

Abstract
Diabetic nephropathy (DN) is the most common cause of end-stage kidney disease worldwide and is associated with increased morbidity and mortality in patients with both type 1 and type 2 diabetes. Recent evidence revealed that dipeptidyl peptidase-4 (DPP-4) inhibitors may exhibit a protective effect against DN. In fact, the kidney is the organ where the DPP-4 activity is the highest level per organ weight. A preclinical analysis revealed that DPP-4 inhibitors also ameliorated kidney fibrosis. In this review, we analyzed recent reports in this field and explore the renoprotective effects and possible mechanism of the DPP-4 inhibitors.
Keywords: Diabetic nephropathy, Dipeptidyl peptidase-4, Kidney protection
Background
Diabetes mellitus has become a major global health issue [1].The number of people with diabetes worldwide is expected to rise from 382 million in 2013 to 592 million by 2035, according to the International Diabetes Federation [2]. Diabetic nephropathy (DN) is one of the most devastating complications of diabetes [3, 4]. The risk of DN is tightly linked to poor glucose control in both type 1 and type 2 diabetes, which is associated with hyperglycemia [5, 6], and the impacts of hyperglycemia are generally mediated through diverse metabolic pathways, including increased reactive oxygen species formation, excessive production of advanced glycation end products (AGEs), and the activation of the polyol, protein kinase C (PKC), and hexosamine pathways [7]. The activation of these pathways leads to a complex dysregulation of various effector molecules, resulting in cellular damage and dysfunction [7]. Experimental studies have shown that some of these pathophysiological mechanisms may be modified by dipeptidyl peptidase-4 (DPP-4) inhibition [8, 9], and preclinical studies also suggest that DPP-4 inhibitors provide renoprotection above and beyond lowering the glucose levels through its protein-protein interactions and proteolytic and antioxidant properties [10]. In this review, we focus on the possible mechanisms by which DPP-4 inhibitors combat diabetic nephropathy, especially about kidney fibrosis.
Biology of DPP-4
DPP-4 is a cell surface aminopeptidase that was originally characterized as a T cell differentiation antigen (CD26). It is a multifunctional protein that exerts diverse biological activities, such as protease activity, association with adenosine deaminase (ADA), interaction with the extracellular matrix, cell surface co-receptor activity to mediate viral entry, and regulation of intracellular signal transduction coupled to the control of cell migration and proliferation [11–15]. DPP-4 is expressed ubiquitously and found in many cell types, including the endothelial cells in multiple vascular beds, rendering the enzyme highly accessible to the peptide substrates circulating through the gut, liver, lung, and kidney [16].
DPP-4 is a member of the serine peptidase/prolyl oligopeptidase gene family. The members of this family are often classified into subgroups according to their structure and function and include: the membrane-bound peptidases: fibroblast activation protein (FAP)/seprase; the resident cytoplasmic enzymes: DPP-8 and DPP-9; and the nonenzymatic peptidases: DPP-6 and DPP-10 [17]. The complexity of DPP-4’s action is amplified by the panoply of bioactive DPP-4 substrates, which, in turn, act as elegant biochemical messengers in multiple tissues, including the immune and neuroendocrine systems. More than 30 peptide substrates for DPP-4 have been identified, including glucagon-like peptide-1(GLP-1), glucose-dependent insulinotropic peptide (GIP) [17], brain natriuretic peptide 1–32 [18, 19], neuropeptide Y [20], high mobility group protein 1 (HMGB1) [21], and others [20, 22, 23].
DPP-4 transmits signals across the cell membranes and interacts with other membrane proteins. The DPP-4 gene encodes a type II transmembrane protein of 766 amino acids, which is anchored to the lipid bilayer by a single hydrophobic segment located at the N-terminus and has a short cytoplasmic tail of six amino acids [24] (29). The extracellular portion of DPP-4 contains a glycosylation domain, a cysteine-rich domain, and a catalytic domain [17]. Mutation studies demonstrated that the C-terminal loop of DPP-4 is essential for both dimer formation and catalytic efficacy [25]. An analysis of the crystal structure revealed that DPP-4 can also form tetramers between two soluble DPP-4 proteins and two membrane-bound DPP-4 proteins. These interactions may influence the efficiency of the entry and cleavage of substrates by the catalytic site or allow cell-cell communication [25]. Catalytically active DPP-4 is liberated from the plasma membrane to produce a soluble circulating form, sDPP-4 (727 aa), which lacks the intracellular tail and transmembrane regions and accounts for a substantial proportion of DPP-4 activity in human serum [26, 27]. Membrane-bound DPP-4 contains residues 1–766, whereas sDPP-4 contains residues 39–766. sDPP-4 lacks the cytoplasmic domain [residues 1–6], transmembrane domain [residues 7–28], and the flexible stalk [residues 29–39] [17, 26] (Fig. 1). sDPP-4 can activate intracellular signaling pathways and increases the proliferation of human lymphocytes, independent of either its catalytic activity [28] or binding to ADA [28]. sDPP-4 impairs insulin-mediated activation of Akt in the human adipocyte, skeletal muscle, and smooth muscle cells in vitro [29]. However, the mechanisms by which sDPP-4 activates signal transduction are poorly understood.
Fig. 1.
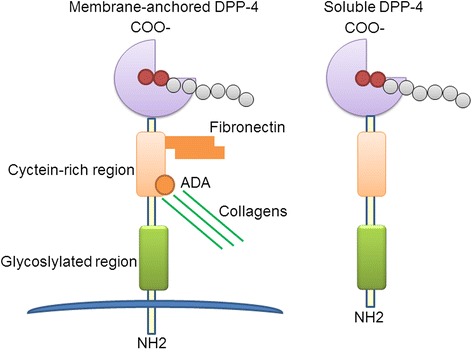
Membrane-anchored DPP-4 and soluble DPP-4. Catalytically active DPP-4 is liberated from the plasma membrane to produce a soluble circulating form, sDPP-4, which lacks the intracellular tail and transmembrane regions and accounts for a substantial proportion of DPP-4 activity. In addition to its exopeptidase activity, DPP-4 also functions as a binding protein which can bind with fibronectin and adenosine deaminase (ADA)
DPP-4 activity is subject to regulation at many levels, including control of gene and protein expression, interactions with its binding partners, and modulation of its enzyme activity. The importance of cytokines in regulating DPP-4 activity was demonstrated in chronic B lymphocytic leukemia cells, leading to the upregulation of both intracellular and cell surface DPP-4 expression, as well as DPP-4 activity [30]. Cell surface and intracellular DPP-4 expression is also highly regulated; it is often expressed at low levels under basal conditions, and then markedly induced by stimulation, such as T cell activation by mitogenic or antigenic stimuli [31]. The control of DPP-4 shedding, which generates sDPP-4, is poorly understood. Lamers et al. found that TNF-α and insulin increased the release of sDPP-4, although there were no changes in the expression of the DPP-4 mRNA in human adipocytes isolated from visceral depots [29]. Some other studies have focused on the origin of sDPP-4 and found that kidney extracts exhibited the highest DPP-4 activity; however, the most important source of sDPP-4 is the bone marrow and not the kidney [32]. These studies highlight our limited understanding of the cell types and tissues that contribute to the generation of sDPP-4 activity in the plasma in vivo. The characterization of these cells, evaluation of additional sources for sDPP-4, and the mechanism of release require additional studies.
