

Abstract
Based on the consistent demonstration of fibrosis of the atrioventricular node surrounded by macrophages and multinucleated giant cells in anti-Ro antibody exposed fetuses dying with heart block, this study focuses on macrophage signaling stimulated by ssRNA associated with the Ro60 protein and the impact of antagonizing innate cell drivers such as TLR7/8. Transcriptome and epigenetic modifications which affect transcription factors, NF-κB and STAT1, were selected to evaluate the phenotype of macrophages in which TLR7/8 was ligated following treatment with either anti-Ro60/Ro60/hY3 RNA immune complexes or transfection with hY3. Based on microarray, TNF and IL6 were among the most highly upregulated genes in both stimulated conditions, each of which was significantly inhibited by preincubation with hydroxychloroquine (HCQ). In contrast, following stimulation of macrophages with either TNF-α or IFN-α, which do not signal through TLR, the resultant gene expression was refractory to HCQ. Ligation of TLR7/8 resulted in increased histone methylation as measured by increased H3K4me2, a requirement for binding of NF-κB at certain promoters, specifically the kB1 region in the TNF promoter (ChIP-qPCR), which was significantly decreased by HCQ. In summary, these results support that the HCQ-sensitive phenotype of hY3 stimulated macrophages reflects the bifurcation of TLR downstream signals involving NF-κB and STAT 1 pathways and for the former dimethylation of H3K4. Accordingly, HCQ may act more as a preventive measure in downregulating the initial production of IFN-α or TNF-α and not affect the resultant autocoid stimulation reflected in TNF-α and IFN-α responsive genes. The beneficial scope of antimalarials in the prevention of organ damage, inclusive of heart block in an anti-Ro offspring or more broadly SLE, may include in part, a mechanism targeting TLR-dependent epigenetic modification.
Keywords: Anti-SSA/Ro autoantibodies, Toll-like receptor 7, Macrophages, Innate immunity, Epigenetics
1. Introduction
The cardiac disease of neonatal lupus (cardiac-NL) represents a pathological readout of passively acquired autoimmunity that occurs during the second trimester of pregnancy and is almost universally associated with maternal antibodies to SSA/Ro ribonucleoproteins [1]. The signature manifestation of cardiac-NL is congenital heart block (CHB) whose histologic correlate is fibrosis of the atrioventricular node surrounded by infiltrating macrophages and giant cells [2,3]. The mechanism by which maternal antibodies initiate and eventuate in cardiac scarring has been challenging to define, in part because the target cardiac antigens are normally sequestered intracellularly. In vitro and in vivo studies suggest that apoptosis may be a key step in facilitating the accessibility of intracellular antigen to extracellular maternal autoantibodies. Previously it was shown that in the cytoplasm Ro60 forms complexes with small non-coding ssRNAs, termed Y RNA, specifically Y3, which are central to intracellular trafficking [4] and cell surface exposure of Ro during apoptosis [5]. Surface accessibility of Ro60 and subsequent binding of maternal autoantibodies generates immune complexes which, when phagocytosed by FcγR on infiltrating macrophages, deliver the Ro-associated ssRNA to the endosomal compartment for ligation with Toll-like receptors (TLRs) 7/8 [6–8] and secretion of proinflammatory and profibrosing cytokines capable of transdifferentiating cardiac fibroblasts [9,10]. Underscoring the importance of macrophage stimulation via TLR ligation in the pathogenesis of disease, both a case control retrospective review of anti-Ro antibody exposed fetuses of mothers with SLE and evaluation of subsequent pregnancies following a cardiac-NL birth suggested that hydroxychloroquine (HCQ), an inhibitor of endosomal TLR ligation, might have a role in both primary and secondary prevention [11,12].
Multiple signal transduction pathways are stimulated upon activation of TLR7/8, leading, for example, to increased expression of TNF, and IL6 and other STAT1-dependent genes. These pathways are classified by their dependence on two transcription factors, NF-κB and STAT1, respectively, which in turn rely on two different post-translational modifications. Binding of NF-κB at certain promoters may require the methylation of histone H3 at lysine 4 (H3K4), which is mediated by Set7 and is associated with promoter activation and release of inflammatory mediators by activated macrophages [13]. STAT1-dependent expression of interferon stimulated genes [14] requires direct modification of STAT1 by histone deacetylase 3 (HDAC3) [15]. While in health, NF-κB and STAT1 prime the innate immune system to mount a “protective” inflammatory response when threatened by viral infection, stimulation by inadvertent anti-Ro60 ssRNA containing immune complexes may result in unwarranted inflammation and scar during remodeling of the fetal heart.
Given that the spectrum of macrophage inflammatory products are a lynchpin of the fetal response to maternal autoantibodies, this study reports the first characterizations of the hY3-stimulated macrophage transcriptome, epigenetic changes involving NFκB and STAT1 and the impact of hydroxychloroquine on these parameters. The identification of hydroxychloroquine’s therapeutic reach may provide a roadmap to extinguish macrophage-derived predisposing factors to cardiac scar and tissue injury more broadly in systemic lupus erythematosus (SLE).
2. Materials and methods
2.1. Preparation of affinity purified anti-Ro60 Ab
Affinity purified anti-Ro60 antibodies were isolated from the serum of a SSA/Ro-positive mother whose child has cardiac-NL. Briefly the Ro60 recombinant protein was coupled to Affigel 10 and an affinity isolate was obtained by affinity column chromatography [16]. Samples are processed by application to Detoxi-Gel Endotoxin Removing Gel (Pierce) to remove any contaminating LPS (<1 pg/ml) [17]. All mothers were enrolled in the Research Registry for Neonatal Lupus and signed informed consent approved by the New York University School of Medicine Institutional Review Board for the use of their sera.
