

Abstract
Background
Intestinal microorganisms affect host physiology, including ageing. Given the difficulty in controlling for human studies of the gut microbiome, mouse models provide an alternative avenue to study such relationships. In this study, we report on the complete genome of “Faecalibaculum rodentium” ALO17, a bacterium that was isolated from the faeces of a 9-month-old female C57BL/6J mouse. This strain will be utilized in future in vivo studies detailing the relationships between the gut microbiome and ageing.
Results
The whole genome sequence of “F. rodentium” ALO17 was obtained using single-molecule, real-time (SMRT) technique on a PacBio instrument. The assembled genome consisted of 2,542,486 base pairs of double-stranded DNA with a GC content of 54.0 % and no plasmids. The genome was predicted to contain 2794 open reading frames, 55 tRNA genes, and 38 rRNA genes. The 16S rRNA gene of ALO17 was 86.9 % similar to that of Allobaculum stercoricanis DSM 13633T, and the average overall nucleotide identity between strains ALO17 and DSM 13633T was 66.8 %. After confirming the phylogenetic relationship between “F. rodentium” ALO17 and A. stercoricanis DSM 13633T, their whole genome sequences were compared, revealing that “F. rodentium” ALO17 contains more fermentation-related genes than A. stercoricanis DSM 13633T. Furthermore, “F. rodentium” ALO17 produces higher levels of lactic acid than A. stercoricanis DSM 13633T as determined by high-performance liquid chromatography.
Conclusion
The availability of the “F. rodentium” ALO17 whole genome sequence will enhance studies concerning the gut microbiota and host physiology, especially when investigating the molecular relationships between gut microbiota and ageing.
Keywords: “Faecalibaculum rodentium” ALO17, Whole genome sequencing, Phylogeny, Comparative genomics
Background
Interactions between animals and their intestinal microorganisms play a crucial role in host physiology [1–3]. For example, perturbations of the intestinal microbiota have been associated with immunological, metabolic, and neurological diseases [4, 5]. The metabolic activities of intestinal microorganisms directly affect food digestion, absorption, and energy production [6, 7]. The composition of the intestinal microbiota is also related to ageing of the host [8–10]. The gut microbiota changes dramatically between early and late stages of life, with a shift from Lactobacillus and Bifidobacterium to Bacteroidetes and Clostridia genera. This shift suggests a change from lactate metabolism to increased short-chain fatty acid (SCFA) production and carbohydrate metabolism as ageing progresses [8, 10–12]. To date, multiple studies have investigated the human intestinal microbiome to understand the relationships between the gut microbiota and host ageing [6, 8, 9]. However, difficulties controlling experimental conditions, including diet, medications, and housing status in complex human systems has contributed to inconsistencies in results from such studies. Mouse studies of the relationships between the microbiome and host ageing have provided better-controlled systems with consistent results. Langile et al. [10] divided female C57BL/6J mice into three age groups based on murine frailty index (FI) scores and reported that Erysipelotrichaceae was one of the dominant bacterial families colonising the guts of middle-aged mice (589 days old). These data are consistent with our unpublished data from investigations of the microbial diversity in mice of different ages. We found the most abundant operational taxonomic units (OTU) of middle-aged mice (18–21 months old) were related to Allobaculum species [13] within the family Erysipelotrichaceae. Using fresh and anoxic mice faeces and anaerobic culture techniques [13, 14], we isolated a strain closely related (99 % 16S rRNA gene sequence similarity) to the most abundant OTUs from the feces of C57BL/6J mice, and designated it “Faecalibaculum rodentium” ALO17 [13]. However, the overall intestinal microbiota, including the dominant strains present in the guts of different aged mice, has not been investigated in precise detail. In this study, the whole genome sequence of “F. rodentium” ALO17 was generated using a PacBio instrument and compared in silico to the previously-reported genome sequence of Allobaculum stercoricanis DSM 13633T.