The role of DPP-4 inhibitors in diabetes and kidney protection
In general, incretins are a group of metabolic hormones that stimulate a decrease in the blood glucose levels, by either increasing insulin release or reducing gastrointestinal absorption. The prototypical incretins are the intestinal GLP-1 and GIP hormones, and both GLP-1 and GIP are rapidly inactivated by the enzyme DPP-4. Therefore, DPP-4 is a well-documented drug target for the treatment of type 2 diabetes [33–35]. The pharmacological inhibition of DPP-4 results in GLP-1 accumulation, which stimulates insulin secretion and contributes to the reduction of postprandial hyperglycemia. Recently, the beneficial pleiotropic effects of DPP-4 inhibitors were reported in both clinical research and preclinical experiments.
Two types of DPP-4 inhibitors have been used in the clinic: dipeptide structure mimetics and non-peptidomimetics. The first type includes sitagliptin (approved by FDA in 2006), vildagliptin (approved by European Medicines Agency in 2007), and saxagliptin (approved by FDA in 2009), while the non-peptidomimetics include linagliptin (xanthine-based, approved by FDA in 2011) and alogliptin (modified pyrimidinedione, approved by FDA in 2013). These are all small molecules that are rapidly absorbed following oral dosing, resulting in greater than 80% inhibition of the DPP-4 activity over a full 24-h period. Typically, these drugs raise the peripheral plasma concentration of the intact forms of both incretins by two- to three-fold. Although this class differs widely in their chemistry, they are all selective for DPP-4 [22]. Clinical proof of concept for DPP-4 inhibition in diabetic patients was first reported in 2002, where a DPP-4 inhibitor significantly reduced the fasting plasma glucose and HbA1c levels in a 4-week study [36]. DPP-4 expression positively correlates with the amount of visceral adipose tissue, adipocyte size, inflammation, and HbA1c levels, and negatively correlates with the glucose infusion rates during euglycemic-hyperinsulinemic clamp [37, 38]. The efficacy of the DPP-4 inhibitors in reducing glycemia is weaker than that of sulfonylureas, insulin, and thiazolidinediones, but they are significantly better tolerated and do not produce weight gain [39–41]. Furthermore, DPP-4 inhibitors not only have benefits for patients who have recently developed diabetes [42] but also in patients with long-standing diabetes [43].
In addition to their glucose-lowering action, DPP-4 inhibitors have been demonstrated to play a protective role in cardiovascular diseases, including hypertension [44], abdominal aortic aneurysm [45], cardiomyopathy [46], atherosclerosis [47], and peripheral vascular disease [48], via both GLP-1-dependent and GLP-1-independent pathways due to their diverse, widely distributed, and pleiotropic actions [49]. Many in vivo and in vitro studies also found that DPP-4 inhibitors can prevent organ fibrosis, including cardiac fibrosis [50, 51], hepatic fibrosis [52], and kidney fibrosis [53]. DPP-4 is localized on the surface of many cell types, including the endothelial cells, kidney epithelial cells, and T cells, where they have a binding partner and transmit intracellular signals [54]. In fact, the kidney expresses the highest levels of DPP-4 per organ weight [20]. Moreover, the mammalian kidney has high concentrations of DPP-4 [20], and the expression of DPP-4 is increased in cultured human renal glomerular epithelial cells during inflammation [55] and in a rat model of type 2 diabetes mellitus [56]. Several candidates for the GLP-1-independent effects of DPP-4 inhibitors in the kidney have been identified and include the known substrates of DPP-4 cleavage, such as HMGB1, Meprin β, neuropeptide Y (NPY), and peptide YY (PYY) [57]. Thus, some researchers have demonstrated that increased DPP-4 activity in the kidney or urine is a hallmark for human glomerular diseases [54, 58]. Some of the DPP-4 inhibitors were analyzed to confirm their role in the kidney. In animal models, a reduction of albuminuria and an improvement in the histological changes in the kidney were observed in T1DM models treated with vildagliptin [59] and in T2DM models treated with sitagliptin [60]. A significant reduction in urinary albumin excretion was also observed in diabetic endothelial nitric oxide synthase knockout mice treated with linagliptin in addition to an Ang II receptor antagonist [8]. In the experimental model of renal ischemia/reperfusion injury treated with vildagliptin, DPP-4 inhibition produced nephroprotective effects that were mediated by antiapoptotic, anti-inflammatory, and anti-oxidative effects [61]. Linagliptin is the recently approved DPP-4 inhibitor and exhibits non-linear pharmacokinetic properties, with a less than dose proportional profile and is almost completely eliminated via the enteric system, with less than 5% found in urine [62]. In humans, it was shown that linagliptin significantly reduced the urinary albumin excretion of patients with T2DM after 24 weeks of treatment [63]. The pharmacokinetic profiles after linagliptin administration showed the accumulation t1/2 of linagliptin following multiple 5 mg/day doses, and there were no significant changes in the control and diabetic subjects, regardless of their renal function [64]. The antifibrotic properties of DPP-4 inhibitors have also been shown in other models of kidney fibrosis, such as the unilateral ureteral obstruction (UUO) model, where the administration of a novel DPP-4 inhibitor, LC15-0444, resulted in a significant decrease in albuminuria, the urinary excretion of 8-isoprostane, and renal fibrosis [65]. In our newest study, we found that linagliptin restored the normal kidney structure of streptozotocin (STZ)-induced diabetic kidney fibrosis in CD-1 mice without altering the blood pressure, body weights, blood sugar levels, or organ weights (of the kidney, liver, and heart) compared with the untreated diabetic mice [53].