2.2. Cells
Peripheral blood mononuclear cells (PBMC) were obtained from white blood cell concentrates from de-identified healthy donors (New York Blood Center, New York, NY) by centrifugation on Ficoll-Hypaque gradients. Monocytes were positively selected using anti-CD14 microbeads (Miltenyi Biotech) and cultured in Teflon beakers (RPMI 1640/10% FBS) for 7 days in the presence of 10 ng/ml GM-CSF to obtain macrophages [7,10]. For in vitro assays, monocyte-derived macrophages (4 × 105/ml) were plated in growth medium and incubated 37 °C for 48 h. The human monocytic cell line, THP-1, was obtained from the ATCC and cultured in RPMI 1640/10% FBS. THP-1 cells (4 × 105/ml) were differentiated into a “macrophage-like” phenotype in 12-well plates with 0.2 μM phorbol-12-myristate-13-acetate (PMA) for 3 days followed by 48 h in growth medium without PMA as described [18].
2.3. In vitro model of anti-Ro60-mediated injury
For the preparation of immune complexes, human Y3 RNA, prepared from hY3 plasmids (kindly provided by Dr. Sandra Wolin, Yale University School of Medicine, CT) as described [7,19], was subjected to 95 °C for 2 min followed by ice 2 min. Native bovine Ro60 (Arotec Diagnostics) in 50 mM Tris–HCl, pH 7.4, 2 mM MgCl2, 150 mM KCl, 1 mM EDTA and 1 mM DTT was added and incubated 1 h at 37 °C. An immune complex (IC) was generated with the addition of affinity purified anti-Ro60 (10 ug/mL). Prior to its use, there was a pre-incubation (time varied) with PBMC-derived macrophages and PMA-differentiated THP-1 cells in serum free RPMI in the presence or absence of IRS661, a TLR7/8 inhibitor (a gift from Dr. Franck Barrat, Dynavax Technologies, Berkeley, CA), hydroxychloroquine (varied dose, 5–100 uM), or curcumin (20 μM). Macrophages were stimulated with immune complexes IC or transfected with hY3, as described [7], for 4 or 18 h as indicated.
2.4. Microarray analysis
RNA from PBMC-derived macrophages from assays described above was isolated using an RNeasy kit (Qiagen) and processed for preparation of complementary DNA (cDNA) and hybridization to filters.
RNA (5 μg) from control and treated cells was used for microarray analysis. The array analysis was performed at the New York University Cancer Institute Genomics Facility. The RNA quality was assessed on a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA). For each sample, 50 ng total RNA was amplified and labeled with the 2-Cycle cDNA Synthesis and the 2-Cycle Target Labeling and Control Reagent packages (Affymetrix, Santa Clara, CA) following the manufacturers recommendations. Biotin labeled fragmented complementary RNA (15 μg) was hybridized to GeneChip human genome U133A 2.0 arrays (Affymetrix). Hybridization occurred at 45 °C for 16 h in Hybridization oven 640 (Affymetrix). Chips were then washed and stained in a Fluidics station 450 and scanned with a GeneChip Scanner 3000. RMAExpress was used for background adjustment, quintile normalization, summarization and quality analysis [20]. Genes were defined as up-regulated if their expression was increased by 50% in the presence of immune complexes or hY3 transfection relative to untreated macrophages. Down-regulated genes were defined as genes that decreased by 50% upon stimulation with immune complexes or hY3 transfection compared to untreated conditions. Annotation and ontology of the regulated genes was obtained using the Database for Annotation, Visualization and Integrated Discovery, DAVID [21]. DAVID provided ‘tables’ containing functional and ontological details of the regulated genes, ‘charts’ containing ontological categories, pathways etc., over-represented in the gene lists, ‘clusters’ of such ontological categories (which identified redundancies and overlaps), transcription factors over-represented in the promoters of the genes, as well as sub-lists of genes specific for each ontological category. Transcription factor binding sites were also evaluated using DAVID, in a separate analysis [22].
2.5. Real-time quantitative polymerase chain reaction for validation of gene expression data
Quantitative PCR was performed under standard conditions using either PBMC-derived macrophages or PMA-differentiated macrophages. Each sample was assessed in triplicate and reverse transcriptase negative control was also tested to exclude any contamination from DNA amplification. The relative expression ratios were calculated using the 2−ΔΔCt method. The expression level of the GAPDH gene was used to normalize for differences in input cDNA.
2.6. Immunofluorescence
In brief, primary human macrophages were seeded on glass coverslips for 48 h and then stimulated with hY3 (above), R848 (10 uM), or TNF-α (10 ng/mL) for 1 h in the presence and absence of hydroxychloroquine. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.1% Triton X-100/PBS for 20 min. For indirect immunofluorescence: mAb α-H3K4me2 (Active Motif) was used at a dilution of 1:600. After addition of anti-rabbit IgG TRITC (1:300, Sigma–Aldrich) the samples were analyzed by indirect immunofluorescence and images captured by digital acquisition. Nuclei were counterstained with Hoechst 33342 (Sigma).
2.7. Immunoblot
Primary human macrophages were seeded at 5 × 105 cells/well in 6-well plates and stimulated with hY3 or R848 for 3 h in the presence and absence of hydroxychloroquine. Cells were lysed in Tris-buffered saline containing 1% Nonidet P-40 and protease inhibitors (Roche) for 30 min on ice. Lysates were subjected to SDSPAGE, transferred to PVDF membranes, and blocked with 0.1% gelatin in PBS for 2 h at room temperature. After three washes (PBS/0.1% Tween 20), membranes were probed with human α-H3K4me2 Ab (1:1000, Active Motif) overnight at 4 °C. Membranes were stained with IRDye 800CW α-Rb IgG conjugate and analyzed on the Odyssey Infrared Imager (LI-COR Biosciences).
