstitial cells (Figure 2G). Collectively, these results suggest that many cell types, but most importantly pDCs, express IFN-I after SARS-CoV infection in mice and likely humans.
Type I Interferon Signaling Regulates Recruitment and Activation of IMMs
Next, we assessed whether IFN-I orchestrated innate cell recruitment in SARS-CoV-infected mice. Total numbers of neutrophils, AMs, natural killer (NK) cells, and pDCs were comparable in SARS-CoV-infected BALB/c and Ifnar −/− lungs at days 1 and 3 p.i. (Figure S3A). In contrast, there was a dramatic increase in numbers of Ly6ChiCD11b+ cells in the lungs of BALB/c mice, and this increase was abrogated in the absence of IFN-I signaling (Figures 3A and 3B). At day 3 p.i. there was an approximately 6-fold increase in the numbers of infiltrating Ly6ChiCD11b+ cells in the lungs of BALB/c compared to Ifnar −/− mice (Figure 3B). Phenotypic examination revealed that these cells expressed CCR2, F4/80, and CD11c, but not Ly6G and Siglec-H, suggesting that they were IMMs (Serbina et al., 2003) (Figure 3C). Furthermore, surface levels of CD69, BST-2, CD80, and CD86 were significantly higher on IMMs harvested from BALB/c compared to Ifnar −/− lungs (Figure 3D), consistent with greater activation. CCL2, CCL7, and CCL12, which are ligands for CCR2, as well as GM-CSF, a macrophage growth factor, are involved in IMM recruitment (Shi and Pamer, 2011, Song et al., 2015). While the expression of CCR2 ligands was significantly reduced at 24 and 72 hr p.i., GM-CSF expression was reduced at 16 hr p.i., but not at later times, in the lungs of Ifnar −/− compared to BALB/c mice (Figure S3B). CCL2 was produced predominantly by IMMs when measured directly ex vivo (Figure S3C), and direct IFN-I signaling was sufficient to induce CCR2 ligand expression by bone marrow cells (Figure S3D). Thus, IFN-I signaling promoted accumulation of highly activated IMMs in SARS-CoV-infected lungs, and this process was amplified by IMM production of CCR2 ligands.
Figure 3.

IFN-I Promotes Accumulation of Inflammatory Monocyte-Macrophages
(A) FACS plots show kinetics of Ly6Chi CD11b+ inflammatory monocyte accumulation in the lungs.
(B) Percentage and total number of Ly6Chi CD11b+ cells in the lungs.
(C) Phenotypic marker expression on inflammatory monocytes from the lungs of BALB/c mice at day 3 p.i.
(D) Cell surface levels of activation markers on lung IMMs at day 3 p.i.
(E) Total number of cytokine positive IMMs in the lungs determined following 7 hr ex vivo incubation in the presence of Golgi plug.
Data are derived from 2–3 independent experiments with 4 mice/group/experiment. Data in (B), (D), and (E) are represented as ±SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S3 and S4.
To determine whether IMMs were also responsible for increased pro-inflammatory cytokine expression, we stained IMMs from BALB/c and Ifnar −/− mice directly ex vivo for intracellular TNF, IL-6, IL-1-β, and iNOS. These analyses revealed significantly higher percentages and numbers of IMMs that produced these pro-inflammatory cytokines in BALB/c compared to Ifnar −/− lungs (Figure 3E). We next assessed whether IMMs were the predominant source of these cytokines in SARS-CoV-infected lungs by depleting IMMs with anti-CCR2 antibody (MC21 mAb) since CCR2 is specifically expressed at high levels on pro-inflammatory monocytes (Mack et al., 2001). MC21 antibody specifically depleted CD11b+Ly6Chi IMMs, reducing the levels to those observed in Ifnar −/− mice (Figures S4A and S4B). MC21 monoclonal antibody treatment significantly reduced levels of CCL2, TNF, and IL-6 in the BALF (Figure S4C), directly establishing IMMs as a major source of inflammatory cytokines/chemokines in infected lungs.