Methods
Strain information
A strictly anaerobic bacterium, ALO17, was previously isolated [13] from the faeces of a 9-month-old female C57BL/6J mouse fed a standard experimental diet (cat. No. 2018S; Harlan Laboratories). The mouse was purchased from DBL Co. Ltd, Korea and housed in the specific pathogen-free facility of Korea Advanced Institute of Science and Technology (KAIST). All animal experiments were performed in accordance with the guidelines and policies for rodent experimentation provided by the Institutional Animal Care and Use Committee (IACUC) of KAIST. This study protocol was approved by the IACUC of KAIST (IACUC-13-140) [13]. The isolate was Gram-stain positive, non-motile, non-spore forming small rod, oxidase and catalase negative. As previously described [13, 15], strictly anaerobic techniques were used for the preparation of the DSM 104 medium (http://www.dsmz.de/microorganisms/medium/pdf/DSMZ_Medium104.pdf) and the cultivation process (the gas atmosphere was 100 % N2). Strain ALO17 was optimally cultivated in the DSM 104 broth at 37 °C and at pH 7 for 3 days under strict anaerobic condition. On the basis of polyphasic taxonomic experiments, we have proposed that the isolate be assigned to the family Erysipelothricaceae with the novel genus and species name, “Faecalibaculum rodentium” [13].
Genome sequencing, assembly and annotation
Genomic DNA was purified from 3L cultures of “F. rodentium” ALO17 as previously described [13, 14]. Extracted DNA samples were sequenced using Pacific Biosciences RS sequencing technology (Pacific Biosciences, Menlo Park, CA), yielding > 50X coverage. Each sample was prepared as a 10-kb insert library using C2 chemistry and sequenced on the PacBio RS II (Pacific Biosciences), according to the manufacturer’s instructions. De novo genome assembly was performed using the CLCbio CLC Genomics Workbench v7.0.4 and PacBio SMRT Analysis 2.2.0 [16]. Annotation was completed using a homology search against the Clusters of Orthologous Groups (COG) and SEED databases [17, 18], respectively. SEED viewer [19] was used for subsystem functional categorization of the predicted ORFs and for visualization [20]. Average nucleotide identity (ANI) values were determined using the BLAST algorithm [21].
Comparative genomics
Comparative genomic analyses were performed using BLAST and a robust pair-wise sequence alignment algorithm. 16S rRNA gene sequences with pairwise similarities >85 % were obtained from the EZtaxon database (http://www.ezbiocloud.net/eztaxon) for nine different species and used for phylogenetic analyses in MEGA6 [22]. Among the nine strains, whole genome sequences for Eubacterium dolichum DSM3991T, Faecalitalea cylindroides ATCC 27803T, Holdemanella biformis DSM 3989T, and Allobaculum stercoricanis DSM 13633T were acquired from the NCBI database (http://www.ncbi.nlm.nih.gov/genome/genomes) and their ANI values [21] to “F. rodentium” ALO17 were calculated. For the calculation of ANI values, the query sequence was randomly cut into fragments of 1020 nucleotides and each was blasted against the subject genome. Following this, a genome tree was constructed using R software, and the most closely related sequences were determined according to the ANI values using the unweighted pair group method. A phylogenetic tree for the five strains was also generated using MEGA6. Fermentation-related genes in the “F. rodentium” ALO17 genome were categorized into functional groups using the annotated genome of A. stercoricanis DSM 13633T.
Measurements of lactic acid concentration
The concentration of lactic acid in the anaerobic DSM 104 broth after 3 days cultivation at 37 °C were determined using an HPLC (1200 Series, Agilent Technology, USA) equipped with an Aminex 87H column (dimensions: 300*7.8, Bio-Rad, USA). The mobile phase was 0.01 M sulfuric acid with 0.5 ml/min flow rate at 40 °C. The average values with error ranges of lactic acid concentration were obtained from two different duplicate experiments.