2.8. Chromatin immunoprecipitation
THP-1 cells were seeded at 1.5 × 107/dish on a 150 × 20-mm dish and differentiated with 0.2 μmol/L phorbol myristate acetate (PMA) for 72 h followed by RPMI/10% FBS without PMA (an additional by 72 h). After stimulation with hY3 for 3 h in the presence and absence of hydroxychloroquine, cells were fixed with formaldehyde and chromatin immunoprecipitations were performed using the ChIP-IT Express Enzymatic kit (Active Motif), following the manufacturer’s protocol. Each ChIP sample was also subjected to PCR with primers to the distal promoter of TNF-α including the kB1 site (5′-CCACAGCAATGGGTAGGAGAATG-3′, 5′-TTCATGAAGCTCTCACTTCTCAG-3′) and the coding region (5′-TCCAGACTTCCTTGAGACAC-3′, 5′-TTGTTCAGCTCCGTTTTCACGG-3′). Antibodies used in the ChIP procedure include α-H3K4me2 and a rabbit IgG isotype control. Immunoprecipitated DNA and input DNA were amplified with gene-specific and GAPDH primers (5′-ACAACTTTGGTATCGTGGAAGG-3′, 5′-GCCATCACGCCACAGTTTC-3′) by qPCR. ChIP data is represented as %input.
2.9. shRNA knockdown of HDAC3
MISSION pLKO.1 vectors encoding shRNAs that target HDAC3 (TRCN0000194993) or scrambled sequences were obtained from Sigma. HEK293T cells were seeded in DMEM/10% FBS without antibiotics in a 150 × 20 mm culture dish and grown to 70% confluency. The pLKO.1 vector (4 μg), psPAX2 packaging plasmid (3 μg), and pMD2.G envelope plasmid (1 μg) were transfected into HEK293T cells using GeneJuice (EMD Millipore) according to the manufacturer’s instructions. After 12 h the transfection media was replaced with 5 ml of fresh DMEM/10% FBS/penicillin/streptomycin and viral supernatant was collected at 24 and 48 h post-transfection. The pooled supernatant was spun and filtered. THP-1 cells (5 × 106 cells/flask) were resuspended in 5 ml viral supernatant and 5 μg/ml Polybrene (Santa Cruz) in a T75 flask and incubated at 37 °C for 24 h and stable clones were selected with puromycin (1.5 mg/ml). As described above, THP-1 cells were differentiated with PMA, seeded onto 6-well plates, and stimulated with hY3. Knockdown of HDAC3 was confirmed with qPCR (5′-GCAAGGCTTCACCAAGAGTCT-3′, 5′-AGATGCGCCTGTGTAACGC-3′).
2.10. Immunohistochemistry of fetal hearts
The protocol for obtaining autopsy tissue and a matched normal tissue involved informed consent and approval of the IRB. A heart from a 26 gestational week fetus dying with CHB and an age matched normal heart obtained after elective termination were interrogated for H3K4me2. For the former, a prior autopsy study showed a lympho-histiocytic infiltrate with giant cells in the interventricular septum [3].
2.11. Statistical analysis
For results of qPCR experiments, the differences between transcript levels in the absence and presence of hydroxychloroquine were determined using ANOVA, followed by the Tukey–Kramer test when findings with the ANOVA model were significant. Analyses were performed using GraphPad InStat version 3.10. Values of p < 0.05 were considered significant.
3. Results
3.1. Characterization of macrophages engulfing Ro60 immune complexes and hY3 RNA
In order to evaluate whether Fcγ receptor-mediated delivery of hY3 ssRNA influences macrophage phenotype, microarray data were obtained on human PBMC-derived macrophages treated with anti-Ro60/Ro60/hY3 RNA IC or transfected using a lipid-based transfection reagent without autoantibodies. Treatment with IC resulted in the increased expression of 1223 genes while hY3 transfection up-regulated 780 genes; 287 being common to both conditions. The 287 in common included genes of IC treated macrophages that significantly correlated with hY3-transfected macrophages (R2 = 0.664, P < 0.0001) (Fig. 1A). TNF was among the 30 most highly upregulated genes in both stimulated conditions (Fig. 2A). To confirm the microarray results, TNF expression was assessed by quantitative real time PCR (qRT-PCR) in the hY3 transfected macrophages in the presence or absence of HCQ (Fig. 3A). Preincubation with HCQ (10 μM) decreased the hY3 stimulated TNF-α expression from 26.5 ± 7.8 the mean -fold induction (fold) to 15.7 ± 4.5 fold (4 h, N = 13, p < 0.05) (Fig. 3A). As confirmed by ELISA, preincubation with HCQ significantly inhibited the induction of TNF-α secretion by macrophages in a dose dependent manner. For example, HCQ (5 μM and 10 μM), decreased TNF-α secretion from 1540 ± 234 pg/ml to 480 ± 63 (P = 0.06) and 374 ± 24.5 pg/ml (p = 0.03), respectively (15 h, N = 3) (Fig 3B).
Fig. 1.

Comparison of gene expression in macrophages by transfection with hY3 RNA and stimulation with immune complexes (IC) comprised of anti-Ro60, Ro60 and hY3 RNA. Total gene expression is reported by each dot representing the same transcript probe in macrophages treated with IC (mean value, N = 2) in the absence (A) and presence (B) of cotreatment with IRS661 and hY3-transfection with (B) or without (A) cotreatment with hydroxychloroquine (n = 1) (log2 relative expression shown). For analysis, inclusion of probes in the RNA array profile was based on values of −1 ≤ log2 (hY3/untreated) ≥ 1; expression was substantially shifted for stimulated macrophages given co-treatment of HCQ and IRS661. While IC-treated human macrophages rely on Fcγ receptor-mediated delivery of hY3 RNA, the phenotype of this condition was similar to macrophages given hY3 RNA, which was transfected directly into macrophages using a lipid-based transfection reagent without Abs.
Fig. 2.

