

Abstract
Three rationally designed glucose-platinum conjugates (Glc-Pts) were synthesized and their biological activities evaluated. The Glc-Pts, 1-3, exhibit high levels of cytotoxicity toward a panel of cancer cells. The subcellular target and cellular uptake mechanism of the Glc-Pts were elucidated. For uptake into cells, Glc-Pt 1 exploits both glucose and organic cation transporters, both widely overexpressed in cancer. Compound 1 preferentially accumulates in and annihilates cancer, compared to normal epithelial, cells in vitro.
Keywords: antitumor agents, glycoconjugates, platinum, glucose transporters, medicinal inorganic chemistry
Graphical Abstract
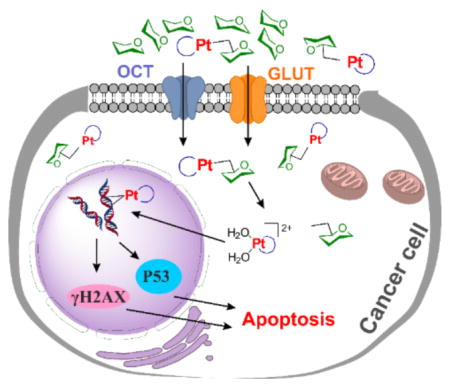
Platinum-based anticancer drugs are among the most widely used of all chemotherapeutic treatments. Three FDA-approved platinum anticancer drugs, cisplatin, carboplatin, and oxaliplatin, have been in the clinic for many years to treat a variety of cancers including testicular, ovarian, cervical, head and neck, non-small-cell lung, and colorectal.[1] Despite their success, platinum compounds have a number of deficiencies originating from a lack of tumor selectivity. Only a small fraction of the total administered platinum accumulates at the tumor site, resulting in sub-optimal drug concentration at the target. Moreover, accumulation of platinum in healthy tissue leads to undesired side effects including nephrotoxicity, myelosuppression, peripheral neuropathy, ototoxicity, and nausea.[1b, 2] These drawbacks need to be addressed when designing next generation platinum drugs. Novel strategies for introducing tumor-targeting properties into platinum anticancer drug candidates are therefore of great interest.[3]
In order to maintain cellular homeostasis, growth, and proliferation, cancer cells significantly increase glucose uptake and the flux of metabolites through glycolysis. This phenomenon, termed “the Warburg effect,” arises from mitochondrial metabolic changes and is one of the hallmarks of cancer.[4] GLUT1, the most common glucose transporter, is widely overexpressed in many human cancers including hepatic, pancreatic, breast, esophageal, brain, renal, lung, cutaneous, colorectal, endometrial, ovarian, and cervical.[5] High GLUT1 expression levels in tumor biopsy samples correlate strongly with poor prognosis. Moreover, several other glucose transporters including GLUT2, GLUT3, GLUT12 and SGLT1/2 are also overexpressed in certain types of cancer cells.[5–6] Therefore glycoconjugation becomes an appealing strategy for targeted delivery of anticancer drugs. The potential of this strategy in diagnosis and therapy has already been realized, but there is much room for improvement.[7]
Examples of glucose-platinum conjugates (Glc-Pts) in which the key structural features of the sugar unit are not perturbed, a prerequisite for optimal transporter recognition, and in which the sugar is linked to the platinum complex via a spacer are scarce.[8] Moreover, these previous studies fail to answer a crucial question in glycoconjugate development, are the conjugates actually taken up by the glucose transporters broadly expressed in cancer cells?
