

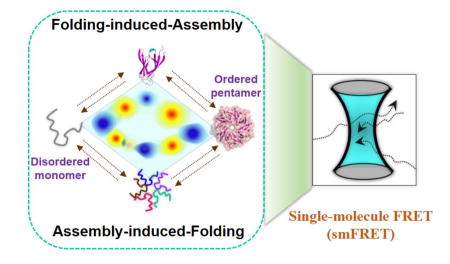
Graphical Abstract
Plastic landscape facilitates functional shape-shifting. Alternative folding-assembly pathways of a conditionally disordered oncogenic protein were revealed by single-molecule and ensemble experiments. Posttranslational modification and partner binding have differential effects on individual steps, and can counteract each other. Tunable mechanistic routes highlight a complex landscape that could efficiently enable its multiple cellular functions.
Keywords: Protein folding, smFRET, coupled folding and binding, kinetics, conformational landscape
Abstract
As for many intrinsically disordered proteins, order-disorder transitions in the N-terminal oligomerization domain of the multi-functional nucleolar protein, nucleophosmin (Npm-N) are central to its function, with phosphorylation and partner-binding acting as regulatory switches. However, the mechanism of this transition and its regulation by these factors remain poorly understood. Here, single-molecule and ensemble experiments revealed pathways with alternative sequence of folding and assembly steps for Npm-N, switchable by altering ionic strength. Phosphorylation resulted in pathway-specific effects, and decoupled folding and assembly steps to facilitate disorder. Conversely, binding to a physiological partner locked Npm-N in ordered pentamers, and counteracted the effects of phosphorylation. Our findings revealed mechanistic plasticity in the Npm-N order-disorder transition, which enabled a complex interplay of phosphorylation and partner binding in modulating its folding landscape.
Many intrinsically disordered proteins (IDPs) experience function altering transitions between ordered and disordered states[1], which can be triggered by post-translational modifications (PTMs) such as phosphorylation[1b], and binding partners[2]. Although recent studies have demonstrated the effects of phosphorylation and ligand association on the conformation of some polymorphic IDPs[1b, 3], detailed mechanistic descriptions of the transition pathways between ordered and disordered states, and how they are modulated by these function altering triggers are generally lacking. Here, we investigated the folding landscape of the N-terminal oligomerization domain (Npm-N) of nucleophosmin (NPM1). NPM1 is a nucleolar phospho-protein with key roles in ribosome biogenesis, centrosome duplication, p53-dependent and independent tumor suppression pathways, apoptosis and cancer[4]. Npm-N interconverts between a globally disordered monomer and an ordered β-sheet rich pentamer. This conformational equilibrium can be tuned by altering the ionic strength of the solution in vitro[1b, 5]. Importantly, the structural polymorphism of Npm-N has been proposed to be a key factor for the multi-functionality of NPM1, and is regulated by phosphorylation and partner binding[1b]. The folded oligomeric form is required for nucleolar localization of NPM1[6]. Strikingly, phosphorylation of a single buried residue in Npm-N, Ser48, by AKT kinase prevents NPM1 oligomerization, causing re-localization of NPM1 and its nucleolar binding partner Arf into the nucleoplasm and activation of the p53 tumor suppressor[7]. However, the molecular mechanism of the order (pentamer) – disorder (monomer) transition of Npm-N remains poorly understood.
To study this mechanism, we first used ensemble CD and Fluorescence Resonance Energy Transfer (FRET) experiments to measure the reaction kinetics, utilizing a change from low to high salt concentration (7.5 to 150 mM) as a trigger for folded pentamer (FP) formation from the disordered monomer (UM). The CD measurements reported on secondary structure formation (folding, Fig-1a; SI Fig-1a&c). Given that FRET between protein monomers labeled with either donor or acceptor dyes can occur only following assembly, ensemble intermolecular FRET provided information about assembly kinetics (Fig-1a; SI Fig-1b&d). At high salt and 10 μM Npm-N concentration, while both folding and assembly were slow, folding (Fig-1b; τF = 1167±7 s) was relatively faster than assembly (Fig-1b; ). Furthermore, the folding time-constant was independent of the Npm-N concentration, while the assembly time-constant decreased monotonically at lower Npm-N concentration and plateaued at a value similar to τF at higher concentration (Fig-1c; concentration range = 2–25 μM). Together these data indicated that in this reaction pathway, the monomer folding step occurs prior to assembly.
Figure 1. Folding-induced-assembly of Npm-N.