Expression of the top genes in macrophages transfected with hY3 RNA and stimulated with immune complexes (IC) comprised of anti-Ro60, Ro60 and hY3 RNA. Each bar represents a transcript of the top 30 up-regulated genes (A) and down-regulated (B) genes in macrophages treated with hY3 and IC. Also shown are plots of the effect of hydroxychloroquine and IRS661, a TLR7 inhibitor, on gene expression of genes that are up-regulated by hY3 and IC treatments, respectively. In addition, shown are expression of genes with a prior link to roles of epigenetic modifications (C). Regarding candidates with a link to epigenetic modifications, there was a trend towards a downregulation of sirtuin 1 (SIRT1), a histone deacetylase.
Fig. 3.

Increased expression of TNF, IL6, and STAT1 in macrophages transfected with hY3 is attenuated by HCQ, a TLR7/8 inhibitor. The upregulation of TNF-α transcript in response to hY3 exposure was attenuated by hydroxychloroquine (N = 13, p < 0.05) (A). hY3 transfection of macrophages resulted in a significant increase in TNF-α secretion compared to untreated cells and this effect was inhibited in a dose dependent manner following pre-incubation with HCQ (B). Preincubation with HCQ also significantly attenuated upregulation of IL-6 (N = 13, p < 0.05) (C) and STAT1 (N = 13, p = 0.003) (D) transcripts. qRT-PCR results are representative of 13 experiments. Bars represent means ± SEM.
For each of the top genes of hY3 treated macrophages following co-treatment with IRS661, a TLR7/8 antagonist, there was attenuation by at least 50% (Not shown). Similar results were obtained with the IC treated cells following co-treatment with IRS661, a TLR7/8 antagonist (Fig. 1B). HCQ was introduced to the hY3 experiments, an approach that was selected because HCQ is clinically relevant. Each of the 287 highest hY3-upregulated genes were inhibited by at least 50% in the presence of HCQ (10 μM) (Fig. 1B). Similar to TNF, preincubation with HCQ resulted in an attenuation of hY3 stimulated IL6 gene expression (147.6 ± 54.5 fold vs 56.7 ± 15.8, N = 13, p < 0.05) (Fig. 3C) and decreased induction of STAT1 mRNA (1.9 ± 0.25 vs 1.2 ± 0.13, respectively, N = 13, p = 0.003) (Fig. 3D). A focus on candidates IL6 and STAT1 was warranted based on the prominence of Stat-dependent genes by an agnostic ranking of categories (Supplemental Table 1). As expected, HCQ did not influence macrophage expression levels of IL6 or STAT1 (Supplemental Fig. 1) following activation by IFN-α, whose signaling occurs independently of TLR7/8.
Compared to the vast number of up-regulated genes, there were fewer down-regulated genes in macrophages treated with IC or transfected with hY3; 161 and 707 respectively; 43 in common (Fig. 1B). Among the 30 most down-regulated genes (Fig. 2B) were transcripts which encoded proteins with functions serving checkpoints to innate immune signals and amplification of inflammation, such as MERTK, an inhibitor of Toll-like receptor (TLR)-mediated innate immune response, and ZNF652, encoding a transcription suppressor.
Given that over 1000 genes were differentially regulated, we used enrichment of categories in the DAVID annotation (Supplemental Table 1) to identify transcription factors, specifically NF-κB and STAT1, as candidates for further evaluation of hY3 dependent epigenetic modifications reflecting enhancement of transcriptional activity.
3.2. Associations of TLR7/8 ligation with broadly based and specific readouts of epigenetic modifications and inhibition of these readouts by HCQ exposure
To profile epigenetic factors and the phenotype of hY3 stimulated macrophages, curcumin, an agent that negatively affects the NF-κB pathway via inhibition of histone lysine acetylase activity was assessed. Treatment of macrophages with hY3 significantly stimulated TNF-α release compared with macrophages alone (2207 ± 227 pg/ml versus 80.5 ± 43.3 pg/ml, respectively, N = 3, p < 0.01) (Fig. 4A). Co-treatment with curcumin (20 μM, with no adverse effects on viability (Not shown)) significantly decreased TNF-α release induced by hY3 (36.2 ± 12.7 pg/ml, N = 3, p = 0.01). Since the decreased secretion of TNF-α observed with curcumin could reflect inhibition of multiple histone deacetylases, an antisense approach was employed to specifically address the dependency on histone deacetylase 3 (HDAC3). Chemical inhibitors of HDAC3 are generally accepted as negative regulators of inflammatory genes [15]. Similar to the results with curcumin, hY3 transfected macrophages resulted in a 27.5-fold induction of STAT1, which was reduced to 13-fold when cells were exposed to HDAC3 shRNA (N = 3, p = 0.03). A similar trend was observed for IL6 (N = 3, p = 0.09) (Fig. 4C). In contrast, TNF expression was not influenced by decreasing HDAC3 (N = 3, p = 0.8) (Fig. 4B). These data are consistent with prior studies demonstrating cross-talk between TLR7/8 and transcriptional activity of STAT1 and expression of IL-6 [23].
Fig. 4.
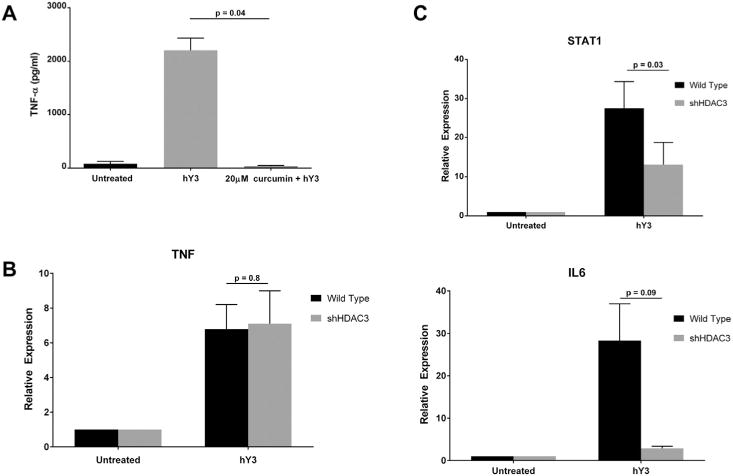
TNF-α protein expression was sensitive to broad inhibition of lysine acetylation while knockdown of histone deacetylase 3 (HDAC3) only affected IL6 and STAT1 expression. Pre-incubation of THP-1 macrophages with curcumin (20 μM), a lysine acetylase inhibitor, attenuated hY3 stimulated increase of TNF-α protein expression (A). shRNA knockdown of histone deacetylase 3 (HDAC3) had no effect on TNF mRNA transcript (qRT-PCR) (B). The hY3 dependent upregulation of STAT1 (p = 0.03) were significantly attenuated, while there was a trend to reduce IL6 (p = 0.09) when levels of HDAC3 were lowered (C). qRT-PCR results are representative of 13 experiments. Bars represent means ± SEM.