In the present work, we report the synthesis, cytotoxicity, and detailed characterization of the cellular uptake mechanism of three novel Glc-Pts 1–3 (Figure 1a). The design of these conjugates was guided by a recently published crystal structure of the bacterial xylose transporter XylE, a GLUT1 homolog.[9] Although a crystal structure of human GLUT1 has also recently been published,[10] in this latter study the protein was captured in the inward open configuration, as opposed to the outward open configuration that a platinum-glucose conjugate would encounter when attempting to enter the cell. The XylE structure with bound D-glucose, on the other hand, exhibits the protein in an outward-facing conformation. This structure reveals that all of the hydroxyl groups of D-glucose except that on C6 are involved in hydrogen-bonding interactions with various amino acid residues of the transporter. We hypothesized that modification at the C6 position of D-glucose should not, therefore, interfere with receptor binding. Previous reports have also suggested that the C6 position of D-glucose can tolerate various functional groups while retaining substrate specificity for, and internalization by, GLUT1.[11] In fact, C6-glucose conjugates of 4-nitrobenzofurazan, ketoprofen, and indomethacin were reported to bind GLUT1 with even higher affinity than unmodified D-glucose.[11a, 11c, 12] This property is highly desirable for a glucose-drug/fluorophore conjugate, which has to compete with the high level of glucose (~ 6 mM) in the blood.[13]
Figure 1.

(a) Structures of Glc-Pts 1-3 and their aglycone 4. (b) The hydrogen-bonding interactions present in the docking model of 1 into XylE (PDB 4GBZ).[9] The protein is shown as grey ribbons with the sidechains of key residues depicted as sticks. Complex 1 is shown as sticks and polar hydrogen atoms are explicitly portrayed. Color code: N blue, O red, H white, C cyan, protein C grey, Pt purple. Hydrogen-bonding interactions are illustrated with dashed yellow lines.
Initial docking studies using a DFT-optimized structure of the C6-glucose-platinum derivative 1 (Figure 1b and S17) suggested that this complex is capable of binding in the cavity of an outward open XylE. The orientation of the sugar moiety in the docked complex differs from that of the glucose unit bound in the crystal structure, but hydrogen-bonding interactions occur with Gln168, Gln288, Tyr298, and Gln175. These residues had all been identified as key glucose-binding units in the XylE structure and either interact directly with the bound D-glucose or indirectly via hydrogen-bonded water molecules.[9] Additionally, Thr28 is capable of interacting with the carboxylate ligand of the platinum moiety.
The synthesis of a C6-Glc-Pt compound has not, to our knowledge, been previously reported. We therefore had to establish feasible routes to Glc-Pts 1-3 (Scheme 1 and see SI for details). All new compounds were unambiguously characterized by NMR (1H, 13C, 195Pt) spectroscopy and electrospray ionization (ESI) mass spectrometry. The purity of the platinum complexes (1–4) was confirmed to be ≥95% by elemental microanalyses and analytical HPLC (Figure S1–S4).
Scheme 1.

Synthetic route for glucose-platinum conjugate 1.
The stability of this class of compounds in water and biological media was evaluated using compound 1. The rate of activation of platinum drugs by dissociation of the leaving group ligand(s) from the platinum center in the presence of biological nucleophiles follows the order dichloride (cisplatin) > oxalate (oxaliplatin) > malonate (carboplatin), suggesting high stability for Glc-Pt 1 because its leaving group ligand is similar to that of carboplatin.[14] Indeed we observed that 1 is highly stable in water as evidenced by no change in the 1H NMR spectrum of 1 after 72 h in D2O (Figure S18). In RPMI medium, which is used routinely for mammalian tissue culture, slow activation of 1 by the nucleophiles present in the medium was observed (Figure S19). This result is consistent with the previously reported activation of structurally similar platinum compounds by nucleophiles.[14a, 14b] No significant decomposition was noticed up to 8 h, and approximately 60% of 1 remained unchanged even after 24 h, suggesting that the compound is highly stable in biological media. As expected, the formation of 5 as a result of activation of 1 was confirmed by ESI-mass spectrometry (Figure S20).