Depletion of IMMs Ameliorates SARS-CoV-Induced Disease
To directly establish a link between augmented IMM infiltration and SARS-CoV-induced lung immunopathology, we depleted IMMs using MC21 mAb. Treatment with MC21 mAb resulted in protection from lethal disease confirming a critical role for IMMs in promoting SARS-CoV-induced morbidity and mortality (Figure 4 A). To confirm these results, we treated another cohort of mice with anti-BST-2-depleting antibody, as BST-2 was highly expressed on activated IMMs (Figure 3D). BST-2 is expressed constitutively on pDCs and is upregulated on IMMs and some stromal cells by inflammatory stimuli (Blasius et al., 2006). BST-2 depletion completely protected BALB/c mice from lethal SARS-CoV challenge, whereas approximately 70% of control Ig-treated mice succumbed to infection (Figure 4C). IMM depletion with either antibody minimally affected lung virus titers (∼2-fold) (Figures 4B and 4D). Histological examination of MC21 or anti-BST-2 mAb-treated lungs showed reduced alveolar edema and bronchial epithelial sloughing with no change in cellular infiltration, compared to control Ig-treated mice (Figure 4E). Additionally, depletion of IMMs also reduced vascular leakage (Figures S2A–S2C). Notably, treatment with MC21 also decreased the numbers of pDCs in lungs (Figure S4D) even though these cells do not express CCR2 (Figure S4E), perhaps by decreasing chemokine expression. As noted above, IMMs express high levels of inflammatory cytokines (Figure 3E), resulting in elevated levels in the lungs (Figure 2C), which could contribute to more severe disease in BALB/c mice. Consistent with this notion, neutralization of a single pro-inflammatory mediator, TNF, which was most increased when BALB/c and Ifnar −/− IMMs were compared (Figure 3E), provided partial, but significant, protection compared to control mice (Figure 4F). Although these results demonstrate a key role for IMMs in severe disease in SARS-CoV-infected mice, neutrophils are often detrimental in respiratory viral infections (Brandes et al., 2013). However, as noted above, neutrophil numbers are marginally higher in BALB/c mice (Figure S3A), and neutrophil depletion had no effect on survival (Figure 4G). Collectively, these results demonstrate roles for IMMs at several steps in the inflammatory process, resulting in immunopathological changes in SARS-CoV-infected mice.
Figure 4.

Depletion of Inflammatory Monocytes Ameliorates SARS-CoV-Induced Lethal Disease
Survival and lung pathology were determined in young BALB/c mice treated with IMM-depleting and control antibodies.
(A and C) Weight curves and survival of SARS-CoV-infected BALB/c mice after control and MC21 (A) or anti-BST-2 (C) antibody treatment.
(B and D) Virus titers in the lungs at days 1 and 3 post control Ig and MC21 (B) or anti-BST-2 mAb (D) treatment.
(E) Histological changes in the lungs of naive, isotype-antibody-treated and IMM-depleted SARS-CoV-challenged BALB/c mice at day 5 p.i.
(F) Weight curves and survival of control and anti-TNF-α antibody-treated BALB/c mice.
(G) Survival of SARS-CoV-infected BALB/c mice after neutrophil depletion.
Data are representative of 2 independent experiments (4–5 mice/group/experiment). Data in (A)–(D) are represented as ±SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. See also Figures S2–S4.
IFN-I Signaling on Hematopoietic Cells Enhances Disease in SARS-CoV-Infected BALB/c Mice
Having identified a key role for IMMs in increasing the severity of SARS, we next assessed whether IFN-I signaling on non-hematopoietic or hematopoietic cells was required to induce IMM migration to the site of infection. To address this, we generated bone marrow chimeras between CD45 disparate BALB/c and Ifnar −/− mice. Engraftment of donor cells was confirmed by flow cytometric analysis of CD45.1 and CD45.2 expression on peripheral blood leukocytes 5 weeks post-transfer (Figure 5 A). 6 weeks after bone marrow reconstitution, chimera mice were challenged with a lethal dose of SARS-CoV. Only 10% of mice in the Ifnar −/−→ Ifnar −/− group and 20% of those in the Ifnar −/−→BALB/c group died after challenge. In contrast, 100% of BALB/c→BALB/c mice and ∼70% of BALB/c→ Ifnar −/− chimera mice succumbed to SARS-CoV infection (Figure 5B). The relative susceptibility of bone marrow chimeric mice to SARS-CoV infection correlated with the numbers of IMMs infiltrating into the lungs at day 3 p.i. (Figures 5C and 5D). Thus, increased IMM infiltration was observed in mice reconstituted with BALB/c bone marrow, while mice that received Ifnar −/− bone marrow were mostly resistant to SARS-CoV infection (Figures 5C and 5D). Together, these results demonstrate that IFN-I signaling, largely on hematopoietic cells, promoted lung immunopathology in SARS-CoV-infected BALB/c mice.
Figure 5.

IFN-I Signaling on Hematopoietic Cells Promotes SARS-CoV-Induced Morbidity and Mortality
(A) PBMCs from uninfected bone-marrow chimera mice (5 weeks post BM transfer) were analyzed by flow cytometry to assess bone-marrow reconstitution.
(B) BM-chimera mice were monitored for survival after SARS-CoV challenge (103 PFU, i.n., 4–5 mice/group, 2 independent experiments).
(C) Lung cell suspensions from SARS-CoV-infected bone marrow chimeric mice were analyzed for IMM infiltration at day 3 p.i.