(a) Cartoon representation of the N-terminal oligomerization domain of NPM1 (Npm-N; 1–130 AAs). The cyan patches indicate two putative nuclear export signaling (NES) motifs (top). Also shown are schematics of different experimental assays that are used to study coupled folding and assembly kinetics of Npm-N (bottom). (b) Representative folding and assembly kinetic traces of Npm-N ([Npm-N] = 10 μM) at high salt (150 mM NaCl). The points are experimental data, lines are fit to appropriate models (See Methods) yielding τF = 1167±7 s, and . (c) Concentration dependence of the folding and assembly time-constants. The dotted line indicates the average value of τF. (d) smFRET histograms showing the folding pathway of Npm-N at 150 mM NaCl. The solid lines represent fitting of the experimental data to a Gaussian model. The peak at zero is due to molecules lacking an active acceptor dye. The time values in the parenthesis indicate uncertainties due to finite data acquisition time.
Next, we used smFRET experiments on freely diffusing molecules to further study the reaction[8] by monitoring intra-molecular (folding) conformational changes during the reaction. Here, we developed an orthogonal labeling scheme (details in SI Note-1), using a combination of known chemistries, to site-specifically label Npm-N with a FRET dye-pair at positions 1 and 104 (termed Npm-N 1*/104). First, the N-terminus of Npm-N was modified to introduce a ketone functional group using pyridoxal-5′-phosphate (PLP)[9], followed by labeling with a hydroxylamine derivative of Alexa488 dye. In the second step, the native C104 was labeled with the Alexa594 dye using thiol-maleimide chemistry. smFRET (intra-molecular) experiments with Npm-N 1*/104 revealed that switching from low to high salt resulted in a transition from the disordered monomer (FRET efficiency (EFRET) ~ 0.45 (Fig-1d; SI Fig-4) to a new, more compact state with EFRET ~ 0.60 (Fig-1d). This transition occurs much faster than the timescales of either secondary-structure or oligomer formation (Fig-1b&c; SI Fig-5) as measured by ensemble CD or inter-molecular FRET experiments, and therefore represents a transition from UM to a more compact but still disordered monomer (IM). Since the net charge of Npm-N at pH 7.0 is −16[10], charge screening by salt is likely to be the cause of the observed collapse of the Npm-N backbone, as previously observed for other charged IDPs[11]. Our smFRET data also showed a slower process, with a new high-FRET peak (EFRET ~ 0.82; F state; SI Fig-3a) that appeared with a concomitant decrease in the IM peak (Fig-1b; SI Fig-3b), consistent with a two-state process. The time constant of this slower process is similar to that of secondary structure formation by CD, together indicating a two-state transition from IM to FM. We further validated these findings with smFRET experiments on another dual-labeled Npm-N construct, which was conventionally labeled with cys-maleimide chemistry (SI Fig-6). Together, the single-molecule and ensemble data suggest a complex folding-induced-assembly mechanism (Fig. 2c) during pentamer formation at high salt, with disordered monomer (UM) first undergoing a relatively rapid collapse to form a disordered intermediate (IM), which then slowly converts to folded monomer (FM), followed by assembly to folded pentamer (FP) (Fig. 4).
Figure 2. Unfolding-induced-disassembly of Npm-N.

(a) Representative unfolding and disassembly kinetic traces of Npm-N at low salt (7.5 mM NaCl). The points are experimental data, lines are fit to appropriate models (See Methods) yielding τU = 691±13 s, and and . (b) smFRET histograms showing the multi-step unfolding-disassembly process. The time values in the parenthesis indicate uncertainties due to finite data acquisition time. (c) Schematic of alternative order-disorder transition pathways. Folding-induced-assembly pathway is followed at high salt (Fig-1), while assembly-induced-folding pathway is favored at low salt (this figure).
Figure 4. A 2D contour diagram showing different states of Npm-N in alternative order-disorder transition pathways.