Quality assurance
Highly purified and intact genomic DNA was obtained from 3L cultures grown in the DSM 104 broth by the modified bead-beating technique [14, 23] and confirmed against the published genome obtained from the NCBI database. The 16S rRNA gene was extracted from the assembled contigs using the RAST annotation system. ANI values were converted into distances between the other genomes analysed.
Results and discussion
The genome of “F. rodentium” ALO17 as assembled here consisted of a single circular DNA chromosome of 2,542,486 base pairs, a GC content of 54.0 %, and no plasmids. The genome contained 2583 predicted open reading frames (ORFs), 55 tRNAs, and 38 rRNAs. Analyses using SEED subsystem categorization and COG functional categorizations are shown in Fig. 1. SEED subsystem categorization predicted 1529 ORFs that encode known functional proteins, whereas 1054 ORFs were of unknown function. Among the ORFs with a predicted function, 274 were predicted to be for carbohydrate synthesis, 200 were predicted to be for amino acid synthesis, 191 for protein metabolism, 140 for RNA metabolism, 122 for DNA metabolism, and 78 for the production of cofactors, vitamins, prosthetic groups, and pigments. Additionally, 40 ORFs belonged to the fermentation category. There are five major roles for genes in this category. A total of 13 ORFs (32.5 %) were similar to genes responsible for the fermentation of acetyl-CoA to butyrate, 12 (30.0 %) were predicted to be involved in fermentations with mixed acids, 9 (22.5 %) in butanol biosynthesis, 4 (10.0 %) in lactate fermentation, and 2 (5.0 %) were predicted to be acetolactate synthase subunits. COG analyses assigned 1566 ORFs (91.0 % of all predicted ORFs) to functional categories. Among these, 815 ORFs (47.39 % of the COG-assigned ORFs) belonged to 5 primary categories: 204 ORFs belonged to Category L (replication, recombination, and repair), 182 to Category G (carbohydrate transport and metabolism), 146 to Category E (amino acid transport and metabolism), 142 to Category K (transcription), and 141 to Category J (translation, ribosomal structure and biogenesis). For comparative genomic analyses, two methods were used: ANI and 16S rRNA gene sequencing. Nine bacterial species with pairwise similarities >85 % for the 16S rRNA gene, compared to “F. rodentium” ALO17, were selected. Among those selected, whole genome sequences for four species were in the NCBI database. A phylogenetic tree was constructed based on the 16S rRNA gene sequences. ANI values calculated between ALO17, E. dolichum DSM3991T, F. cylindroides ATCC 27803T, H. biformis DSM 3989T, A. stercoricanis DSM 13633T, were 63.9, 65.8, 66.2 and 66.8 %, respectively. Strain ALO17 clustered with strain DSM 13633T in the phylogenetic tree, which was