(D) Percentage and total number of IMMs in chimeric mice at day 3 p.i.
For (C) and (D), Data are representative of 2–3 independent experiments (2–3 mice/group/experiment). Data in (D) are represented as ±SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
IFN-I-Mediated Inflammatory Response Impairs Virus-Specific T Cell Responses in SARS-CoV-Infected BALB/c Mice
Since a robust T cell response is required for SARS-CoV clearance (Channappanavar et al., 2014, Zhao et al., 2010), we next evaluated T cell responses in infected BALB/c, Ifnar −/−, and MC21-treated mice at day 6 p.i. The total numbers of virus-specific CD8 (epitope S366) and CD4 (epitope N353) T cells in lungs were significantly lower in BALB/c mice compared to Ifnar −/− or MC21-treated mice (Figures 6 A–6D). Previous work has associated a suboptimal T cell response with impaired respiratory dendritic cell (rDC) migration to draining lymph nodes (Zhao et al., 2011, Zhao et al., 2009). Therefore, to examine whether diminished rDC migration contributed to sub-optimal virus-specific T cell responses, BALB/c and Ifnar −/− mice were intranasally instilled with CFSE 6 hr prior to SARS-CoV infection and analyzed for rDC migration to DLN at 18 hr p.i. Although the percentage of CFSE+ rDCs migrating to the DLN was lower in Ifnar −/− mice, similar numbers of CFSE+ rDCs migrated to DLNs (Figures 6E and 6F) in BALB/c and Ifnar −/− mice, suggesting that the defective T cell response did not reflect poor rDC migration.
Figure 6.

IFN-I Signaling Impairs T Cell Response
Lung T cell responses were measured in SARS-CoV-challenged BALB/c, Ifnar−/−, and rat IgG and MC21 antibody-treated BALB/c mice at day 6 p.i.
(A and B) FACS plots (A) and bar graphs (B) show percentage and total number of virus-specific CD4 and CD8 T cells in the lungs of BALB/c and Ifnar−/− mice.
(C and D) FACS plots (C) and bar graphs (D) show percentage and total number of virus-specific CD4 and CD8 T cells in the lungs of rat Ig and MC21 antibody-treated BALB/c mice.
(E and F) Mediastinal lymph node cell suspensions were prepared and analyzed at 18 hr p.i. for CFSE-labeled rDC migration. Representative FACS plots (E) and percentages and total numbers (F) are shown.
(G and H) Percentage of apoptotic CD4 and CD8 T cells in SARS-CoV-infected lungs at day 5 p.i. Representative histograms (G) and percentage of apoptotic T cells (H) are shown.
Data are representative of 2 independent experiments (4–5 mice/group/experiment). Data in (B), (D), (F), and (H) are represented as ±SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. See also Figure S5.
Since IFN-I is known to sensitize T cells to apoptosis (Carrero et al., 2004, Welsh et al., 2012), we measured the level of T cell apoptosis in SARS-CoV-infected BALB/c and Ifnar −/− mice. As shown in Figures 6G and 6H, higher percentages of total CD8 and CD4 T cells were apoptotic in SARS-CoV-infected BALB/c compared to Ifnar −/− mice. To investigate possible mechanisms of T cell apoptosis, we measured levels of Fas and DR5, the receptors for FasL and TNF-related apoptosis-inducing ligand (TRAIL), respectively, on T cells (Fujikura et al., 2013, Kayagaki et al., 1999). Both Fas, on T cells and its ligand, and FasL, on IMMs and neutrophils, were upregulated in BALB/c compared to Ifnar −/− mice (Figures S5A and S5B). TRAIL and DR5 expression was similar in both groups of mice (Figures S5D and S5E). However, blocking FasL-Fas or TRAIL-DR5 interactions did not diminish the amount of apoptosis (Figures S5C–S5F). Moreover, we observed no differences in expression of molecules associated with the intrinsic apoptosis pathway (Figure S5G). By contrast, neutralization of TNF decreased the amount of T cell apoptosis (Figures S5H and S5I), indicating a direct or indirect role for TNF in this process.
Discussion
The results described here indicate that the rapid kinetics of SARS-CoV replication and relative delay in IFN-I signaling promoted inflammatory monocyte-macrophage accumulation, extensive vascular leakage, and impaired virus-specific T cell responses resulting in severe disease in infected BALB/c mice. These results parallel findings obtained in SARS patients. Fatal outcomes in humans were often accompanied by high initial viral loads (Chu et al., 2004) and immunopathological disease with elevated IFN-I and cytokine levels (Cameron et al., 2007). Increased numbers of macrophages in the lungs were characteristic of patients who had severe SARS (Franks et al., 2003, Nicholls et al., 2003). Exogenous IFN-I delivered prior to peak virus titers abrogates this cascade of events and ameliorates immunopathological disease (Figures 2D and 2E).