Given that knockdown of HDAC3 did not affect hY3 stimulated expression of TNF-α, other potential points of epigenetic control, such as the methylation state of histones at the TNF promoter were assessed. The TNF gene is closely linked to Set7, a 41 kDa lysine-specific SET-domain methyltransferase promoting methylation at lysine 4 of histone H3 (H3K4), a modification commonly associated with transcriptional activation. An increase of H3K4me2 expression was observed in hY3 transfected THP-1 macrophages, compared with no treatment (Red immunofluorescence, Fig. 5A vs B). Similarly, hY3 transfected macrophages demonstrated increased expression of histone dimethylation in the nucleus compared to untreated cells (Fig. 5C vs D, respectively). Consistent with the in vitro data, immunostaining of the AV nodal region of an affected heart from a fetus dying with CHB revealed several infiltrating mononuclear cells intensely stained with antibody recognizing H3K4me2, while the vascular smooth muscle cells did not stain. Not unexpectedly, H3K4me2 staining was also detected in glycogenated myocytes of the AV node in an otherwise healthy heart from an electively terminated fetus (Fig. 5E, F).
Fig. 5.
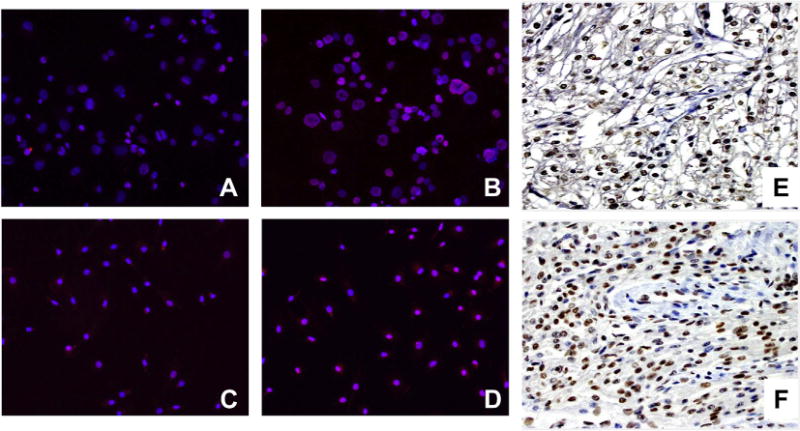
Expression of H3K4me2 was observed in stimulated human macrophages and the AV node of a fetus suffering from CHB. Differential in vitro and in vivo expression of H3K4me2 antigen in THP-1 cells (A, B), human macrophages (C, D), and autopsy heart tissue (E, F). Resting (A, C) and stimulated cells (B, D) were probed with an α-H3K4me2 antibody (red) and nuclei counterstain (blue). Note that from merged images, magenta indicates an overlap of H3K4me2 and the nucleus. Panels E & F correspond to expression of H3K4me2 in normal and autoimmune CHB cardiac tissue, respectively. Macrophage immunofluorescence result is representative of three experiments run in triplicate. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As predicted, preincubation with various concentrations of HCQ resulted in a dose-dependent inhibition of hY3 stimulated expression of H3K4 dimethylation (Fig. 6A) in THP-1 cells. The HCQ effect was specific to TLR7/8 as demonstrated by the fact that the histone marker H3K4me2 was also up-regulated by TNF-α-stimulated human macrophages but not inhibited by HCQ (Fig. 6B). These results were confirmed by immunoblot. Lysates from macrophages transfected with hY3 or treated with the imidazoquinoline, R848, revealed an increase in H3K4me2 compared to resting cells which in both cases was inhibited by preincubation with HCQ (ratio of intensities, 2.43 vs 0.91, respectively, N = 3, p = 0.03) (Fig. 6C). Total histone 3, used as a loading control, was unchanged.
Fig. 6.