Transforming growth factor β (TGFβ) is the primary cytokine that drives fibrosis in the kidney and other organs that are susceptible to fibrotic injury, such as the lung and liver. Members of the TGFβ superfamily transduce intracellular signals through Smad proteins. A study demonstrates that TGFβ signals can mediate renal fibrosis through Smad2/3 [66]. DPP-4 inhibitors may ameliorate diabetic nephropathy and reduce the overproduction of TGF-β1. Renoprotection is attributed to the inhibition of DPP-4 activity, which mimics incretin action, and the activation of the GLP-1R [59]. In our previous study, STZ-induced diabetic mice treated with the DPP-4 inhibitor linagliptin exhibited a suppression of DPP-4 activity/protein expression and an amelioration of kidney fibrosis associated with the inhibition of the endothelial-to-mesenchymal transition (EndMT) and TGF-β2-induced Smad3 phosphorylation [53]. EndMT was first discovered in heart development [9], which was confirmed to be crucially important in forming the valves and septa of the heart during embryogenesis [67, 68]. EndMT contributes to the accumulation of activated fibroblasts and myofibroblasts in kidney fibrosis [69], fibroblasts are key mediators of fibrosis in the kidney and other organs [70]. Chronic kidney disease (CKD) is associated with an increase in circulating angiogenesis and NO inhibitors, which impact proliferation and apoptosis of cardiac endothelial cells and promote EndMT, leading to cardiac fibrosis and capillary rarefaction [71]. EndMT is not only involved in kidney fibrosis progress, Zeisberg et al. confirmed that EndMT also involved in heart and tumor progression [72–77]. In this regard, linagliptin could exhibit an antifibrotic effect through a mechanism that specifically targets endothelial cells [53, 78, 79]. DPP-4 is essential for TGF-β-induced receptor hetero-dimerization and subsequent intracellular signal transduction, including the levels of TGF-βRs and the protein-protein interactions of TGF-βRs, both of which are critical for TGF-β signal transduction. We found that the TGF-β2-induced formation of the TGF-βR1/2 heterodimer was suppressed in the DPP-4 siRNA-transfected endothelial cells compared with the cells transfected with a control siRNA [79]. In a UUO model, a DPP-4 inhibitor, LC15-0444, reduced the levels of inflammatory and fibrotic markers, such as phosphorylated Smad2/3, TGF-β1, toll-like receptor 4, HMGB1, NADPH oxidase4, and nuclear factor kappa B [65]. These results suggest that the activation of DPP-4 in the kidney has an important role in TGF-β signaling and the progression of renal disease and that targeted therapy that inhibits DPP-4 may prove to be a useful new approach in the management of progressive renal disease, including kidney fibrosis.
Interaction of DPP-4 and integrin β1 in kidney fibrosis
Integrins exist as αβ heterodimers that are formed from 18 α- and 8 β-subunits, each of which exhibits different ligand binding and signaling properties [80]. Each integrin subunit consists of an extracellular domain, which determines the ligand binding properties, a transmembrane domain, and a short cytoplasmic tail that binds to multiple cytosolic and transmembrane proteins to form focal adhesions (with the exception of β4) [81]. Integrins bind to extracellular matrix (ECM) glycoproteins, including collagens, fibronectins, and laminins, and cellular receptors, such as vascular cell adhesion molecule-1 (VCAM-1) and members of the intercellular cell adhesion molecule (ICAM) family [82, 83]. In addition, integrins also play key roles in the assembly of the actin cytoskeleton as well as in modulating the signal transduction pathways that control biological and cellular functions, including cell adhesion, migration, proliferation, cell differentiation, and apoptosis [84]. Integrins are able to transduce signals intracellularly following ligand binding (“outside-in” signaling) [85]. Unlike most other cell receptors, integrins can shift between high- and low-affinity conformations for ligand binding (“inside-out” signaling) [86]. Depending on the cell type, integrins can be either basally activated, as is the case in most adherent cells that are attached to a basement membrane, or basally inactive, as is the case with platelets or leukocytes that freely circulate until they are activated to undergo platelet aggregation or mediate an inflammatory response, respectively. Integrins themselves have no kinase activity, but instead provide a connection between the ECM and the actin cytoskeleton. This connection allows integrins to regulate the cytoskeletal organization and cell motility, as well as to alter the fluxes of many intracellular-signaling pathways, including cell survival, cell proliferation, cell shape, and angiogenesis [86, 87]. Thus, integrins are critical for maintaining cellular homeostasis, triggering a number of signaling pathways under normal conditions; under pathological conditions, integrins are associated with a wide variety of renal pathologies, including obstructive nephropathy and DN [88–91].
Among the members of the integrin family, the β1-integrin is the most critical, given that β1-integrin can pair with different α-subunits, making it a receptor for many types of stimuli [92–95] (Fig. 2). Integrin β1 is ubiquitously expressed and can bind to multiple partners, and thus it is not surprising that knockout of β1-integrin results in embryonic lethality due to a complete inhibition of preimplantation development. In contrast, knockouts of the α1, α2, α10, and α11 integrin subunits, which each exclusively heterodimerize with β1 to function as primary collagen receptors, are all viable and fertile, but possess distinct characteristic abnormalities [96]. Focal adhesion kinase (FAK) is the most essential intracellular integrator in the integrin β1-FAK signaling pathway, and the abnormal redistribution and decreased expression of integrin β1 and FAK are important molecular events that regulate the functions of podocytes under abnormal hemodynamic conditions [97]. Expression of integrin β1 by fibroblasts is required for fibrogenesis. Blocking integrin β1 signaling can diminish the progression of cutaneous fibrosis [98]. Yeh et al. found that the expression of the β1-integrin mRNA and protein was significantly upregulated in UUO mice, which was accompanied by a corresponding elevation in the tubular expression of TGF-β1 [90]. The inhibition of β1-integrin signals reduced the TGF-β1 levels and ameliorated fibrosis, demonstrating strong correlations between the expression of β1-integrin within the tubulointerstitium and the presence of tubulointerstitial fibrosis [90]. Hamzeh et al. also found that in the absence of β1-integrin, human proximal tubular cells fail to activate the signaling cascade that would lead to the synthesis of profibrotic proteins and, ultimately, to the development of renal fibrosis. They also showed that cyclic stretch-induced TGF-β1 and fibronectin expression is mediated by β1-integrin through c-Src- and STAT3-dependent pathways in renal epithelial cells [91].
Fig. 2.