supported by a high bootstrap value and ANI dendrogram (Fig. 2). Due to their high level of relatedness, additional detailed genomic analyses were performed between “F. rodentium” ALO17 and other 4 reference of A. stercoricanis DSM 13633T, H. biformis DSM 3989T, F. cylindroides ATCC 27803T and E. dolichum DSM3991T. This comparison of the fermentation related genes is summarised in Table 1. “F. rodentium” ALO17 had a single homologue predicted to encode a protein that stimulates sugar/maltose fermentation. Additionally, subsystems for acetolactate synthase subunits, the fermentation of acetyl-CoA to butyrate, butanol biosynthesis, lactate fermentation, and mixed acid fermentation were present in both “F. rodentium” ALO17 and A. stercoricanis DSM 13633T genomes. The numbers of genes in “F. rodentium” ALO17 and A. stercoricanis DSM 13633T predicted to be involved in fermentation subsystems were 40 and 23, respectively. “F. rodentium” ALO17 had more genes predicted to be involved in specific functions, compared to A. stercoricanis DSM 13633T. Such genes were predicted to encode for 3-hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157), 3-hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157), electron transfer flavoprotein (alpha and beta subunit), 3-hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157), pyruvate formate-lyase (EC 2.3.1.54), L-lactate dehydrogenase (EC 1.1.1.27), pyruvate formate-lyase (EC 2.3.1.54), and pyruvate formate-lyase activating enzyme (EC1.97.1.4). These data suggest that this isolate has stronger fermentation activity than A. stercoricanis DSM 13633T. To test this hypothesis, the lactic acid concentrations were measured in the growth medium of “F. rodentium” ALO17 and A. stercoricanis DSM 13633T by high-performance liquid chromatography. After three days of incubation at 37 °C, the lactic acid concentrations were observed to be 9.5 ± 0.6 mM (strain ALO17) and 4.9 ± 0.8 mM (A. stercoricanis DSM 13633T), respectively. Previous study of A. stercoricanis DSM 13633T showed the level of lactic acid was 3.5 mM [24]. In summary, despite their phylogenetic clustering, “F. rodentium” ALO17 differed from A. stercoricanis DSM 13633T in lactic acid production. We hypothesize that “F. rodentium” ALO17 is an obligate anaerobe that replaces Lactobacilli and Bifidobacterium in middle-aged mice. Lactobacilli and Bifidobacterium are primary lactic acid producers in young mice; however, as the gut becomes strictly anaerobic with age, “F. rodentium” may become dominant. Thus, dominance of lactate producers and lactate production in the animal gut might be inversely related to ageing [10].
Fig. 1.
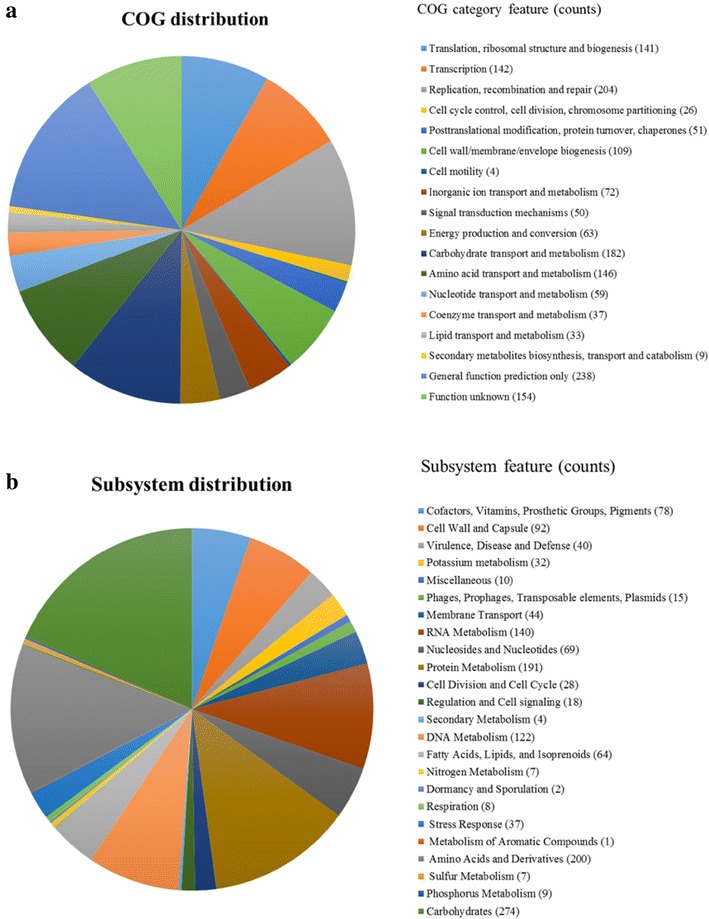
Statistics of annotated genes for “F. rodentium” Alo17 based on a COG and b SEED databases
Fig. 2.