Although the antiviral and immunomodulatory effects of IFN-I are well recognized as protective against viral infections, it has become increasingly evident that IFN-I also promotes detrimental inflammatory responses in some bacterial, fungal, and chronic virus infections through a complex array of mechanisms that tend to be pathogen specific (Carrero, 2013, McNab et al., 2015, Trinchieri, 2010). Thus, IFN-I promotes pathological effects during intracellular bacterial infections (e.g., M. tuberculosis, L. monocytogenes) by suppressing neutrophil and monocyte production of key effector molecules (TNF and IL-1), allowing uncontrolled bacterial replication (Auerbuch et al., 2004, Mayer-Barber et al., 2014). In a second mechanism, IFN-I induces fatal immunopathology during some fungal infections (e.g., Candida albicans) by promoting IM and neutrophil-mediated kidney immunopathology (Majer et al., 2012). Furthermore, during persistent viral infections such as those caused by lymphocytic choriomeningitis virus or SIV, IFN-I promotes chronic immune activation and upregulates the expression of inhibitory molecules such as PD-1 and LAG-3, resulting in impaired T cell responses (Sandler et al., 2014, Teijaro et al., 2013, Wang et al., 2012, Wilson et al., 2013). Lack of IFN-I signaling during PR8/IAV infection results in accumulation of CXCL1-producing Ly6Cint monocytes with subsequent neutrophil influx, resulting in lung pathology (Seo et al., 2011). In contrast, infection of some mouse strains with IAV was lethal, and this lethality was ameliorated in the absence of IFN-I signaling (Davidson et al., 2014). Unlike severe IAV infections, lack of IFN-I signaling during a highly lethal SARS-CoV infection did not promote lung neutrophil infiltration but rather decreased IMM accumulation (Figures 3 and S3A).
SARS-CoV infection of BALB/c mice is characterized by rapid virus replication, with peak titers reached between 16 hr and 2 days p.i. (Figure 2A). While rapid virus replication probably contributes to poor outcomes, virus-mediated delayed IFN-I induction and relative resistance of SARS-CoV to IFN-I and its downstream effectors likely play a role. SARS-CoV abortively infected human macrophages and dendritic cells, minimally inducing IFN-I (Cheung et al., 2005, Law et al., 2005). Additionally, SARS-CoV encodes proteins that inhibit IFN induction or signaling or the function of downstream effector molecules (Totura and Baric, 2012).
An elevated IFN-I response was accompanied by CCR2 ligand release (Figures 2C and S3B–S3D), resulting in influx of IMMs in SARS-CoV-infected mice, which further amplified production of IFN-I, CCL2, and other inflammatory mediators (Figures 2 and S3C). Although IMMs are pathogenic in SARS-CoV-infected BALB/c mice, they may be either protective or pathogenic, dependent upon the pathogen in question. In another model of SARS in which STAT1−/− mice are infected with SARS-CoV, macrophages are pathogenic, but in this instance pathological changes in the lungs are caused by conversion of a M1 (classical) to a M2 (alternatively activated) phenotype contributing to increased fibrosis and poor outcomes. In these mice, abrogation of STAT-1 signaling specifically in monocyte-macrophage subsets promoted lung pathology (Frieman et al., 2010, Page et al., 2012, Zornetzer et al., 2010). During respiratory syncytial virus (RSV), herpes simplex virus (HSV), and West Nile virus (WNV) infections, IMMs mediate virus clearance and are protective (Goritzka et al., 2015, Terry et al., 2012). In contrast, in mice infected with the pathogenic PR8 strain of IAV, IMMs have dual effects: promoting lung pathology as well as being required for optimal virus-specific T cell responses (Aldridge et al., 2009, Lin et al., 2008).
Despite massive IMM accumulation in SARS-CoV-infected lungs, the mechanisms by which these cells promote fatal disease remain unclear. Our results show that TNF contributes to lethal disease since its neutralization enhances survival (Figure 4F), but it is likely that other cytokines expressed by IMM such as IL-6, IL-1β, and iNOS, among others, have detrimental effects. IFN-I-induced pro-inflammatory cytokines induce TRAIL-DR5 or FasL-Fas expression on myeloid and T cells involved in T cell apoptosis (Boonnak et al., 2014, Daigneault et al., 2012, Kayagaki et al., 1999). We identified increased T cell apoptosis in SARS-CoV-infected BALB/c compared to Ifnar −/− mice, but unlike earlier studies (Boonnak et al., 2014) we found that neither FasL-Fas or TRAIL-DR5 pathways nor intrinsic apoptosis pathways were involved in this process (Figure S5). Augmented T cell apoptosis likely contributed to suboptimal T cell responses (Figure 6). Since T cells dampen cytokine storms by suppressing the innate response (Kim et al., 2007), suboptimal T cell responses may result in unchecked innate immune responses, causing prolonged production of pro-inflammatory mediators and subsequent immunopathology.