Cellular uptake studies in A2780, DU145 and A549 cells revealed that, of the three Glc-Pts, 1 is taken up most efficiently (Figure S5). We also observed a consistent decrease in uptake with increasing length of the linker joining the glucose and platinum moieties. It has been proposed that, upon binding to substrate in its outward open conformation, the GLUT1 transporter undergoes a conformational change in which the extracellular entrance to the cavity is occluded and an opening to the cytoplasmic side of the membrane forms, allowing the substrate to enter into the cell.[15] Steric hindrance caused by an overly long substrate could block this conformational change, and we propose that this phenomenon is responsible for our observation that glucose-platinum conjugates with longer linkers display reduced cellular uptake. In this respect, we note that when the structure of GLUT1 in the inward open form is aligned with that of XylE in the outward open form into which the Glc-Pts have been docked, significant steric clashes are observed for 3 but not 1 (Figure S17).[9–10]
We next evaluated the cytotoxicity of 1–3 and their aglycone 4 against a panel of human cancer cells of different origin using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, a standard assay for cytotoxicity evaluation. The IC50 (concentration required to reduce 50% cell viability) values derived from dose-response curves are summarized in Table S1. The cytotoxicity of the Glc-Pt compounds is generally comparable to those of aglycone 4, but greater than that of cisplatin. Ovarian cancer A2780 cells were the most sensitive to the Glc-Pt compounds (IC50 = 0.15–0.22 μM). The relatively tight distribution of IC50 values for 1–3 suggests that, whereas the length of the spacer between the glucose and platinum moieties significantly influences their cellular uptake, it is not the primary determinant of the IC50 values of these compounds.
Although the previously described cellular uptake and cytotoxicity data may appear inconsistent, it is important to note that, for technical reasons, these initial assays were performed on different time scales. We subsequently investigated the effect of incubation time on the outcomes of these assays. Whereas the Glc-Pts were designed to be taken up by facilitated diffusion, the passive diffusion of the aglycone 4 will be impacted significantly by its lipophilicity and consequent ability to traverse the cellular membrane. We found that, even though the lipophilicity of 4 is approximately one log P unit higher than that of 1 (Figure S10a), the accumulation of 1 was significantly higher than that of 4 when cells were incubated with either compound for 8 h (Figure S11). This result highlights the importance of the glucose moiety of 1 in its cellular uptake. In contrast to the comparable activity of 1 and 4 observed in the 72 h incubation MTT assay (Table S1 and Figure S13b), an 8 h incubation MTT assay revealed 1 to be more cytotoxic than 4 in both A2780 and DU145 cells (Figure S12, S13a & S13b), which is again consistent with the observed cellular uptake differences between 1 and 4 in an 8 h assay (Figure S11). We propose that the initial rate of accumulation of 1 in cells is faster than that of 4, but that this protein-mediated transport becomes saturated at longer time scales. On the other hand, the passive uptake of 4 is slower but does not saturate. As a result, prolonged incubation with 4 allows the levels of cellular platinum accumulation and cytotoxcitity to approach that of 1. The difference in the cellular uptake between 1 and 4 diminishes with increased incubation time, monitored from 8 h to 17 h (Figure S13c).
In order to obtain insight into possible subcellular targets of the Glc-Pts, we studied the intracellular distribution of 1 and 2 as representatives of this class of compound in A2780 cells. As shown in Figure S6, detection of platinum in the nucleus points to nuclear DNA as one potential target.[1a] Analysis of DNA platination levels (Figure S7a) revealed that 1 and 2 platinate nuclear DNA, the extent of which is 764 ± 57 Pt adducts/104 nucleotides for 1, 483 ± 79 Pt adducts/104 nucleotides for 2, and 685 ±17 adducts/104 nucleotides for oxaliplatin, which was included as a positive control. Increases in the expression levels of γH2AX, phos-p53, and phos-CHK2, which are canonical DNA damage biomarkers,[16] were also observed when cells were treated with increasing concentrations of 1 or 2 (Figure S7b). As expected for DNA-targeting platinum compounds,[1a] cell cycle arrest at G2/M phase and induction of apoptosis were observed when A2780 cells were treated with compounds 1 or 2 and then analyzed by flow cytometry (Figure S8 & S9). Taken together, these results are consistent with the proposal that Glc-Pts target genomic DNA, the platination of which leads to apoptosis.