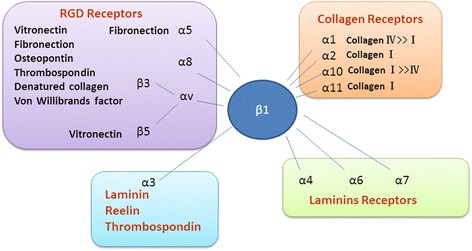
Integrin β1 receptors and their ligands. In the integrin family, the β1-integrin is the most critical, given that β1-integrin can pair with different α-subunits, making it a receptor for many types of stimuli
The loss of DPP-4 cell surface expression has shown to be associated with decreased phosphorylation of integrin β1 at the S785 residue, which has a key role in cellular adhesion to ECM [99]. In our most recent published paper, we identified a new profibrotic molecular mechanism that was associated with the interaction between DPP-4 and integrin β1 [79] (Fig. 3). The DPP-4-associated EndMT was inhibited by integrin β1 deletion. In addition, DPP-4 or integrin β1 deficiency resulted in the inhibition of TGF-β2-stimulated hetero-dimerization of TGF-βRs. Finally, the interaction between DPP-4 and integrin β1 induced vascular endothelial growth factor-receptor (VEGF-R)1 expression, with the concomitant suppression of VEGF-R2 levels [79]. This is relevant because VEGF is the most prominent stimulus for endothelial cells and angiogenesis. The endothelial cell responses toward VEGF are modulated by distinct VEGF receptors, as VEGFR1 favors the EndMT, whereas VEGFR2 counteracts the EndMT [100]. These results indicate that the interaction between DPP-4 and integrin β1 may be a therapeutic target for kidney fibrosis in diabetes [79].
Fig. 3.
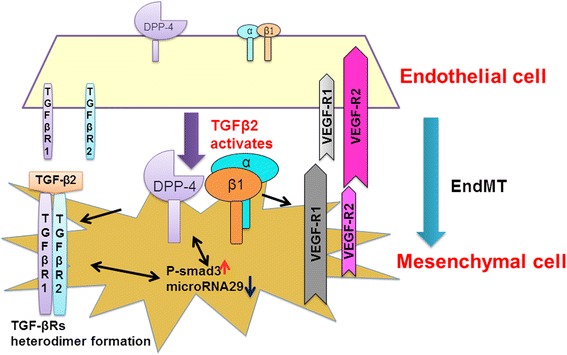
Interaction of DPP-4 and integrin β1 in the endothelial cells. Interaction between DPP-4 and integrin β1 displays key role in the TGF-β-induced signal transductions and VEGF-induced survival signaling in endothelial cells as well as in the subsequent induction of EndMT, which is associated with regulation of miR-29s
MicroRNAs and DPP-4 in the kidney
The cumulative effects of hyperglycemia, inflammatory cytokines, proteinuria, ageing, high blood pressure, and hypoxia result in alterations of the miRNA expression profiles. The altered miRNA levels initiate a transition program in the normal kidney that ultimately leads to fibrosis. MicroRNAs (miRs) were discovered 20 years ago. The actions and synthesis of miRs are tightly regulated. The key antifibrotic miRs miR-let-7s and miR-29s are involved in suppression and are important for understanding the fibrotic mechanism in the diabetic kidney [53, 101, 102]. According to the prediction of microRNA targets by TargetScan (http://www.targetscan.org/vert_60/), we identified the miR29 bind site in 3′UTR of DPP-4 [53]. By cloning and utilizing the reporter vector containing 3′UTR legends of human DPP-4 mRNA, we have confirmed that miR29 binding site in DPP-4 3′UTR negatively regulated DPP-4 gene expression. In diabetic kidney, the increased DPP-4 levels were associated with the suppression of miR29s when compared with the normoglycemic kidney [53] (Fig. 4). Linagliptin, a DPP-4 inhibitor, ameliorates the kidney functions by inducing miR-29 expression in the diabetic kidney model [53]. A quantitative analysis revealed that microRNAs 29 a, b, and c were suppressed in the diabetic kidney compared with the control kidneys, and linagliptin restored the diabetes-suppressed microRNA 29s levels. Similarly, the TGF-β2-suppressed microRNA29s levels were restored by linagliptin in vitro (Fig. 5). These molecules exhibited similar antifibrotic mechanisms, such as anti-EndMT and anti-TGF-β/Smad signaling effects [53]. A microRNA array analysis of the kidney samples revealed that the expression of the mmu-let-7 family of microRNAs was suppressed in the diabetic kidney [101]. Blockade of FGF signaling induced an EndMT program that can be mimicked by let-7b or let-7c miRNA inhibition [103, 104], and the FGF receptor-microRNA let-7 family axis can suppress the TGF-β receptor I levels [101]. DPP-4 inhibitors have been shown to inhibit the EndMT, and thus may regulate expression levels of miR-let-7s in the diabetic kidney. Additionally, microRNA23 and microRNA 21 have been shown to have an important role in the EndMT and kidney fibrosis [105, 106]; the regulation of these microRNAs by DPP-4 inhibitors must be analyzed to determine the detailed mechanism of kidney fibrosis.
Fig. 4.
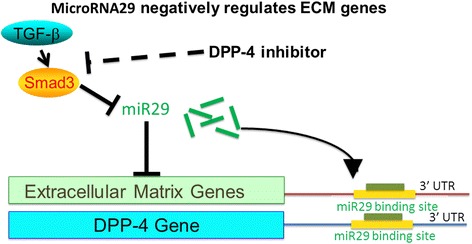
DPP-4 3′UTR and microRNA 29. TGF-β2-stimulated luciferase activity of 3′UTR fragment of DPP-4, where microRNA 29 binding site was involved, DPP-4 inhibitor may restore the miR29 levels by inhibiting TGF-β/Smad3 signaling
Fig. 5.
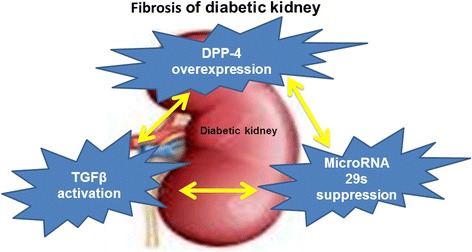
Fibrosis of diabetic kidney. Diabetic kidney fibrosis is associated with suppression of microRNA29s, which targets both DPP-4 protein levels and TGFβ-activating process
Perspective
In a Zucker Diabetic Fatty rat model, Takai et al. found there was no significant difference in the blood glucose and plasma insulin concentrations between the sitagliptin- and linagliptin-treated groups, but the DPP-4 activity in the plasma and vascular tissues of the linagliptin-treated group was significantly lower than those in the sitagliptin-treated group [108]. Another study found that CD26/DPP-4 was localized to the nucleus, and its nuclear translocation was enhanced by an anti-CD26 monoclonal antibody, suggesting that DPP-4 inhibition helps the DPP-4 on the cell surface move into the nucleus [109]. These data suggest that DPP-4 can be expressed on the membrane and in the nucleus. Although every DPP-4 inhibitor displays similar role in suppressing DPP-4 activity in the plasma and other tissues, each DPP-4 inhibitor might exert unique, drug-specific effects. Indeed, we have recently reported that Linagliptin can suppress all of the following: DPP-4 activity and protein level, integrin β1 protein levels, EndMT, DPP-4 3’UTR activity, and VEGF-R1 induction/-R2 suppression; Sitagliptin, inhibited none of these [110]. Future studies need to focus on the molecular mechanisms of the DPP-4 inhibitors in different organs and cells.