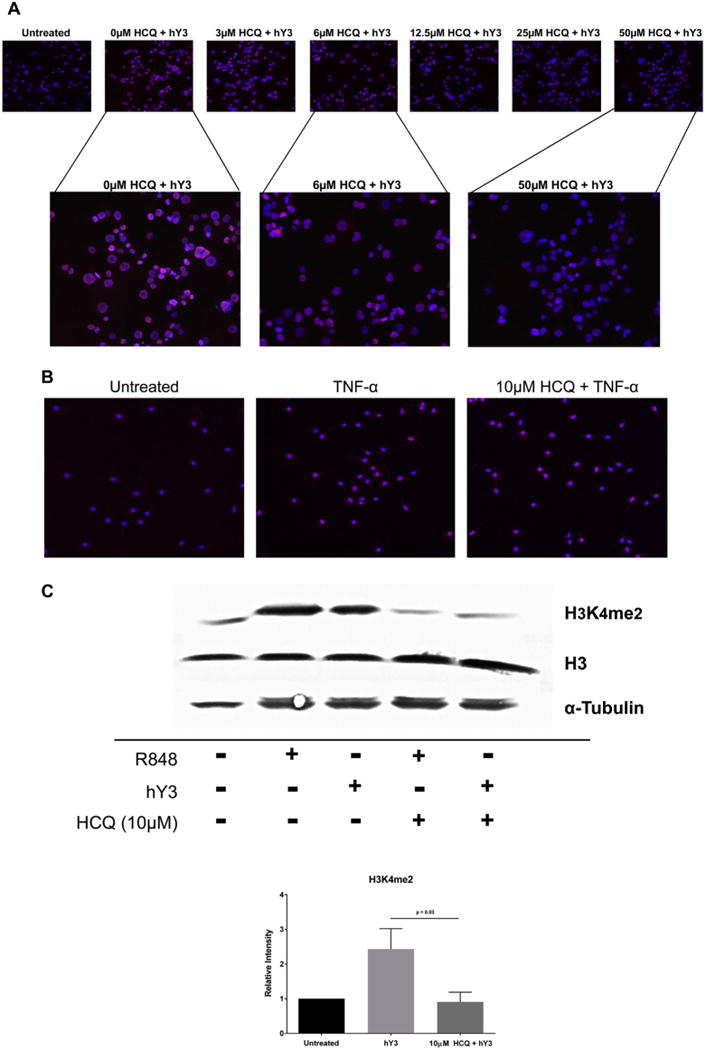
Increased expression of H3K4me2 by THP-1 cells stimulated with hY3 or R848 is TLR7/8 dependent. H3K4me2 expression was assessed by immunofluorescence in resting THP-1 cells and cells stimulated with hY3 (A) or TNF-α (B) in the presence and absence of HCQ. Permeabilized cells were probed with an α-H3K4me2 antibody (red) and nuclei counterstain (blue). Expression was confirmed by Western blot (C). For the latter, pixel intensities were measured and normalized to untreated. α-tubulin and H3 served as loading controls. Results are representative of three experiments run in triplicate. Bars represent means ± SEM. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To address whether H3K4me2, which is associated with increased gene expression, is enriched at the TNF promoter in an NF-κB binding region, ChIP-qPCR was applied [24]. When compared to total input DNA, there was a significant enrichment of H3K4me2 at the TNF promoter at the kB1 region, an enrichment that was inhibited by pre-incubation with HCQ (11.2 ± 1.2% vs 6.6 ± 0.7%, N = 3, p = 0.03, hY3 vs hY3 + HCQ, respectively, Fig. 7).
Fig. 7.

Use of ChIP-qPCR with α-H3K4me2 demonstrates an hY3 dependent and hydroxychloroquine sensitive-enrichment of H3K4me2 at the TNF-α Promoter in an NF-κB Binding Region, kB1. THP-1 cells were stimulated with hY3 with or without HCQ pre-incubation as described in Fig. 4 and then fixed with formaldehyde. ChIP-qPCR with primers in the kB1 distal region of the TNF-α promoter was used to interrogate the presence of H3K4me2. When compared to total input DNA, for the hY3 test condition, there was a significant enrichment of H3K4me2 at the TNF-α promoter at the region kB1. This enrichment was inhibited by pre-incubation with hydroxychloroquine (A). Conventional PCR was used with the same primer set to illustrate loss of H3K4me2 enrichment at the TNF-α promoter in HCQ treated cells (B). The middle lanes show that the immunoprecipitations performed with the rabbit IgG isotype control were negative. The right most lane highlights the increased enrichment of H3K4me2 as quantified in the graph on the left. A rabbit IgG served as an isotype control (C). Results are representative of three experiments. Bars represent means ± SEM.
4. Discussion
Toll-like receptor 7/8 and transcription factors play key roles in the macrophage release of inflammatory mediators, yet information regarding regulatory interactions between these signals remains limited. Such molecular insight might be leveraged for therapeutic consideration in antibody mediated diseases resulting from inadvertent ssRNA-TLR ligation, such as CHB. The studies presented address the pathological role of hY3 by categorizing the pathways invoked by its ligation of TLRs. Exposure of human macrophages to IC comprised of anti-Ro60 Ab, Ro60 protein and its associated hY3 RNA and transfection with hY3 resulted in the increased expression of numerous proinflammatory genes including TNF and IL6. In addition, macrophages also demonstrated increased expression of STAT1-dependent genes which are specific for RNA such as the exonuclease ISG20 (degrades ssRNA) and IFI44L (antiviral activity against Hepatitis C, a ssRNA virus). Expression of TNF and STAT1 transcripts are clearly linked to epigenetic modifications of histones and transcription factors [13,15,25]. Regarding the former, there was an increase H3K4me2 expression by ligation of TLR7/8 in macrophages, e.g. at the kB1 region in the TNF promoter (ChIP-qPCR). These findings, along with the profile of H3K4me2 expression, which was reported in cardiac tissue of a subject with fetal demise, add to a vast body of literature implicating TNF-α as a plausible contributor to the pathogenic scenario at atrioventricular node and endocardium [6,7,26]. HCQ reduced hY3-stimulated H3K4me2 expression and TNF-α release in a dose-dependent manner.
The cross-talk of STAT1 and TLR signaling transduction resulting in inflammatory mediator release has been recently reported [23]. A putative activity of central importance to STAT1-dependent gene expression involves a non-canonical post-translational modification of STAT1 conferred by HDAC3. STAT1 activity, in part, depends on “cycling” through sequential modifications beginning with tyrosine phosphorylation to promote its dimerization and nuclear translocation. In the nuclear compartment, STAT1 undergoes acetylation [27] and this form of STAT1 is vulnerable to dephosphorylation by the tyrosine phosphatase TCP45, leading to termination of STAT signaling. Acetylated but not dephosphorylated STAT1 exits the nucleus prior to its deacetylation by HDAC3 [28,29] to yield a latent STAT1 available for reactivation. The contribution of HDAC3 to TLR-induced inflammatory responses is also supported by the report that genes up-regulated in LPS-treated bone marrow macrophages were significantly reduced in HDAC deficient cells including IL6, IL15, and CXCL11 [15], which is in keeping with the data presented herein, i.e. hY3 dependent up-regulation of IL-6 which was substantially attenuated when levels of HDAC3 were lowered.