As described earlier, one crucial question in glycoconjugate chemistry is whether or not the sugar-conjugated molecule is actually transported by the targeted sugar transporters. To address this issue, we carried out a series of experiments to investigate the details of the mechanism by which 1-3 are taken up by cells. Glc-Pts 1–3 are very hydrophilic (log P ~ −2) rendering cellular internalization via passive diffusion through the cellular lipid membrane highly unlikely. Furthermore, the lack of correlation between the log P values and cellular uptake is consistent with a protein-mediated transport mechanism (Figure S10a). The ovarian cancer cell line A2780 was chosen to evaluate the cellular uptake mechanism of the Glc-Pts because of its high level of GLUT1 expression,[17] confirmed by immunoblotting analyses (Figure S10b). Cellular uptake was first monitored in the absence and presence of an exofacial GLUT1 inhibitor 4,6-O-ethylidene-α-D-glucose (EDG),[18] and the results are presented in Figures 2a and S14c. A 50% reduction in cellular uptake of 1 was measured in the presence of 100 mM EDG. Under similar conditions, the reduction in the cellular uptake of 2 and 3 was 38% and 30%, respectively. The cellular uptake of cisplatin did not change significantly in the presence of the inhibitor. Because cisplatin can be taken up via passive diffusion, this result matches well with our expectations. The inhibitor did, however, cause a 24% decrease in the cellular uptake of the aglycone 4. Because energy-dependent organic cation transporters (OCTs) contribute, at least in part, to the cellular uptake of 4 (Figure 2d, vide infra), we propose that the differential uptake induced by the presence of EDG most likely arises from the energy-depleted conditions produced by glucose transport inhibition. The extent of cellular uptake inhibition of the Glc-Pts in the presence of EDG is in the order 1>2>3 and this trend tracks with the cellular uptake of these compounds (Figure S5), providing further support for the proposal that the GLUT1 translocation efficiencies for C6-glucose conjugates decrease with increasing linker length. Cumulatively, these results suggest that the cellular uptake of 1 is not only superior to, but is also more glucose-transporter-specific than, that of either 2 or 3. As a consequence, only 1 was used in the subsequent cellular uptake experiments.
Figure 2.

(a) Effect of GLUT1 inhibitor EDG on the cellular uptake of 1–4 and cisplatin (10 μM compounds, 17 h). (b) Effect of externally added D-glucose and L-glucose (10 μM compounds, 17 h). (c) Effect of EDG on the IC50 values (72 h assay). (d) Effect of EDG, Ctd, and their mixture on the cellular up-take of 1, 4 and oxaliplatin (10 μM compounds, 8 h). All experiments were done in A2780 cells and cellular uptake in absence of inhibitor was normalized to 100%. Data represent the mean ± SD of at least three or more replicates. The asterisks denote differences are statistically significant (*p < 0.01, **p < 0.001), ns = not statistically significant.
Similarly to co-treatment with EDG, a 50% reduction in the cellular uptake of 1 was observed when a structurally and functionally different glucose transport inhibitor, phloretin, was used (Figure S14b). Moreover, given that D-glucose is the main substrate of GLUT1 and other glucose transporters, it should compete with and inhibit the protein-mediated uptake of the Glc-Pts. When probed, D-glucose, but not L-glucose, exhibited a weak but statistically significant (p < 0.01) inhibitory effect on the uptake of 1 (Figure 2b). The poor inhibitory effect (ca. 30% reduction in uptake) exerted by D-glucose can be attributed to the high binding affinity of 1 to glucose transporters, a phenomenon previously reported for other C6-glucose conjugates and GLUT1.[11b, 11c, 12] We also tested the effect of D-glucose on the cellular uptake of the aglycone 4 and found the uptake to be unaffected. Furthermore, in cytotoxicity assays carried out in the presence of EDG, the IC50 value of 1 increased 19-fold (Figure 2c). We note that EDG does not affect the ability of 1 to platinate DNA in vitro (Figure S16). In contrast to the results with 1, only a 6-fold increase in IC50 value was observed during cotreatment with the control aglycone 4 and EDG. The slight increase in IC50 value of 4 mirrors the observed decrease in cellular uptake of 4 in the presence of glucose transport inhibitors, which we attribute to energy depletion. In order probe Glc-Pt uptake through glucose transporters in an orthogonal manner, we capitalized on the fact that hypoxia causes stimulation of glucose transport and metabolism in cancer cells.[19] As shown in Figure S14a, cellular uptake of 1 increased by 50% when A549 cells were treated with the hypoxia-inducing agent cobalt(II) chloride.[20] No significant difference in the uptake of 4 was observed under similar conditions. In summary, the uptake assays support the hypothesis that glucose transporters, such as GLUT1, are at least partially involved in the cellular entry mechanism of 1.