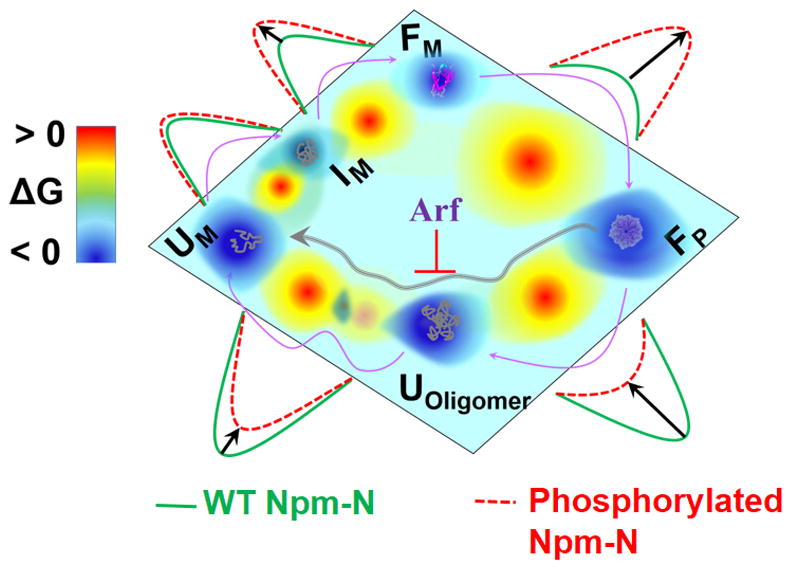
The purple arrows are guides to the eye for the direction of transition among different states under high salt/partner binding (folding-induced-assembly) and low salt (unfolding-induced-disassembly). The effect of phosphor-mutation in each step is shown by a relative activation energy barrier. Also shown is the effect of Arf in arresting Npm-N in ordered pentamer.
We next used the same methods to study the disassembly of pentameric Npm-N at low salt. Intriguingly, we observed that an alternative pathway was followed. While unfolding (by CD, τU = 691±13 s; Fig-2a; SI Fig-1c; SI Fig-7a) was monophasic, decay of intermolecular FRET was observed to have at least 3 phases (Fig-2a; SI Fig-7b). The relatively fast time-constant (τ1D) is comparable to τU from CD, while the other two time-constants (τ2D and τ3D) were ~ 3 and 16 fold slower than τU (SI Table-2). Since the slowest step could be attributed to the transition to monomers, the two relatively faster steps must correspond to the formation of disordered, oligomeric states, with the fastest step consistent with unfolding within pentamers (though other oligomeric species are also possible). smFRET experiments further validated this alternative transition pathway as a multi-step process. Fig-2b shows that the F state initially transitioned to an intermediate state (I*/UOligomer) with EFRET ~ 0.55 at a time-scale comparable to that of unfolding from CD measurements (Fig-2a). Subsequently, the I* state converted relatively slowly into a somewhat lower EFRET state, consistent with the slower phases leading to UM observed in our ensemble inter-molecular FRET data. Both unfolding and disassembly processes were reasonably independent of [Npm-N], as expected for unimolecular processes (SI Table-3). Taken together, our data suggest a multi-step unfolding-induced-disassembly mechanism for Npm-N at low salt, with unfolding occurring within oligomers before multi-step dissociation into monomers. We can infer from the above results and the principle of microscopic reversibility[12] that an assembly-induced-folding mechanism is operative for the forward reaction under this condition (Fig-2c), though we note this reaction occurs only to a very small extent under these conditions that strongly favor disordered monomers.
Once we had established the mechanism of the order-disorder transition pathways of Npm-N, we next examined the effects of phosphorylation. These sites in Npm-N can be grouped as surface exposed and buried[1a]. We first studied the effects of phosphorylation at surface-exposed sites using the phospho-mimicking mutants T95D, S125E and T95D/S125E[1b], and observed significant alterations in the kinetics of order-disorder transitions for the T95D and T95D/S125E mutants, and less pronounced changes for the S125E mutant. All phospho-mutants impeded the rate of assembly more than folding at high salt (SI Table-2; SI Fig-8a&b), thereby temporally decoupling folding and assembly of Npm-N. Thus, the relative time that Npm-N samples the intermediate folded monomeric state (FM) is increased in these mutants, compared to WT protein. We observed that T95D has a more dramatic effect than S125E (SI Fig-8a,b; SI Table-2), which is likely due to their respective locations in pentameric Npm-N[1b, 13]. T95 is part of the folded core while S125 is located in a flexible disordered region, relatively remote from the β-sheet core. Next, we investigated another phospho-mutant, S48E, representing phosphorylation at a buried site which favors the disordered monomeric form[1b]. CD experiments indicated signatures of random coil instead of folded β-sheet structure (F state) for the S48E mutant (SI Fig-9) at high salt, consistent with the severe thermodynamic instability of its pentameric state as estimated previously[1a]. Remarkably, smFRET experiments revealed that even though the FM state was not populated, S48E mutant still formed and remained trapped in the folding intermediate (IM state) at high salt (Fig-3a). Therefore, the effect of phospho-mutation, in general, is manifested by an increase of the activation energy for the 2nd step of the Npm-N folding reaction (IM to FM conversion), and more severely in the subsequent assembly process (Fig-4; discussion in SI Note-2).