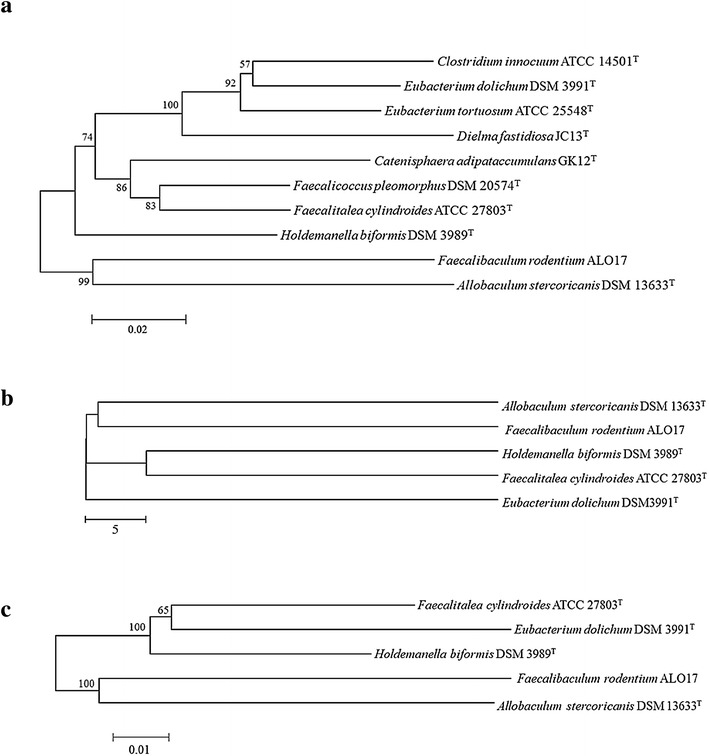
a Phylogenetic tree analysis of 10 strains using 16S rRNA sequence method (pairwise similarity >85 %). b Phylogenetic tree analysis of 5 strains using ANI (average nucleotide identity) methods. c Phylogenetic tree analysis of 5 strains using 16S rRNA sequence method. Bootstrap values (expressed as percentages of 1000 replication, >50 %) are shown at branching points. Bar 0.02 substitution per nucleotide position
Table 1.
Comparison of fermentation related organism between “F. rodentium” ALO17 and other 4 references of A. stercoricanis DSM 13633T, H. biformis DSM 3989T, F. cylindroides ATCC 27803T and E. dolichum DSM3991T
| Subsystem | Role description | Number of genes | ||||
|---|---|---|---|---|---|---|
| Alo17 | 13633T | 3989T | 27803T | 3991T | ||
| Butanol biosynthesis | Alcohol dehydrogenase (EC 1.1.1.1) | 1 | 1 | 1 | 1 | 1 |
| Butanol biosynthesis | NADH-dependent butanol dehydrogenase A (EC 1.1.1.-) | 1 | 1 | 1 | 0 | 1 |
| Butanol biosynthesis | Pyruvate formate-lyase (EC 2.3.1.54) | 3 | 1 | 5 | 4 | 1 |
| Butanol biosynthesis | 3-Hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157) | 3 | 1 | 1 | 1 | 1 |
| Butanol biosynthesis | Acetyl-CoA acetyltransferase (EC 2.3.1.9) | 1 | 1 | 2 | 2 | 1 |
| Fermentations: mixed acid | Alcohol dehydrogenase (EC 1.1.1.1) | 1 | 1 | 1 | 1 | 1 |
| Fermentations: mixed acid | l-lactate dehydrogenase (EC 1.1.1.27) | 2 | 1 | 2 | 2 | 1 |
| Fermentations: mixed acid | Sugar/maltose fermentation stimulation protein homolog | 1 | 0 | 0 | 0 | 0 |
| Fermentations: mixed acid | Pyruvate formate-lyase (EC 2.3.1.54) | 3 | 1 | 5 | 4 | 1 |
| Fermentations: mixed acid | Pyruvate formate-lyase activating enzyme (EC 1.97.1.4) | 3 | 1 | 4 | 4 | 1 |
| Fermentations: mixed acid | Phosphate acetyltransferase (EC 2.3.1.8) | 1 | 1 | 1 | 0 | 1 |
| Fermentations: Mixed acid | Acetate kinase (EC 2.7.2.1) | 1 | 1 | 1 | 1 | 1 |
| Acetolactate synthase subunits | Acetolactate synthase large subunit (EC 2.2.1.6) | 1 | 1 | 0 | 0 | 0 |
| Acetolactate synthase subunits | Acetolactate synthase small subunit (EC 2.2.1.6) | 1 | 1 | 0 | 0 | 0 |
| Fermentations: lactate | l-Lactate dehydrogenase (EC 1.1.1.27) | 2 | 1 | 2 | 2 | 1 |
| Fermentations: lactate | Phosphate acetyltransferase (EC 2.3.1.8) | 1 | 1 | 1 | 0 | 1 |
| Fermentations: lactate | Acetate kinase (EC 2.7.2.1) | 1 | 1 | 1 | 1 | 1 |
| Acetyl-CoA fermentation to butyrate | 3-Hydroxybutyryl-CoA dehydratase (EC 4.2.1.55) | 1 | 1 | 1 | 1 | 0 |
| Acetyl-CoA fermentation to butyrate | Electron transfer flavoprotein, beta subunit | 2 | 1 | 1 | 1 | 0 |