Since endosomal TLR activity is dependent on the acidic environment of the endosome, pharmacologic approaches to attenuate TLR-dependent readouts have utilized bafilomycin, a macrolide antibiotic inhibitor of vacuolar-type H+-ATPase, causing an increase in the pH of endosomal compartments, and antimalarials, nonspecific inhibitors of endosomal acidification [30,31]. The mechanism of the latter was recently challenged in a study demonstrating that the inhibitory effect of HCQ is secondary to its direct binding to nucleic acids when concentrated within the endosome, thereby preventing ligands from binding to TLRs [32,33].
The translational connection between HCQ and TLR is also evident in SLE where the cytokine of interest is Type I interferon produced by plasmacytoid dendritic cells [30]. Elevations in IFN-α responsive genes may track disease activity and it has been posited that HCQ may inhibit this signature. However, this effect would be due to attenuation of the cellular production of IFN-α mediated by TLR ligation but not the downstream effects resulting from subsequent stimulation by IFN-α. Indeed in the studies presented herein, HCQ had no effect on the upregulation of IL-6 by exogenous IFN-α. Consistent with this concept, it has been reported that plasmacytoid dendritic cells isolated from lupus patients taking HCQ compared to those not on this drug, produce less IFN-α and TNF-α upon stimulation with TLR9 and TLR7 agonists [34]. Accordingly, treatment with anti-malarials may act more as a preventive measure than one capable of extinguishing the burden of the downstream effects reflected in IFN-α requent SLE flares. Importantly, the in vitro activities of HCQ have been demonstrated at concentrations similar to those found in patients compliant with HCQ therapy (>1000 ng/ml) [35]. In pregnant patients receiving 400 mg of HCQ, levels of 1500 ng/ml (5 μM) are readily achieved (JB personal communication).
This study represents a first-time evaluation of hY3 signaling and the macrophage transciptome with evidence to support a major contribution by NF-κB and STAT1. Both pathways were inhibited by HCQ. These data support a link between TLR7/8 activation and dimethylation of lysine 4 of histone 3 (H3K4me2), which has been shown to increase binding of NF-κB at inflammatory gene promoters. Signaling by TNF-α and IFN-α exposed macrophages, which in part involves NF-κB and STAT1, were refractory to hydroxychloroquine suggesting that hydroxychloroquine may act more as a preventive measure than one capable of extinguishing the transcriptome and epigenetic burden of the downstream effects reflected in TNF-α and INF-α responsive genes. The scope of the beneficial effects of antimalarials, in the prevention of organ damage, inclusive of heart block in an anti-Ro offspring, may include in part a mechanism targeting TLR-dependent epigenetic modification.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health Merit Award (R37 AR042455, 3R37AR042455-21S1, 3R37AR042455-21S2 (J.P.B.)), the Research Registry for Neonatal Lupus (N01-AR-4-2220 (J.P.B.)), a Lupus Foundation of America Lifeline grant (J.P.B.) and the National Institutes of Health (R03 HD069986, 1 R01 HD079951-01A1 (J.P.B.)) and the Australian National Health and Medical Research Council (grant 595989 (J.H.R.)).
Footnotes
Disclosure statement
The authors declare no conflict of interests.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jaut.2015.09.003.
References
- 1.Buyon JP, Friedman DM. Neonatal lupus. In: Lahita RG, Tsokos G, Buyon JP, Koike T, editors. Systemic Lupus Erythematosus. fifth. Academic Press; San Diego: 2011. pp. 541–567. [Google Scholar]
- 2.Clancy RM, Kapur RP, Molad Y, Askanase AD, Buyon JP. Immunohistologic evidence supports apoptosis, IgG deposition, and novel macrophage/fibroblast crosstalk in the pathologic cascade leading to congenital heart block. Arthritis Rheum. 2004;50:173–182. doi: 10.1002/art.11430. [DOI] [PubMed] [Google Scholar]
- 3.Llanos C, Friedman DM, Saxena A, Izmirly PM, Tseng CE, Dische R, Abellar RG, Halushka M, Clancy RM, Buyon JP. Anatomical and pathological findings in hearts from fetuses and infants with cardiac manifestations of neonatal lupus. Rheumatol Oxf. 2012;51:1086–1092. doi: 10.1093/rheumatology/ker515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sim S, Weinberg DE, Fuchs G, Choi K, Chung J, Wolin SL. The subcellular distribution of an RNA quality control protein, the Ro autoantigen, is regulated by noncoding Y RNA binding. Mol Biol Cell. 2009;20:1555–1564. doi: 10.1091/mbc.E08-11-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reed JH, Sim S, Wolin SL, Buyon JP, Clancy RM. A Point Mutation in the SSA/Ro60 autoantigen which prevents Y RNA Binding attenuates a requisite signal for cell surface expression and TLR-dependent inflammation. [abstract] Arthritis Rheum. 2011;63(Suppl):772. [Google Scholar]
- 6.Miranda-Carus ME, Askanase AD, Clancy RM, Di Donato F, Chou TM, Libera MR, Chan EK, Buyon JP. Anti-SSA/Ro and anti-SSB/La autoantibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of TNF-alpha by macrophages. J Immunol. 2000;165:5345–5351. doi: 10.4049/jimmunol.165.9.5345. [DOI] [PubMed] [Google Scholar]