It is well documented that organic cation transporter 2 (OCT2) plays important roles in the cellular accumulation and consequent cytotoxicity of platinum complexes containing the (1R,2R)-cyclohexane-1,2-diamine (DACH) ligand.[21] OCT2-mediated cellular uptake has been suggested as a leading factor responsible for the sensitivity of colorectal cancer to oxaliplatin.[21] Because the Glc-Pts reported here bear the chelating DACH ligand, we investigated the potential of 1 to undergo translocation via OCT2, a transporter overexpressed in certain types of cancer cells and tumor samples from patients.[21–22] Expression of OCT2 in a panel of cancer cell lines was confirmed by Western blotting analysis (Figure S10b). A2780 cells were incubated with 10 μM 1 for 8 h in the presence or absence of EDG and/or the OCT2 inhibitor cimetidine (Ctd); oxaliplatin was employed as a positive control. In the presence of EDG, uptake of 1 was reduced by 50%, whereas the uptake levels of 4 and oxaliplatin were reduced by only 25% and 30%, respectively (Figure 2d). The OCT2 inhibitor Ctd reduces the uptake of the positive control compound oxaliplatin by 70%. Assays with Ctd revealed reductions of 45% and 35% in the cellular uptake of 1 and 4, respectively. These results support the involvement of OCT2 in the cellular internalization of both 1 and 4. The uptake of 1 was further decreased by 20% (p < 0.001) following treatment with a mixture of EDG and Ctd, compared to treatment with Ctd alone. These results further confirm the involvement of glucose transporters in the cellular uptake of 1 and indicate that OCT2 facilitates the cellular accumulation of 1 as well.
An ideal anticancer compound should be selective for cancer cells over normal healthy cells, thereby mitigating undesired toxic side effects associated with chemotherapy. We therefore evaluated the selectivity of Glc-Pt 1 using DU145 prostate and A498 kidney cancer cells and matched normal prostate epithelial (RWPE2) and kidney epithelial (CCD1105 KIDTr) cells. Both of the cancerous cell lines have high levels of GLUT1 expression as compared to the normal epithelial cells (Figure S10b). Strikingly, as presented in Figure 3, the accumulation of 1 was significantly higher in the cancer cells as compared to the matched normal cells. Notably, the cellular uptake of 1 in DU145 cells is four-fold higher than RWPE2 cells, and it can be inhibited by the potent glucose transport inhibitor Cytochalasin B or the OCT2 inhibitor Ctd (Figure S15), suggesting that the cognate transporters mediate, at least in part, the preferential uptake of 1 by cancer cells. Similarly, 1 reduced the viability of cancer cells more efficiently as compared to normal epithelial cells (Figure 3c and d).
Figure 3.

(a and b) Preferential accumulation of Glc-Pt 1 in prostate and kidney cancer cells as compared to matched normal epithelial cells (20 μM, 8 h). (c and d) Effect of Glc-Pt 1 on the viability of cancer and matched normal cells. The asterisks denote differences that are statistically significant (*p<0.001, **p<0.02, ***p<0.01, ****p<0.002).