In summary, we demonstrate that delayed expression of IFN-I orchestrates the induction of an inappropriate inflammatory response and consequent lung immunopathology during SARS-CoV infection. Moreover, minimal differences in lung virus loads in Ifnar −/− and BALB/c mice and complete protection after IMM depletion suggest that IFN-I-dependent immunopathological events, largely independent of virus replication, promote SARS-CoV morbidity and mortality. The protective effect of early, but not late, IFN-I administration in SARS-CoV-infected mice (Figure 2D; Haagmans et al., 2004, Kumaki et al., 2011) and pathogenic effects of a delayed IFN-I response suggest that IFN-I should be used judiciously in infected patients. Further, in addition to reducing initial viral load through anti-viral interventions, therapeutic approaches that moderate immunopathology may help reduce the high case fatality rates associated with emerging coronaviruses and perhaps other high pathogenic virus infections.
Experimental Procedures
Mice and Viruses
Specific pathogen-free young (7–9 weeks) and middle-aged (8–9 months) female BALB/c mice were purchased from the National Cancer Institute and Charles River Laboratories International or bred at the University of Iowa. Both groups were equivalently susceptible to infection with mouse-adapted SARS-CoV. Ifnar −/− mice on a BALB/c background were obtained from Dr. Joan Durbin (Rutgers-New Jersey Medical School). CD45.1 (Ly5.1) mice on a BALB/c background were provided by Dr. Lyse Norian (University of Iowa). Mice were bred and maintained in the University of Iowa animal care facility. All animal experiments were approved by the University of Iowa Institutional Animal Care and Use Committee. Mouse-adapted SARS-CoV (MA15 strain), obtained from Dr. Kanta Subbarao (NIH, Bethesda, MD), was propagated on Vero E6 cells. Mice were intranasally infected with 3 × 104 PFU SARS-CoV, 5 × 104 PFU MHV-1, or 500 TCID PR8 strain of IAV in 50 μl DMEM. All work with SARS-CoV was conducted in the University of Iowa Biosafety Level 3 (BSL3) Laboratory.
Virus Titers
Lung virus titers were obtained as previously described (Zhao et al., 2011).
Vascular Leakage
Infected mice were intravenously injected with 200 μl of Evan’s blue dye (1.0% in PBS) at day 5 p.i. After 30 min, mice were anesthetized, and lungs were perfused with 10 ml intracardial injection of PBS and harvested. Extravascular Evan’s blue was then extracted by overnight incubation in formamide at 56°C and quantified by spectrophotometric analysis.
Lung Cell Preparation for FACS Analysis and Intracellular Cytokine Staining
Mice were perfused via the right ventricle with 10 ml PBS. Cells were prepared from lungs as previously described (Zhao et al., 2011). In some cases, cells were analyzed for intracellular cytokine expression. All flow cytometry data were acquired on a BD FACSVerse (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Apoptosis Detection
106 cells were stained for Annexin V using an apoptosis detection kit (Annexin V Staining with Surface and Intracellular Staining kit, eBioscience).
In Situ CFSE Staining
CFSE (8 mM, Molecular Probes) was administered i.n. (50 μl) 6 hr before infection. Draining lymph nodes were harvested 18 hr p.i., and the percentage and total number of rDCs were determined (Legge and Braciale, 2005, Zhao et al., 2011).
Mouse Bone Marrow Chimera
Bone marrow cells were extracted from femurs and tibia of BALB/c, Ifnar −/−, and CD45.1 BALB/c mice (all 6 weeks old) and filtered through a 70 μm nylon filter, and red blood cells were lysed using ACK buffer. Isolated bone marrow cells (1 × 107 cells) were adoptively transferred into lethally irradiated (750 rads) BALB/c or Ifnar −/− mice. Chimeric mice (BALB/c[Ly5.2]→BALB/c[Ly5.1], BALB/c[Ly5.1]→ Ifnar −/−[Ly5.2], Ifnar −/−[Ly5.2]→ BALB/c[Ly5.1], and Ifnar −/−[Ly5.2]→ Ifnar −/−[Ly5.2]) were maintained on water supplemented with antibiotics for 4 weeks to prevent opportunistic infections. Reconstitution was verified 5 weeks after bone marrow transfer by FACS analysis of PBMCs. 6 weeks after bone marrow transfer, mice were infected with SARS-CoV (103 PFU) and monitored for weight loss and survival. IMM accumulation in lungs was enumerated in chimera mice at day 3 p.i.