Conclusions
The present review describes various aspects and possible mechanisms by which DPP-4 inhibitors combat kidney fibrosis. The activation of DPP-4 in the kidney has an important role in TGF-β signaling, and the progression of renal disease by regulating the microRNA29s levels, and that targeted the inhibition of DPP-4 may prove to be a useful new approach in the management of progressive renal disease, including kidney fibrosis.
Acknowledgements
This work was partially supported by grants from the Japan Society for the Promotion of Science to KK (23790381) and DK (25282028 and 25670414) and research grants from the Japan Research Foundation for Clinical Pharmacology to K.K. (2011). This work was partially supported by a Grant for Collaborative Research awarded to DK (C2011-4, C2012-1) and a Grant for Promoted Research awarded to KK (S2011-1, S2012-5) from Kanazawa Medical University. S Shi was supported by foreign scholar grants from Kanazawa Medical University. KK and DK received lecture fees from Boehringer Ingelheim and Eli Lilly. Both Boehringer Ingelheim and Eli Lilly donated funds to Kanazawa Medical University and were not directly associated with this project. Boehringer Ingelheim, Mitsubishi Tanabe Pharma, and Ono Pharmaceutical contributed funds to establish the Division of Anticipatory Molecular Food Science and Technology. KK is under a consulting contract with Boehringer Ingelheim.
Abbreviations
- ADA
adenosine deaminase
- AGEs
advanced glycation end products
- DN
diabetic nephropathy
- DPP-4
dipeptidyl peptidase-4
- ECM
extracellular matrix
- EndMT
endothelial-to-mesenchymal transition
- FAK
focal adhesion kinase
- FAP
fibroblast activation protein
- GIP
glucose-dependent insulinotropic peptide
- GLP-1
glucagon-like peptide-1
- HMGB1
high-mobility group protein 1
- ICAM
intercellular cell adhesion molecule
- NPY
neuropeptide Y
- PKC
protein kinase C
- TGF-β
transforming growth factor-β
- UUO
unilateral ureteral obstruction
- VEGF-R
vascular endothelial growth factor-receptor
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
SS designed the study, wrote the review manuscript, and was involved in the discussion. DK made intellectual contributions. KK conceived of the project, provided intellectual contribution, and guided the manuscript writing and editing. All authors read and approved of the final manuscript.
References
- 1.Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes care. 2011;34(6):1249–57. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi Y, Hu FB. The global implications of diabetes and cancer. Lancet. 2014;383(9933):1947–8. doi: 10.1016/S0140-6736(14)60886-2. [DOI] [PubMed] [Google Scholar]
- 3.Parving HH. Diabetic nephropathy: prevention and treatment. Kidney international. 2001;60(5):2041–55. doi: 10.1046/j.1523-1755.2001.00020.x. [DOI] [PubMed] [Google Scholar]
- 4.Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in patients with type 2 diabetes. The New England journal of medicine. 2002;346(15):1145–51. doi: 10.1056/NEJMcp011773. [DOI] [PubMed] [Google Scholar]
- 5.Mazze RS, Bergenstal R, Ginsberg B. Intensified diabetes management: lessons from the diabetes control and complications trial. Int J Clin Pharmacol Ther. 1995;33(1):43–51. [PubMed] [Google Scholar]
- 6.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. Bmj. 2000;321(7258):405–12. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satchell SC, Tooke JE. What is the mechanism of microalbuminuria in diabetes: a role for the glomerular endothelium? Diabetologia. 2008;51(5):714–25. doi: 10.1007/s00125-008-0961-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alter ML, Ott IM, von Websky K, Tsuprykov O, Sharkovska Y, Krause-Relle K, et al. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney & blood pressure research. 2012;36(1):119–30. doi: 10.1159/000341487. [DOI] [PubMed] [Google Scholar]
- 9.Yamagishi S, Fukami K, Matsui T. Crosstalk between advanced glycation end products (AGEs)-receptor RAGE axis and dipeptidyl peptidase-4-incretin system in diabetic vascular complications. Cardiovasc Diabetol. 2015;14:2. doi: 10.1186/s12933-015-0176-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haluzik M, Frolik J, Rychlik I. Renal effects of DPP-4 inhibitors: a focus on microalbuminuria. Int J Endocrinol. 2013;2013:895102. doi: 10.1155/2013/895102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambeir AM, Durinx C, Scharpe S, De Meester I. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci. 2003;40(3):209–94. doi: 10.1080/713609354. [DOI] [PubMed] [Google Scholar]
- 12.Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science. 1993;261(5120):466–9. doi: 10.1126/science.8101391. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7(10):800–8. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 14.Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013;500(7461):227–31. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahne T, Lendeckel U, Wrenger S, Neubert K, Ansorge S, Reinhold D. Dipeptidyl peptidase IV: a cell surface peptidase involved in regulating T cell growth (review) Int J Mol Med. 1999;4(1):3–15. doi: 10.3892/ijmm.4.1.3. [DOI] [PubMed] [Google Scholar]
- 16.Deacon CF. What do we know about the secretion and degradation of incretin hormones? Regul Pept. 2005;128(2):117–24. doi: 10.1016/j.regpep.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 17.Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. 2014;35(6):992–1019. doi: 10.1210/er.2014-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boerrigter G, Costello-Boerrigter LC, Harty GJ, Lapp H, Burnett JC., Jr Des-serine-proline brain natriuretic peptide 3–32 in cardiorenal regulation. Am J Physiol Regul Integr Comp Physiol. 2007;292(2):R897–901. doi: 10.1152/ajpregu.00569.2006. [DOI] [PubMed] [Google Scholar]
- 19.Brandt I, Lambeir AM, Ketelslegers JM, Vanderheyden M, Scharpe S, De Meester I. Dipeptidyl-peptidase IV converts intact B-type natriuretic peptide into its des-SerPro form. Clin Chem. 2006;52(1):82–7. doi: 10.1373/clinchem.2005.057638. [DOI] [PubMed] [Google Scholar]
- 20.Mentlein R. Dipeptidyl-peptidase IV, (CD26)—role in the inactivation of regulatory peptides. Regul Pept. 1999;85(1):9–24. doi: 10.1016/S0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- 21.Marchetti C, Di Carlo A, Facchiano F, Senatore C, De Cristofaro R, Luzi A, et al. High mobility group box 1 is a novel substrate of dipeptidyl peptidase-IV. Diabetologia. 2012;55(1):236–44. doi: 10.1007/s00125-011-2213-6. [DOI] [PubMed] [Google Scholar]