| Acetyl-CoA fermentation to butyrate | Electron transfer flavoprotein, alpha subunit | 2 | 1 | 1 | 1 | 0 |
| Acetyl-CoA fermentation to butyrate | 3-Hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157) | 3 | 1 | 1 | 1 | 0 |
| Acetyl-CoA fermentation to butyrate | 3-Hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35) | 3 | 1 | 1 | 1 | 0 |
| Acetyl-CoA fermentation to butyrate | Acetyl-CoA:acetoacetyl-CoA transferase, alpha subunit (EC 2.8.3.8) | 1 | 1 | 2 | 1 | 0 |
| Acetyl-CoA fermentation to butyrate | Acetyl-CoA Acetyltransferase (EC 2.3.1.9) | 1 | 1 | 2 | 2 | 0 |
Initial findings
The genome of “F. rodentium” ALO17 contained 2583 predicted open reading frames (ORFs), 55 tRNAs, and 38 rRNAs. Among them, 40 and 23 genes in “F. rodentium” ALO17 and A. stercoricanis DSM 13633T predicted to be involved in fermentation subsystems. This result suggest that “F. rodentium” ALO17 has more fermentation activity than A. stercoricanis DSM 13633T. The lactic acid concentrations were measured by high-performance liquid chromatography and “F. rodentium” ALO17 produces higher levels of lactic acid than A. stercoricanis DSM 13633T.
Future directions
This is the first report on the complete genome sequence of “F. rodentium” ALO17. This bacterium was isolated from a 9-month-old laboratory mouse and its genome was sequenced using PacBio SMRT technology. “F. rodentium” ALO17 is phylogenetically related to A. stercoricanis DSM 13633T, which belongs to the family Erysipelotrichaceae, and this family of bacterium is dominant in the gut of middle-aged mice [10]. Considering the robust production of lactic acid in this isolate, further analyses will provide useful information regarding the relationships between gut microbiota, lactate metabolism, and host ageing.
Availability of supporting data
The genome sequence of “F. rodentium” ALO17 was deposited in the Genbank under the accession number of CP011391.
Authors’ contributions
SY Lim and BC Kim designed the study. DH Chang and SR Ahn performed the experiments. SY Lim analyzed and conducted whole genome sequencing comparison. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the Research Program for Agricultural Science and Technology Development (Project No. PJ010168) and was partially supported by Grants from the National Research Foundation of Korea (NRF) (2008-2004721 and NRF-2013M3A9A5076601 and 2015M3C9A4053394), the KRIBB Research Initiative Programs (KGS4121551) and by a Grant from of the Korea Health Technology R&D Project (HI14C0368).
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Sooyeon Lim, Email: limsooy@kribb.re.kr.
Dong-Ho Chang, Email: dark2000@kribb.re.kr.
Sharon Ahn, Email: sharon@kribb.re.kr.