Given that neuronal cells express high levels of GLUT1 transporters, adverse off-target neurological effects could arise with these glycoconjugates. We therefore evaluated the cytotoxicity of 1 in a murine hippocampal derived Neuro-2A neuronal cell line, which is known to express the GLUT1 transporter,[23] using short (8 h) and long (72 h) term assay. As shown in Figure S21, 1 has several fold higher IC50 values in Neuro-2A cells (IC50 = 13.6±0.7 μM and 2.3±0.3 μM at 8 h and 72 h, respectively) when compared to the most sensitive overian cancer A2780 cells (IC50 = 2.2±0.1 μM and 0.15±0.06 μM for 8 h and 72 h assays, respectively). This result indicates that 1 is much more potent in ovarian cancer cells compared to neuronal Neuro-2A cells in vitro. Encouragingly, neurotoxicity has not yet been observed in vivo for glycoconjugated drugs tested thus far.[7a] Moreover, no neurological adverse side effects were observed during a phase II clinical study of glufosamide, a glucose conjugated DNA alkylating agent ifosamide mustard.[24] Finally, we note that the significant body of work showing preferential accumulation of 18FDG in tumors, used to diagnose malignancies, underscores the potential of glucose to become a powerful molecular tag for targeting cancer cells.[7d, 25] [7a]
In summary, novel C6-Glc-Pts were rationally designed and synthesized, and their mechanism of uptake was evaluated. Genomic DNA was confirmed to be one of the intracellular targets of the Glc-Pts. Among the Glc-Pts investigated, 1 most readily translocates through glucose transporters. The translocation efficiency and subsequent cellular accumulation were reduced with increasing size of the conjugate linker. Strikingly, 1 preferentially accumulates in and annihilates cancer cells while showing reduced accumulation and low toxicity in noncancerous cells in vitro. These results clearly demonstrate the potential of glycoconjugation for selective destruction of cancer cells by platinum compounds. To our knowledge, 1 represents the first glucose-platinum conjugate where a glucose-transporter-mediated cellular uptake mechanism has been rigorously established. Furthermore, in addition to glucose transporters, OCT2 was identified as an additional transporter involved in the protein-mediated transport of 1, demonstrating the potential of 1 to exploit these two transporters, commonly overexpressed on the surface of tumor cells.
Supplementary Material
Acknowledgments
This work is supported by the NCI under grant CA034992. We thank Dr. Samuel G. Awuah for help with flow cytometry.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Wang D, Lippard SJ. Nat Rev Drug Discovery. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]; b) Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 2.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Toxins. 2010;2:2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Guo Z. Chem Soc Rev. 2013;42:202–224. doi: 10.1039/c2cs35259a. [DOI] [PubMed] [Google Scholar]
- 4.a) Vander Heiden MG, Cantley LC, Thompson CB. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 5.a) Szablewski L. Biochim Biophys Acta. 2013;1835:164–169. doi: 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]; b) Calvo MB, Figueroa A, Pulido EG, Campelo RG, Aparicio LA. Int J Endocrinol. 2010;2010:14. doi: 10.1155/2010/205357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Guo GF, Cai YC, Zhang B, Xu RH, Qiu HJ, Xia LP, Jiang WQ, Hu PL, Chen XX, Zhou FF, Wang F. Med Oncol. 2011;28:197–203. doi: 10.1007/s12032-010-9696-8. [DOI] [PubMed] [Google Scholar]; b) Ishikawa N, Oguri T, Isobe T, Fujitaka K, Kohno N. Jpn J Cancer Res. 2001;92:874–879. doi: 10.1111/j.1349-7006.2001.tb01175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvaresi EC, Hergenrother PJ. Chem Sci. 2013;4:2319–2333. doi: 10.1039/C3SC22205E.and references therein; Kim WH, Lee J, Jung DW, Williams DR. Sensors. 2012;12:5005–5027. doi: 10.3390/s120405005.Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, Oster ZH, Sacker DF, Shiue CY, Turner H, Wan CN, Wolf AP, Zabinski SV. J Nucl Med. 1980;21:670–675.Ben-Haim S, Ell P. J Nucl Med. 2009;50:88–99. doi: 10.2967/jnumed.108.054205.