- 22.Kirby M, Yu DM, O'Connor S, Gorrell MD. Inhibitor selectivity in the clinical application of dipeptidyl peptidase-4 inhibition. Clinical science. 2010;118(1):31–41. doi: 10.1042/CS20090047. [DOI] [PubMed] [Google Scholar]
- 23.Gorrell MD, Gysbers V, McCaughan GW. CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol. 2001;54(3):249–64. doi: 10.1046/j.1365-3083.2001.00984.x. [DOI] [PubMed] [Google Scholar]
- 24.De Meester I, Korom S, Van Damme J, Scharpe S. CD26, let it cut or cut it down. Immunology today. 1999;20(8):367–75. doi: 10.1016/S0167-5699(99)01486-3. [DOI] [PubMed] [Google Scholar]
- 25.Chien CH, Huang LH, Chou CY, Chen YS, Han YS, Chang GG, et al. One site mutation disrupts dimer formation in human DPP-IV proteins. J Biol Chem. 2004;279(50):52338–45. doi: 10.1074/jbc.M406185200. [DOI] [PubMed] [Google Scholar]
- 26.Durinx C, Lambeir AM, Bosmans E, Falmagne JB, Berghmans R, Haemers A, et al. Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur J Biochem. 2000;267(17):5608–13. doi: 10.1046/j.1432-1327.2000.01634.x. [DOI] [PubMed] [Google Scholar]
- 27.Cordero OJ, Salgado FJ, Nogueira M. On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol Immunother. 2009;58(11):1723–47. doi: 10.1007/s00262-009-0728-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu DM, Slaitini L, Gysbers V, Riekhoff AG, Kahne T, Knott HM, et al. Soluble CD26 / dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand J Immunol. 2011;73(2):102–11. doi: 10.1111/j.1365-3083.2010.02488.x. [DOI] [PubMed] [Google Scholar]
- 29.Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60(7):1917–25. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauvois B, Djavaheri-Mergny M, Rouillard D, Dumont J, Wietzerbin J. Regulation of CD26/DPPIV gene expression by interferons and retinoic acid in tumor B cells. Oncogene. 2000;19(2):265–72. doi: 10.1038/sj.onc.1203292. [DOI] [PubMed] [Google Scholar]
- 31.Mattern T, Reich C, Duchrow M, Ansorge S, Ulmer AJ, Flad HD. Antibody-induced modulation of CD26 surface expression. Immunology. 1995;84(4):595–600. [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Grigo C, Steinbeck J, von Horsten S, Amann K, Daniel C. Soluble DPP4 originates in part from bone marrow cells and not from the kidney. Peptides. 2014;57:109–17. doi: 10.1016/j.peptides.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3(3):153–65. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Drucker DJ. Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes. Expert opinion on investigational drugs. 2003;12(1):87–100. doi: 10.1517/13543784.12.1.87. [DOI] [PubMed] [Google Scholar]
- 35.Fleischer B. CD26: a surface protease involved in T-cell activation. Immunology today. 1994;15(4):180–4. doi: 10.1016/0167-5699(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 36.Ahren B, Simonsson E, Larsson H, Landin-Olsson M, Torgeirsson H, Jansson PA, et al. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes care. 2002;25(5):869–75. doi: 10.2337/diacare.25.5.869. [DOI] [PubMed] [Google Scholar]
- 37.Sell H, Bluher M, Kloting N, Schlich R, Willems M, Ruppe F, et al. Adipose dipeptidyl peptidase-4 and obesity: correlation with insulin resistance and depot-specific release from adipose tissue in vivo and in vitro. Diabetes care. 2013;36(12):4083–90. doi: 10.2337/dc13-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong J, Maiseyeu A, Davis SN, Rajagopalan S. DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circulation research. 2015;116(8):1491–504. doi: 10.1161/CIRCRESAHA.116.305665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karagiannis T, Paschos P, Paletas K, Matthews DR, Tsapas A. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: systematic review and meta-analysis. Bmj. 2012;344:e1369. doi: 10.1136/bmj.e1369. [DOI] [PubMed] [Google Scholar]
- 40.Wu D, Li L, Liu C. Efficacy and safety of dipeptidyl peptidase-4 inhibitors and metformin as initial combination therapy and as monotherapy in patients with type 2 diabetes mellitus: a meta-analysis. Diabetes, obesity & metabolism. 2014;16(1):30–7. doi: 10.1111/dom.12174. [DOI] [PubMed] [Google Scholar]
- 41.Liu SC, Tu YK, Chien MN, Chien KL. Effect of antidiabetic agents added to metformin on glycaemic control, hypoglycaemia and weight change in patients with type 2 diabetes: a network meta-analysis. Diabetes, obesity & metabolism. 2012;14(9):810–20. doi: 10.1111/j.1463-1326.2012.01606.x. [DOI] [PubMed] [Google Scholar]
- 42.Richter B, Bandeira-Echtler E, Bergerhoff K, Lerch CL. Dipeptidyl peptidase-4 (DPP-4) inhibitors for type 2 diabetes mellitus. The Cochrane database of systematic reviews. 2008;2:CD006739. doi: 10.1002/14651858.CD006739.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar KV, Gupta AK. Clinical audit of patients using DPP4 inhibitors in longstanding type 2 diabetes. Diabetes & metabolic syndrome. 2014 doi: 10.1016/j.dsx.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 44.Ferreira L, Teixeira-de-Lemos E, Pinto F, Parada B, Mega C, Vala H, et al. Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat) Mediators Inflamm. 2010;2010:592760. doi: 10.1155/2010/592760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu HY, Huang CY, Shih CM, Chang WH, Tsai CS, Lin FY, et al. Dipeptidyl peptidase-4 inhibitor decreases abdominal aortic aneurysm formation through GLP-1-dependent monocytic activity in mice. PloS one. 2015;10(4):e0121077. doi: 10.1371/journal.pone.0121077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54(1):146–51. doi: 10.2337/diabetes.54.1.146. [DOI] [PubMed] [Google Scholar]
- 47.Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation. 2011;124(21):2338–49. doi: 10.1161/CIRCULATIONAHA.111.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CY, Shih CM, Tsao NW, Lin YW, Huang PH, Wu SC, et al. Dipeptidyl peptidase-4 inhibitor improves neovascularization by increasing circulating endothelial progenitor cells. Br J Pharmacol. 2012;167(7):1506–19. doi: 10.1111/j.1476-5381.2012.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cernea S, Raz I. Therapy in the early stage: incretins. Diabetes care. 2011;34(Suppl 2):S264–71. doi: 10.2337/dc11-s223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bostick B, Habibi J, Ma L, Aroor A, Rehmer N, Hayden MR, et al. Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of Western diet induced obesity. Metabolism: clinical and experimental. 2014;63(8):1000–11. doi: 10.1016/j.metabol.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirakawa H, Zempo H, Ogawa M, Watanabe R, Suzuki J, Akazawa H, et al. A DPP-4 inhibitor suppresses fibrosis and inflammation on experimental autoimmune myocarditis in mice. PloS one. 2015;10(3):e0119360. doi: 10.1371/journal.pone.0119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaji K, Yoshiji H, Ikenaka Y, Noguchi R, Aihara Y, Douhara A, et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J Gastroenterol. 2013 doi: 10.1007/s00535-013-0783-4. [DOI] [PubMed] [Google Scholar]