Byoung-Chan Kim, Phone: +82-42-860-4628, Email: bckim@kribb.re.kr.
References
- 1.Vaughan EE, de Vries MC, Zoetendal EG, Ben-Amor K, Akkermans AD, de Vos WM. The intestinal LABs. Antonie Van Leeuwenhoek. 2002;82(1–4):341–352. doi: 10.1023/A:1020672724450. [DOI] [PubMed] [Google Scholar]
- 2.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291(5505):881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 3.Sommer F, Backhed F. The gut microbiota [mdash] masters of host development and physiology. Nat Rev Micro. 2013;11(4):227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 4.Su CG, Judge TA, Lichtenstein GR. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31(1):307–327. doi: 10.1016/S0889-8553(01)00019-X. [DOI] [PubMed] [Google Scholar]
- 5.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 7.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 8.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vázquez-Baeza Y, Jansson JK, Gordon JI, Knight R. Meta-analyses of studies of the human microbiota. Genome Res. 2013;23(10):1704–1714. doi: 10.1101/gr.151803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langille MG, Meehan CJ, Koenig JE, Dhanani AS, Rose RA, Howlett SE, Beiko RG. Microbial shifts in the aging mouse gut. Microbiome. 2014;2(1):50. doi: 10.1186/s40168-014-0050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Tongeren SP, Slaets JP, Harmsen H, Welling GW. Fecal microbiota composition and frailty. Appl Environ Microbiol. 2005;71(10):6438–6442. doi: 10.1128/AEM.71.10.6438-6442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mariat D, Firmesse O, Levenez F, Guimarăes V, Sokol H, Dore J, Corthier G, Furet J. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9(1):123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang D-H, Rhee M-S, Ahn S, Bang B-H, Oh JE, Lee HK, Kim B-C. “Faecalibaculum rodentium” gen. nov., sp. nov., isolated from the faeces of a laboratory mouse. Antonie Van Leeuwenhoek. 2015;108(Pt6):1309–1318. doi: 10.1007/s10482-015-0583-3. [DOI] [PubMed] [Google Scholar]
- 14.Lee J-H, Kumar S, Lee G-H, Chang D-H, Rhee M-S, Yoon M-H, Kim B-C. Methanobrevibacter boviskoreani sp. nov., isolated from the rumen of Korean native cattle. Int J Syst Evol Microbiol. 2013;63(Pt 11):4196–4201. doi: 10.1099/ijs.0.054056-0. [DOI] [PubMed] [Google Scholar]
- 15.Bang B-H, Rhee M-S, Chang D-H, Park D-S, Kim B-C. Erysipelothrix larvae sp. nov., isolated from the larval gut of the rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae) Antonie Van Leeuwenhoek. 2015;107(2):443–451. doi: 10.1007/s10482-014-0342-x. [DOI] [PubMed] [Google Scholar]
- 16.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 17.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278(5338):631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 18.Disz T, Akhter S, Cuevas D, Olson R, Overbeek R, Vonstein V, Stevens R, Edwards RA. Accessing the SEED genome databases via Web services API: tools for programmers. BMC Bioinformatics. 2010;11(1):319. doi: 10.1186/1471-2105-11-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42(D1):D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M. The RAST server: rapid annotations using subsystems technology. BMC Genom. 2008;9(1):75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 22.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujimoto S, Nakagami Y, Kojima F. Optimal bacterial DNA isolation method using bead-beating technique. Memoirs Kyushu Univ Dep Of Health Scis Of Medical Sch. 2004;3:33–38. [Google Scholar]
- 24.Greetham HL, Gibson GR, Giffard C, Hippe H, Merkhoffer B, Steiner U, Falsen E, Collins MD. Allobaculum stercoricanis gen. nov., sp. nov., isolated from canine feces. Anaerobe. 2004;10(5):301–307. doi: 10.1016/j.anaerobe.2004.06.004. [DOI] [PubMed] [Google Scholar]