Lung Histology and Immunohistochemistry
Lungs were removed, fixed in zinc formalin, and paraffin embedded prior to staining with H&E. Viral antigen was detected using rabbit anti-N protein (1:1,000) (IMG548; IMGENEX) followed by labeling with biotinylated goat anti-rabbit IgG (1:200). Samples were developed with 3,3'-diaminobenzidine for 3 min.
Statistical Analysis
Data were analyzed using Student’s t test. Results in the graphs are represented as mean ± SEM, unless otherwise mentioned. ∗p ≤ 0.05, ∗∗ p ≤ 0.01, and ∗∗∗ p ≤ 0.001.
Author Contributions
R.C. and S.P. conceived the concept, planned the experiments, and wrote the manuscript with contributions from all authors. R.C., A.R.F., R.V., and J.Z. performed the experiments. D.K.M. analyzed histological and immunochemistry data. M.M. provided reagents.
Acknowledgments
We thank Dr. S. Varga for careful review of this manuscript. We thank Dr. Lyse Norian for providing Ly5.1 BALB/c mice and Dr. Steven Varga and Stacey Hartwig for providing anti-TNF antibody. This work was supported in part by grants from the N.I.H. (PO1 AI060699, RO1 AI091322).
Published: February 10, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2016.01.007.
Supplemental Information
References
- Aldridge J.R., Jr., Moseley C.E., Boltz D.A., Negovetich N.J., Reynolds C., Franks J., Brown S.A., Doherty P.C., Webster R.G., Thomas P.G. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci. USA. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbuch V., Brockstedt D.G., Meyer-Morse N., O’Riordan M., Portnoy D.A. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius A.L., Giurisato E., Cella M., Schreiber R.D., Shaw A.S., Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- Brandes M., Klauschen F., Kuchen S., Germain R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonnak K., Vogel L., Feldmann F., Feldmann H., Legge K.L., Subbarao K. Lymphopenia associated with highly virulent H5N1 virus infection due to plasmacytoid dendritic cell-mediated apoptosis of T cells. J. Immunol. 2014;192:5906–5912. doi: 10.4049/jimmunol.1302992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron M.J., Ran L., Xu L., Danesh A., Bermejo-Martin J.F., Cameron C.M., Muller M.P., Gold W.L., Richardson S.E., Poutanen S.M., Canadian SARS Research Network Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 2007;81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron M.J., Bermejo-Martin J.F., Danesh A., Muller M.P., Kelvin D.J. Human immunopathogenesis of severe acute respiratory syndrome (SARS) Virus Res. 2008;133:13–19. doi: 10.1016/j.virusres.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero J.A. Confounding roles for type I interferons during bacterial and viral pathogenesis. Int. Immunol. 2013;25:663–669. doi: 10.1093/intimm/dxt050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero J.A., Calderon B., Unanue E.R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragan L., Züst R., Weber F., Spiegel M., Lang K.S., Akira S., Thiel V., Ludewig B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragán L., Kalinke U., Züst R., König M., Reizis B., López-Macías C., Thiel V., Ludewig B. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J. Immunol. 2009;182:1099–1106. doi: 10.4049/jimmunol.182.2.1099. [DOI] [PubMed] [Google Scholar]
- Channappanavar R., Zhao J., Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014;59:118–128. doi: 10.1007/s12026-014-8534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Subbarao K. The Immunobiology of SARS. Annu. Rev. Immunol. 2007;25:443–472. doi: 10.1146/annurev.immunol.25.022106.141706. [DOI] [PubMed] [Google Scholar]
- Cheung C.Y., Poon L.L., Ng I.H., Luk W., Sia S.F., Wu M.H., Chan K.H., Yuen K.Y., Gordon S., Guan Y., Peiris J.S. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol. 2005;79:7819–7826. doi: 10.1128/JVI.79.12.7819-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.M., Poon L.L., Cheng V.C., Chan K.S., Hung I.F., Wong M.M., Chan K.H., Leung W.S., Tang B.S., Chan V.L. Initial viral load and the outcomes of SARS. CMAJ. 2004;171:1349–1352. doi: 10.1503/cmaj.1040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W., Fan Y., Wu W., Zhang F., Wang J.Y., Ni A.P. Expression of lymphocytes and lymphocyte subsets in patients with severe acute respiratory syndrome. Clin. Infect. Dis. 2003;37:857–859. doi: 10.1086/378587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigneault M., De Silva T.I., Bewley M.A., Preston J.A., Marriott H.M., Mitchell A.M., Mitchell T.J., Read R.C., Whyte M.K., Dockrell D.H. Monocytes regulate the mechanism of T-cell death by inducing Fas-mediated apoptosis during bacterial infection. PLoS Pathog. 2012;8:e1002814. doi: 10.1371/journal.ppat.1002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S., Crotta S., McCabe T.M., Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014;5:3864. doi: 10.1038/ncomms4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks T.J., Chong P.Y., Chui P., Galvin J.R., Lourens R.M., Reid A.H., Selbs E., McEvoy C.P., Hayden C.D., Fukuoka J. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum. Pathol. 2003;34:743–748. doi: 10.1016/S0046-8177(03)00367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M.B., Chen J., Morrison T.E., Whitmore A., Funkhouser W., Ward J.M., Lamirande E.W., Roberts A., Heise M., Subbarao K., Baric R.S. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog. 2010;6:e1000849. doi: 10.1371/journal.ppat.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikura D., Chiba S., Muramatsu D., Kazumata M., Nakayama Y., Kawai T., Akira S., Kida H., Miyazaki T. Type-I interferon is critical for FasL expression on lung cells to determine the severity of influenza. PLoS ONE. 2013;8:e55321. doi: 10.1371/journal.pone.0055321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X.Y., Li J.L., Yang X.L., Chmura A.A., Zhu G., Epstein J.H., Mazet J.K., Hu B., Zhang W., Peng C. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goritzka M., Makris S., Kausar F., Durant L.R., Pereira C., Kumagai Y., Culley F.J., Mack M., Akira S., Johansson C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015;212:699–714. doi: 10.1084/jem.20140825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski L.E., Baric R.S. Molecular pathology of emerging coronavirus infections. J. Pathol. 2015;235:185–195. doi: 10.1002/path.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Gong E., Zhang B., Zheng J., Gao Z., Zhong Y., Zou W., Zhan J., Wang S., Xie Z. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005;202:415–424. doi: 10.1084/jem.20050828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagmans B.L., Kuiken T., Martina B.E., Fouchier R.A., Rimmelzwaan G.F., van Amerongen G., van Riel D., de Jong T., Itamura S., Chan K.H. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat. Med. 2004;10:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Yamaguchi N., Nakayama M., Eto H., Okumura K., Yagita H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999;189:1451–1460. doi: 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killip M.J., Fodor E., Randall R.E. Influenza virus activation of the interferon system. Virus Res. 2015;209:11–22. doi: 10.1016/j.virusres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.D., Zhao J., Auh S., Yang X., Du P., Tang H., Fu Y.X. Adaptive immune cells temper initial innate responses. Nat. Med. 2007;13:1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki Y., Ennis J., Rahbar R., Turner J.D., Wandersee M.K., Smith A.J., Bailey K.W., Vest Z.G., Madsen J.R., Li J.K., Barnard D.L. Single-dose intranasal administration with mDEF201 (adenovirus vectored mouse interferon-alpha) confers protection from mortality in a lethal SARS-CoV BALB/c mouse model. Antiviral Res. 2011;89:75–82. doi: 10.1016/j.antiviral.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law H.K., Cheung C.Y., Ng H.Y., Sia S.F., Chan Y.O., Luk W., Nicholls J.M., Peiris J.S., Lau Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106:2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legge K.L., Braciale T.J. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity. 2005;23:649–659. doi: 10.1016/j.immuni.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Lin K.L., Suzuki Y., Nakano H., Ramsburg E., Gunn M.D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- Mack M., Cihak J., Simonis C., Luckow B., Proudfoot A.E., Plachý J., Brühl H., Frink M., Anders H.J., Vielhauer V. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- Mahlakõiv T., Ritz D., Mordstein M., DeDiego M.L., Enjuanes L., Müller M.A., Drosten C., Staeheli P. Combined action of type I and type III interferon restricts initial replication of severe acute respiratory syndrome coronavirus in the lung but fails to inhibit systemic virus spread. J. Gen. Virol. 2012;93:2601–2605. doi: 10.1099/vir.0.046284-0. [DOI] [PubMed] [Google Scholar]
- Majer O., Bourgeois C., Zwolanek F., Lassnig C., Kerjaschki D., Mack M., Müller M., Kuchler K. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog. 2012;8:e1002811. doi: 10.1371/journal.ppat.1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber K.D., Andrade B.B., Oland S.D., Amaral E.P., Barber D.L., Gonzales J., Derrick S.C., Shi R., Kumar N.P., Wei W. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F., Mayer-Barber K., Sher A., Wack A., O’Garra A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Eisfeld A.J., Schäfer A., Josset L., Sims A.C., Proll S., Fan S., Li C., Neumann G., Tilton S.C. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. MBio. 2014;5 doi: 10.1128/mBio.01174-14. e01174–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Yount B.L., Jr., Debbink K., Agnihothram S., Gralinski L.E., Plante J.A., Graham R.L., Scobey T., Ge X.Y., Donaldson E.F. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015;21:1508–1513. doi: 10.1038/nm.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls J.M., Poon L.L., Lee K.C., Ng W.F., Lai S.T., Leung C.Y., Chu C.M., Hui P.K., Mak K.L., Lim W. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page C., Goicochea L., Matthews K., Zhang Y., Klover P., Holtzman M.J., Hennighausen L., Frieman M. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J. Virol. 2012;86:13334–13349. doi: 10.1128/JVI.01689-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Guan Y., Yuen K.Y. Severe acute respiratory syndrome. Nat. Med. 2004;10(12, Suppl):S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A., Deming D., Paddock C.D., Cheng A., Yount B., Vogel L., Herman B.D., Sheahan T., Heise M., Genrich G.L. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007;3:e5. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler N.G., Bosinger S.E., Estes J.D., Zhu R.T., Tharp G.K., Boritz E., Levin D., Wijeyesinghe S., Makamdop K.N., del Prete G.Q. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S.U., Kwon H.J., Ko H.J., Byun Y.H., Seong B.L., Uematsu S., Akira S., Kweon M.N. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. 2011;7:e1001304. doi: 10.1371/journal.ppat.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina N.V., Salazar-Mather T.P., Biron C.A., Kuziel W.A., Pamer E.G. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C., Lee J.S., Gilfillan S., Robinette M.L., Newberry R.D., Stappenbeck T.S., Mack M., Cella M., Colonna M. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med. 2015;212:1869–1882. doi: 10.1084/jem.20151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M., Schneider K., Weber F., Weidmann M., Hufert F.T. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 2006;87:1953–1960. doi: 10.1099/vir.0.81624-0. [DOI] [PubMed] [Google Scholar]
- Swiecki M., Wang Y., Riboldi E., Kim A.H., Dzutsev A., Gilfillan S., Vermi W., Ruedl C., Trinchieri G., Colonna M. Cell depletion in mice that express diphtheria toxin receptor under the control of SiglecH encompasses more than plasmacytoid dendritic cells. J. Immunol. 2014;192:4409–4416. doi: 10.4049/jimmunol.1303135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro J.R., Ng C., Lee A.M., Sullivan B.M., Sheehan K.C., Welch M., Schreiber R.D., de la Torre J.C., Oldstone M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry R.L., Getts D.R., Deffrasnes C., van Vreden C., Campbell I.L., King N.J. Inflammatory monocytes and the pathogenesis of viral encephalitis. J. Neuroinflammation. 2012;9:270. doi: 10.1186/1742-2094-9-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totura A.L., Baric R.S. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012;2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Type I interferon: friend or foe? J. Exp. Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brand J.M., Haagmans B.L., van Riel D., Osterhaus A.D., Kuiken T. The pathology and pathogenesis of experimental severe acute respiratory syndrome and influenza in animal models. J. Comp. Pathol. 2014;151:83–112. doi: 10.1016/j.jcpa.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Swiecki M., Cella M., Alber G., Schreiber R.D., Gilfillan S., Colonna M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe. 2012;11:631–642. doi: 10.1016/j.chom.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh R.M., Bahl K., Marshall H.D., Urban S.L. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog. 2012;8:e1002352. doi: 10.1371/journal.ppat.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2016). Middle East respiratory syndrome coronavirus Update. http://www.who.int/emergencies/mers-cov/en/.
- Wilson E.B., Yamada D.H., Elsaesser H., Herskovitz J., Deng J., Cheng G., Aronow B.J., Karp C.L., Brooks D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.K., Lam C.W., Wu A.K., Ip W.K., Lee N.L., Chan I.H., Lit L.C., Hui D.S., Chan M.H., Chung S.S., Sung J.J. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004;136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen Y.T., Liao F., Hsiao C.H., Kao C.L., Chen Y.C., Wu-Hsieh B.A. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J. Virol. 2006;80:2684–2693. doi: 10.1128/JVI.80.6.2684-2693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhao J., Zhao J., Van Rooijen N., Perlman S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. 2009;5:e1000636. doi: 10.1371/journal.ppat.1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Zhao J., Perlman S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. 2010;84:9318–9325. doi: 10.1128/JVI.01049-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Zhao J., Legge K., Perlman S. Age-related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Invest. 2011;121:4921–4930. doi: 10.1172/JCI59777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zornetzer G.A., Frieman M.B., Rosenzweig E., Korth M.J., Page C., Baric R.S., Katze M.G. Transcriptomic analysis reveals a mechanism for a prefibrotic phenotype in STAT1 knockout mice during severe acute respiratory syndrome coronavirus infection. J. Virol. 2010;84:11297–11309. doi: 10.1128/JVI.01130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Hui D.S., Perlman S. Middle East respiratory syndrome. Lancet. 2015;386:995–1007. doi: 10.1016/S0140-6736(15)60454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.