- 53.Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63(6):2120–31. doi: 10.2337/db13-1029. [DOI] [PubMed] [Google Scholar]
- 54.Sun AL, Deng JT, Guan GJ, Chen SH, Liu YT, Cheng J, et al. Dipeptidyl peptidase-IV is a potential molecular biomarker in diabetic kidney disease. Diabetes & vascular disease research. 2012;9(4):301–8. doi: 10.1177/1479164111434318. [DOI] [PubMed] [Google Scholar]
- 55.Stefanovic V, Ardaillou N, Vlahovic P, Placier S, Ronco P, Ardaillou R. Interferon-gamma induces dipeptidylpeptidase IV expression in human glomerular epithelial cells. Immunology. 1993;80(3):465–70. [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Campitelli J, Hu G, Lin Y, Luo J, Xue C. Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life sciences. 2007;81(4):272–9. doi: 10.1016/j.lfs.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 57.Panchapakesan U, Mather A, Pollock C. Role of GLP-1 and DPP-4 in diabetic nephropathy and cardiovascular disease. Clinical science. 2013;124(1):17–26. doi: 10.1042/CS20120167. [DOI] [PubMed] [Google Scholar]
- 58.Mitic B, Lazarevic G, Vlahovic P, Rajic M, Stefanovic V. Diagnostic value of the aminopeptidase N, N-acetyl-beta-D-glucosaminidase and dipeptidylpeptidase IV in evaluating tubular dysfunction in patients with glomerulopathies. Renal failure. 2008;30(9):896–903. doi: 10.1080/08860220802359048. [DOI] [PubMed] [Google Scholar]
- 59.Liu WJ, Xie SH, Liu YN, Kim W, Jin HY, Park SK, et al. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in streptozotocin-induced diabetic rats. J Pharmacol Exp Ther. 2012;340(2):248–55. doi: 10.1124/jpet.111.186866. [DOI] [PubMed] [Google Scholar]
- 60.Mega C, de Lemos ET, Vala H, Fernandes R, Oliveira J, Mascarenhas-Melo F, et al. Diabetic nephropathy amelioration by a low-dose sitagliptin in an animal model of type 2 diabetes (Zucker diabetic fatty rat) Experimental diabetes research. 2011;2011:162092. doi: 10.1155/2011/162092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glorie LL, Verhulst A, Matheeussen V, Baerts L, Magielse J, Hermans N, et al. DPP4 inhibition improves functional outcome after renal ischemia-reperfusion injury. American journal of physiology Renal physiology. 2012;303(5):F681–8. doi: 10.1152/ajprenal.00075.2012. [DOI] [PubMed] [Google Scholar]
- 62.Sortino MA, Sinagra T, Canonico PL. Linagliptin: a thorough characterization beyond its clinical efficacy. Frontiers in endocrinology. 2013;4:16. doi: 10.3389/fendo.2013.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Groop PH, Cooper ME, Perkovic V, Emser A, Woerle HJ, von Eynatten M. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes care. 2013;36(11):3460–8. doi: 10.2337/dc13-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graefe-Mody U, Giessmann T, Ring A, Iovino M, Woerle HJ. A randomized, open-label, crossover study evaluating the effect of food on the relative bioavailability of linagliptin in healthy subjects. Clinical therapeutics. 2011;33(8):1096–103. doi: 10.1016/j.clinthera.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 65.Min HS, Kim JE, Lee MH, Song HK, Kang YS, Lee MJ, et al. Dipeptidyl peptidase IV inhibitor protects against renal interstitial fibrosis in a mouse model of ureteral obstruction. Lab Invest. 2014;94(6):598–607. doi: 10.1038/labinvest.2014.50. [DOI] [PubMed] [Google Scholar]
- 66.Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology. 2005;10(1):48–56. doi: 10.1111/j.1440-1797.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 67.Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circulation research. 1995;77(1):1–6. doi: 10.1161/01.RES.77.1.1. [DOI] [PubMed] [Google Scholar]
- 68.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. The American journal of pathology. 2011;179(3):1074–80. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. Journal of the American Society of Nephrology: JASN. 2008;19(12):2282–7. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strutz F, Zeisberg M. Renal fibroblasts and myofibroblasts in chronic kidney disease. Journal of the American Society of Nephrology : JASN. 2006;17(11):2992–8. doi: 10.1681/ASN.2006050420. [DOI] [PubMed] [Google Scholar]
- 71.Charytan DM, Padera R, Helfand AM, Zeisberg M, Xu X, Liu X, et al. Increased concentration of circulating angiogenesis and nitric oxide inhibitors induces endothelial to mesenchymal transition and myocardial fibrosis in patients with chronic kidney disease. Int J Cardiol. 2014;176(1):99–109. doi: 10.1016/j.ijcard.2014.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67(21):10123–8. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 73.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13(8):952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 74.Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99(9):1375–9. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu X, Friehs I, Zhong Hu T, Melnychenko I, Tampe B, Alnour F, et al. Endocardial fibroelastosis is caused by aberrant endothelial to mesenchymal transition. Circulation research. 2015;116(5):857–66. doi: 10.1161/CIRCRESAHA.116.305629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu X, Tan X, Tampe B, Nyamsuren G, Liu X, Maier LS, et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc Res. 2015;105(3):279–91. doi: 10.1093/cvr/cvv015. [DOI] [PubMed] [Google Scholar]
- 77.Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail is a direct target of hypoxia-inducible factor 1alpha (HIF1alpha) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem. 2015;290(27):16653–64. doi: 10.1074/jbc.M115.636944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zeisberg M, Zeisberg EM. Evidence for antifibrotic incretin-independent effects of the DPP-4 inhibitor linagliptin. Kidney international. 2015;88(3):429–31. doi: 10.1038/ki.2015.175. [DOI] [PubMed] [Google Scholar]
- 79.Shi S, Srivastava SP, Kanasaki M, He J, Kitada M, Nagai T, et al. Interactions of DPP-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney international. 2015;88(3):479–89. doi: 10.1038/ki.2015.103. [DOI] [PubMed] [Google Scholar]
- 80.Pozzi A, Zent R. Extracellular matrix receptors in branched organs. Curr Opin Cell Biol. 2011;23(5):547–53. doi: 10.1016/j.ceb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pozzi A, Zent R. Integrins: sensors of extracellular matrix and modulators of cell function. Nephron Exp Nephrol. 2003;94(3):e77–84. doi: 10.1159/000072025. [DOI] [PubMed] [Google Scholar]