- 8.a) Chen Y, Heeg MJ, Braunschweiger PG, Xie W, Wang PG. Angew Chem Int Ed. 1999;38:1768–1769. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1768::AID-ANIE1768>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; b) Liu P, Lu Y, Gao X, Liu R, Zhang-Negrerie D, Shi Y, Wang Y, Wang S, Gao Q. Chem Commun. 2013;49:2421–2423. doi: 10.1039/c3cc38589b. [DOI] [PubMed] [Google Scholar]; c) Möker J, Thiem J. Eur J Org Chem. 2009;2009:4842–4847. [Google Scholar]; d) Hartinger CG, Nazarov AA, Ashraf SM, Dyson PJ, Keppler BK. Curr Med Chem. 2008;15:2574–2591. doi: 10.2174/092986708785908978. [DOI] [PubMed] [Google Scholar]; e) Li H, Gao X, Liu R, Wang Y, Zhang M, Fu Z, Mi Y, Wang Y, Yao Z, Gao Q. Eur J Med Chem. 2015;101:400–408. doi: 10.1016/j.ejmech.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. Nature. 2012;490:361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- 10.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 11.a) Speizer L, Haugland R, Kutchai H. Biochim Biophys Acta. 1985;815:75–84. doi: 10.1016/0005-2736(85)90476-6. [DOI] [PubMed] [Google Scholar]; b) Kumar P, Shustov G, Liang H, Khlebnikov V, Zheng W, Yang XH, Cheeseman C, Wiebe LI. J Med Chem. 2012;55:6033–6046. doi: 10.1021/jm2017336. [DOI] [PubMed] [Google Scholar]; c) Gynther M, Ropponen J, Laine K, Leppänen J, Haapakoski P, Peura L, Järvinen T, Rautio J. J Med Chem. 2009;52:3348–3353. doi: 10.1021/jm8015409. [DOI] [PubMed] [Google Scholar]
- 12.Barros LF, Bittner CX, Loaiza A, Ruminot I, Larenas V, Moldenhauer H, Oyarzún C, Alvarez M. J Neurochem. 2009;109:94–100. doi: 10.1111/j.1471-4159.2009.05885.x. [DOI] [PubMed] [Google Scholar]
- 13.König M, Bulik S, Holzhütter HG. PLoS Comput Biol. 2012;8:e1002577. doi: 10.1371/journal.pcbi.1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Buß I, Kalayda G, Lindauer A, Reithofer M, Galanski M, Keppler B, Jaehde U. J Biol Inorg Chem. 2012;17:699–708. doi: 10.1007/s00775-012-0889-9. [DOI] [PubMed] [Google Scholar]; b) Rajković S, Ašanin DP, Živković MD, Djuran MI. Inorg Chim Acta. 2013;395:245–251. [Google Scholar]; c) Summa N, Soldatović T, Dahlenburg L, Bugarčić ŽD, Eldik Rv. J Biol Inorg Chem. 2007;12:461–475. doi: 10.1007/s00775-006-0200-z. [DOI] [PubMed] [Google Scholar]
- 15.Quistgaard EM, Löw C, Moberg P, Trésaugues L, Nordlund P. Nat Struct Mol Biol. 2013;20:766–768. doi: 10.1038/nsmb.2569. [DOI] [PubMed] [Google Scholar]
- 16.a) Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]; c) Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Wang Y, Dong L, Huang Y, Yuan J, Ben W, Yang Y, Ning N, Lu M, Guan Y. Cancer Sci. 2013;104:1690–1696. doi: 10.1111/cas.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnett JE, Holman GD, Chalkley RA, Munday KA. Biochem J. 1975;145:417–429. doi: 10.1042/bj1450417a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denko NC. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 20.Kim TR, Cho EW, Paik SG, Kim IG. FEBS Lett. 586:303–309. doi: 10.1016/j.febslet.2011.12.036. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, Chen Y, Komori T, Gray JW, Chen X, Lippard SJ, Giacomini KM. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burger H, Zoumaro-Djayoon A, Boersma AWM, Helleman J, Berns E, Mathijssen RHJ, Loos WJ, Wiemer EAC. Br J Pharmacol. 2010;159:898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salto R, Vílchez JD, Girón MD, Cabrera E, Campos N, Manzano M, Rueda R, López-Pedrosa JM. PLoS ONE. 2015;10:e0135614. doi: 10.1371/journal.pone.0135614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bent MJvd, Grisold W, Frappaz D, Stupp R, Desir JP, Lesimple T, Dittrich C, Jonge MJAd, Brandes A, Frenay M, Carpentier AF, Chollet P, Oliveira J, Baron B, Lacombe D, Schuessler M, Fumoleau P. Ann Oncol. 2003;14:1732–1734. doi: 10.1093/annonc/mdg491. [DOI] [PubMed] [Google Scholar]
- 25.Bronstein Y, Tummala S, Rohren E. Clin Nuc Med. 2011;36:96–100. doi: 10.1097/RLU.0b013e318203bb0e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.