Figure 3. Effect of phosphorylation and binding partner on the order-disorder/assembly-disassembly transitions of Npm-N.

(a) smFRET histograms for the phospho-mimicking variant S48E at low-salt, and at two different time points after the addition of 150 mM NaCl. (b) smFRET histograms of Npm-N showing three state (UM, IM and FM) folding pathway induced by Arf6. (c) Various Arf fragments arrest Npm-N in the pentameric state at low salt, which otherwise leads to complete disassembly. (d) Arf fragments counteract the unfolding-disassembly of the phospho-variants at low salt. The horizontal line represents FRET signal observed in absence of any Arf. The time values in the parenthesis in (a) and (b) indicate uncertainties due to finite data acquisition time.
In the reverse reaction, although the identical pathway was followed by these mutants, the rates of unfolding and disassembly were substantially faster relative to WT Npm-N (SI Fig-8c,d; SI Table-2). Intriguingly, the kinetics of unfolding are more severely affected than disassembly, which is the reverse of the effects of phosphorylation on the folding-assembly pathway. The net result was a substantial increase of life-times of the intermediate states (Uoligomer/I*). We suggest that the observed asymmetric effects of phosphorylation on the unfolding-disassembly process are inherent to the alternative pathway that is followed under this condition, as discussed using a model in SI Note-2.
Finally, we used linear binding motifs of Arf tumor suppressor protein to investigate the kinetic effects of the binding partners. NPM1 is known to function in stabilizing Arf in the nucleolus by forming high molecular weight complexes. Binding of a six-residue R-rich fragment of Arf, corresponding to the N-terminus of Arf (Arf6), mediated a disorder-to-order transition of Npm-N via a folding-induced-assembly pathway (SI Fig-10a,b). Furthermore, smFRET experiments revealed an identical three state folding mechanism as observed at high salt (Fig-3b), indicating that the I state is a conserved intermediate species in this pathway. Switching to low salt in the presence of Arf6, and more complex Arf motifs (Arf14 or Arf37) locked Npm-N in the pentameric state (Fig-3c), suggesting that Arf binding in the inter-subunit grooves lined by acidic A1 tracts[1b] within the Npm-N pentamer blocks the F to I* transition. Strikingly, when applied to phospho-mutants, a similar action of the Arf fragments (Fig-3d) was observed, indicating that partner binding can even counteract the effects of phosphorylation. These observations provide a molecular basis for the negative effect of expression of the Arf tumor suppressor which opposes the shuttling activity of NPM1, and leads to altered intracellular localization and partial impairment of its functions[14].
In summary, our study demonstrates that the folding and assembly of Npm-N are slow but coupled processes. While future studies can focus on understanding the mechanistic basis of such sluggish kinetics of this important protein system, we speculate that it could be a consequence of a relatively high energy barrier for proline cis/trans isomerization[15] (each Npm-N monomer has two cis prolines in its pentameric form: P97 and P108). Our data also demonstrate tunable order-disorder transition pathways of Npm-N (Fig-4), which may provide a molecular basis for regulating the multi-functional behavior of NPM1 [4a]. We speculate that the unfolding-induced-disassembly mechanism can be manipulated by phosphorylation as a cellular switch for controlling intracellular shuttling activity of NPM1[16]. Phosphorylation substantially enhances unfolding, but not disassembly, thereby decoupling these two processes. Upon unfolding, putative nuclear export signaling motifs[17], buried within the pentameric core in Npm-N, become accessible for binding to nuclear export factors for shuttling of oligomeric NPM1. Moreover, our proposed kinetic model suggests that the differential effects of phosphorylation are manifested by the distinct mechanistic routes that are followed at low vs. high ionic strength, accounting for the observed asymmetry. Interestingly, partner binding can overcome the effects of phosphorylation, providing a molecular basis for the altered Npm-N function and localization as a result Arf expression. Overall, our findings provide a foundation for further exploration of the mechanism of NPM1 function and disease association at the molecular and cellular levels.