- 82.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–87. doi: 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 83.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275(29):21785–8. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 84.Park EJ, Yuki Y, Kiyono H, Shimaoka M. Structural basis of blocking integrin activation and deactivation for anti-inflammation. J Biomed Sci. 2015;22:51. doi: 10.1186/s12929-015-0159-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ratnikov BI, Partridge AW, Ginsberg MH. Integrin activation by talin. J Thromb Haemost. 2005;3(8):1783–90. doi: 10.1111/j.1538-7836.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- 86.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arnaout MA, Goodman SL, Xiong JP. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol. 2007;19(5):495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zent R, Yan X, Su Y, Hudson BG, Borza DB, Moeckel GW, et al. Glomerular injury is exacerbated in diabetic integrin alpha1-null mice. Kidney international. 2006;70(3):460–70. doi: 10.1038/sj.ki.5000359. [DOI] [PubMed] [Google Scholar]
- 89.Chen X, Abair TD, Ibanez MR, Su Y, Frey MR, Dise RS, et al. Integrin alpha1beta1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol Cell Biol. 2007;27(9):3313–26. doi: 10.1128/MCB.01476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yeh YC, Wei WC, Wang YK, Lin SC, Sung JM, Tang MJ. Transforming growth factor-{beta}1 induces Smad3-dependent {beta}1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. The American journal of pathology. 2010;177(4):1743–54. doi: 10.2353/ajpath.2010.091183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hamzeh MT, Sridhara R, Alexander LD. Cyclic stretch-induced TGF-beta1 and fibronectin expression is mediated by beta1-integrin through c-Src- and STAT3-dependent pathways in renal epithelial cells. American journal of physiology Renal physiology. 2015;308(5):F425–36. doi: 10.1152/ajprenal.00589.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang XH, Huang SM, Tan SQ, Ma YL. Effect of rosiglitazone on integrin beta1 expression and apoptosis of proximal tubular cell exposed to high glucose. Sichuan Da Xue Xue Bao Yi Xue Ban. 2007;38(2):291–4. [PubMed] [Google Scholar]
- 93.Glynne PA, Picot J, Evans TJ. Coexpressed nitric oxide synthase and apical beta(1) integrins influence tubule cell adhesion after cytokine-induced injury. Journal of the American Society of Nephrology : JASN. 2001;12(11):2370–83. doi: 10.1681/ASN.V12112370. [DOI] [PubMed] [Google Scholar]
- 94.Elias BC, Mathew S, Srichai MB, Palamuttam R, Bulus N, Mernaugh G, et al. The integrin beta1 subunit regulates paracellular permeability of kidney proximal tubule cells. J Biol Chem. 2014;289(12):8532–44. doi: 10.1074/jbc.M113.526509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lal H, Verma SK, Foster DM, Golden HB, Reneau JC, Watson LE, et al. Integrins and proximal signaling mechanisms in cardiovascular disease. Front Biosci (Landmark Ed) 2009;14:2307–34. doi: 10.2741/3381. [DOI] [PubMed] [Google Scholar]
- 96.Srichai M, Zent R. Integrin structure and function. In: Zent R, Pozzi A, editors. Cell-Extracellular Matrix Interactions in Cancer. New York: Springer; 2010. pp. 19–41. [Google Scholar]
- 97.Yuan X, Wang W, Wang J, Yin X, Zhai X, Wang L, et al. Down-regulation of integrin beta1 and focal adhesion kinase in renal glomeruli under various hemodynamic conditions. PloS one. 2014;9(4):e94212. doi: 10.1371/journal.pone.0094212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu S, Kapoor M, Denton CP, Abraham DJ, Leask A. Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis and rheumatism. 2009;60(9):2817–21. doi: 10.1002/art.24801. [DOI] [PubMed] [Google Scholar]
- 99.Sato T, Yamochi T, Yamochi T, Aytac U, Ohnuma K, McKee KS, et al. CD26 regulates p38 mitogen-activated protein kinase-dependent phosphorylation of integrin beta1, adhesion to extracellular matrix, and tumorigenicity of T-anaplastic large cell lymphoma Karpas 299. Cancer Res. 2005;65(15):6950–6. doi: 10.1158/0008-5472.CAN-05-0647. [DOI] [PubMed] [Google Scholar]
- 100.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16(12):1400–6. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagai T, Kanasaki M, Srivastava S, Nakamura Y, Ishigaki Y, Kitada M et al. N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-Mesenchymal transition. BioMed research international. 2014;2014. doi:10.1155/2014/696475. [DOI] [PMC free article] [PubMed]
- 102.Srivastava SP, Koya D, Kanasaki K. MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. BioMed research international. 2013;2013:125469. doi: 10.1155/2013/125469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell reports. 2012;2(6):1684–96. doi: 10.1016/j.celrep.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheng MF, Chen LJ, Wang MC, Hsu CT, Cheng JT. Decrease of FGF receptor (FGFR) and interstitial fibrosis in the kidney of streptozotocin-induced diabetic rats. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2014;46(1):1–7. doi: 10.1055/s-0033-1349090. [DOI] [PubMed] [Google Scholar]
- 105.Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. Journal of the American Society of Nephrology : JASN. 2011;22(9):1668–81. doi: 10.1681/ASN.2010111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lagendijk AK, Goumans MJ, Burkhard SB, Bakkers J. MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production. Circulation research. 2011;109(6):649–57. doi: 10.1161/CIRCRESAHA.111.247635. [DOI] [PubMed] [Google Scholar]
- 107.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348(6232):aaa2151. doi: 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takai S, Sakonjo H, Jin D. Significance of vascular dipeptidyl peptidase-4 inhibition on vascular protection in Zucker diabetic fatty rats. J Pharmacol Sci. 2014;125(4):386–93. doi: 10.1254/jphs.14052FP. [DOI] [PubMed] [Google Scholar]
- 109.Yamada K, Hayashi M, Du W, Ohnuma K, Sakamoto M, Morimoto C, et al. Localization of CD26/DPPIV in nucleus and its nuclear translocation enhanced by anti-CD26 monoclonal antibody with anti-tumor effect. Cancer Cell Int. 2009;9:17. doi: 10.1186/1475-2867-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shi S, Kanasaki K, Koya D. Linagliptin but not Sitagliptin inhibited transforming growth factor-β2-induced endothelial DPP-4 activity and the endothelial-mesenchymal transition. Biochem Biophys Res Commun. 2016 Jan 27. pii: S0006-291X(16)30154-1. doi:10.1016/j.bbrc.2016.01.154. [DOI] [PubMed]