Supplementary Material
Footnotes
This work was supported by US NIH (grants R01 GM066833 to A.A.D. and 1R01GM115634, 2R01GM083159, 2R01CA082491 to R.W.K.), US NSF (grant MCB1121959 to A. A. D.), US NCI Cancer Center (support grant P30CA21765 at St. Jude Children’s Research Hospital to R.W.K.) and ALSAC (to R.W.K.), and by the AHA (postdoctoral fellowship 15POST22520013 to P.R.B.).
References
- 1.a) Mitrea DM, Kriwacki RW. Pacific Symposium on Biocomputing. 2012:152–163. [PubMed] [Google Scholar]; b) Mitrea DM, Grace CR, Buljan M, Yun MK, Pytel NJ, Satumba J, Nourse A, Park CG, Madan Babu M, White SW, Kriwacki RW. Proc Natl Acad Sci U S A. 2014;111:4466–4471. doi: 10.1073/pnas.1321007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rogers JM, Oleinikovas V, Shammas SL, Wong CT, De Sancho D, Baker CM, Clarke J. Proc Natl Acad Sci U S A. 2014;111:15420–15425. doi: 10.1073/pnas.1409122111. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jakob U, Kriwacki R, Uversky VN. Chemical reviews. 2014;114:6779–6805. doi: 10.1021/cr400459c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD. Nature. 2015;519:106–109. doi: 10.1038/nature13999. [DOI] [PubMed] [Google Scholar]
- 4.a) Lindstrom MS. Biochemistry research international. 2011;2011:195209. doi: 10.1155/2011/195209. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bertwistle D, Sugimoto M, Sherr CJ. Molecular and cellular biology. 2004;24:985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrera JE, Correia JJ, Jones AE, Olson MO. Biochemistry. 1996;35:2668–2673. doi: 10.1021/bi9523320. [DOI] [PubMed] [Google Scholar]
- 6.a) Jian Y, Gao Z, Sun J, Shen Q, Feng F, Jing Y, Yang C. Oncogene. 2009;28:4201–4211. doi: 10.1038/onc.2009.275. [DOI] [PubMed] [Google Scholar]; b) Enomoto T, Lindstrom MS, Jin A, Ke H, Zhang Y. J Biol Chem. 2006;281:18463–18472. doi: 10.1074/jbc.M602788200. [DOI] [PubMed] [Google Scholar]
- 7.Hamilton G, Abraham AG, Morton J, Sampson O, Pefani DE, Khoronenkova S, Grawenda A, Papaspyropoulos A, Jamieson N, McKay C, Sansom O, Dianov GL, O’Neill E. Oncotarget. 2014;5:6142–6167. doi: 10.18632/oncotarget.2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Schuler B, Eaton WA. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Deniz AA, Mukhopadhyay S, Lemke EA. J R Soc Interface. 2008;5:15–45. doi: 10.1098/rsif.2007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew Chem Int Ed Engl. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 10.Gasteiger HCE, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: The Proteomics Protocols Handbook. Walker JM, editor. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 11.Muller-Spath S, Soranno A, Hirschfeld V, Hofmann H, Ruegger S, Reymond L, Nettels D, Schuler B. Proc Natl Acad Sci U S A. 2010;107:14609–14614. doi: 10.1073/pnas.1001743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day R, Daggett V. J Mol Biol. 2007;366:677–686. doi: 10.1016/j.jmb.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HH, Kim HS, Kang JY, Lee BI, Ha JY, Yoon HJ, Lim SO, Jung G, Suh SW. Proteins. 2007;69:672–678. doi: 10.1002/prot.21504. [DOI] [PubMed] [Google Scholar]
- 14.a) Yu Y, Maggi LB, Jr, Brady SN, Apicelli AJ, Dai MS, Lu H, Weber JD. Molecular and cellular biology. 2006;26:3798–3809. doi: 10.1128/MCB.26.10.3798-3809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brady SN, Yu Y, Maggi LB, Jr, Weber JD. Molecular and cellular biology. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wedemeyer WJ, Welker E, Scheraga HA. Biochemistry. 2002;41:14637–14644. doi: 10.1021/bi020574b. [DOI] [PubMed] [Google Scholar]
- 16.a) Maggi LB, Jr, Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, Pandolfi PP, Weber JD. Molecular and cellular biology. 2008;28:7050–7065. doi: 10.1128/MCB.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Cell. 1989;56:379–390. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 17.Hingorani K, Szebeni A, Olson MO. J Biol Chem. 2000;275:24451–24457. doi: 10.1074/jbc.M003278200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.