- 7.Clancy RM, Alvarez D, Komissarova E, Barrat FJ, Swartz J, Buyon JP. Ro60-associated single-stranded RNA links inflammation with fetal cardiac fibrosis via ligation of TLRs: a novel pathway to autoimmune-associated heart block. J Immunol. 2010;184:2148–2155. doi: 10.4049/jimmunol.0902248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reed JH, Sim S, Wolin SL, Clancy RM, Buyon JP. Ro60 Requires Y3 RNA for cell surface exposure and inflammation associated with cardiac manifestations of neonatal lupus. J Immunol. 2013;191:110–116. doi: 10.4049/jimmunol.1202849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clancy RM, Askanase AD, Kapur RP, Chiopelas E, Azar N, Miranda-Carus ME, Buyon JP. Transdifferentiation of cardiac fibroblasts, a fetal factor in anti-SSA/Ro-SSB/La antibody-mediated congenital heart block. J Immunol. 2002;169:2156–2163. doi: 10.4049/jimmunol.169.4.2156. [DOI] [PubMed] [Google Scholar]
- 10.Alvarez D, Briassouli P, Clancy RM, Zavadil J, Reed JH, Abellar RG, Halushka M, Fox-Talbot K, Barrat FJ, Buyon JP. A novel role of endothelin-1 in linking toll-like receptor 7-mediated inflammation to fibrosis in congenital heart block. J Biol Chem. 2011;286:30444–30454. doi: 10.1074/jbc.M111.263657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izmirly PM, Kim MY, Llanos C, Le PU, Guerra MM, Askanase AD, Salmon JE, Buyon JP. Evaluation of the risk of anti-SSA/Ro-SSB/La antibody-associated cardiac manifestations of neonatal lupus in fetuses of mothers with systemic lupus erythematosus exposed to hydroxychloroquine. Ann Rheumatic Dis. 2010;69:1827–1830. doi: 10.1136/ard.2009.119263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izmirly PM, Costedoat-Chalumeau N, Pisoni CN, Khamashta MA, Kim MY, Saxena A, Friedman D, Llanos C, Piette JC, Buyon JP. Maternal use of hydroxychloroquine is associated with a reduced risk of recurrent anti-SSA/Ro-antibody-associated cardiac manifestations of neonatal lupus. Circulation. 2012;126:76–82. doi: 10.1161/CIRCULATIONAHA.111.089268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Reddy MA, Miao F, Shanmugam N, Yee JK, Hawkins D, Ren B, Natarajan R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem. 2008;283:26771–26781. doi: 10.1074/jbc.M802800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Au-Yeung N, Mandhana R, Horvath CM. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. Jak-Stat. 2013;2:e23931. doi: 10.4161/jkst.23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109:E2865–E2874. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clancy RM, Neufing PJ, Zheng P, O’Mahony M, Nimmerjahn F, Gordon TP, Buyon JP. Impaired clearance of apoptotic cardiocytes is linked to anti-SSA/Ro and -SSB/La antibodies in the pathogenesis of congenital heart block. J Clin Invest. 2006;116:2413–2422. doi: 10.1172/JCI27803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miranda-Carus ME, Boutjdir M, Tseng CE, DiDonato F, Chan EK, Buyon JP. Induction of antibodies reactive with SSA/Ro-SSB/La and development of congenital heart block in a murine model. J Immunol. 1998;161:5886–5892. [PubMed] [Google Scholar]
- 18.Maess MB, Sendelbach S, Lorkowski S. Selection of reliable reference genes during THP-1 monocyte differentiation into macrophages. BMC Mol Biol. 2010;11:90. doi: 10.1186/1471-2199-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien CA, Wolin SL. A possible role for the 60-kD Ro autoantigen in a discard pathway for defective 5S rRNA precursors. Genes Dev. 1994;8:2891–2903. doi: 10.1101/gad.8.23.2891. [DOI] [PubMed] [Google Scholar]
- 20.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 21.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 22.Lee DD, Zavadil J, Tomic-Canic M, Blumenberg M. Comprehensive transcriptional profiling of human epidermis, reconstituted epidermal equivalents, and cultured keratinocytes using DNA microarray chips. Methods Mol Biol. 2010;585:193–223. doi: 10.1007/978-1-60761-380-0_15. [DOI] [PubMed] [Google Scholar]
- 23.Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. 2014;92:761–769. doi: 10.1038/icb.2014.51. [DOI] [PubMed] [Google Scholar]
- 24.Matsushita N, Endo Y, Sato K, Kurumizaka H, Yamashita T, Takata M, Yanagi S. Direct inhibition of TNF-alpha promoter activity by Fanconi anemia protein FANCD2. PloS One. 2011;6:e23324. doi: 10.1371/journal.pone.0023324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 26.Clancy RM, Backer CB, Yin X, Kapur RP, Molad Y, Buyon JP. Cytokine polymorphisms and histologic expression in autopsy studies: contribution of TNF-alpha and TGF-beta 1 to the pathogenesis of autoimmune-associated congenital heart block. J Immunol. 2003;171:3253–3261. doi: 10.4049/jimmunol.171.6.3253. [DOI] [PubMed] [Google Scholar]
- 27.Ginter T, Bier C, Knauer SK, Sughra K, Hildebrand D, Munz T, Liebe T, Heller R, Henke A, Stauber RH, Reichardt W, Schmid JA, Kubatzky KF, Heinzel T, Kramer OH. Histone deacetylase inhibitors block IFNgammainduced STAT1 phosphorylation. Cell Signal. 2012;24:1453–1460. doi: 10.1016/j.cellsig.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 28.Hancock WW, Akimova T, Beier UH, Liu Y, Wang L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann Rheumatic Dis. 2012;71(Suppl. 2):i46–54. doi: 10.1136/annrheumdis-2011-200593. [DOI] [PubMed] [Google Scholar]
- 29.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 30.Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 31.Kelly KM, Zhuang H, Nacionales DC, Scumpia PO, Lyons R, Akaogi J, Lee P, Williams B, Yamamoto M, Akira S, Satoh M, Reeves WH. “Endogenous adjuvant” activity of the RNA components of lupus autoantigens Sm/RNP and Ro 60. Arthritis Rheum. 2006;54:1557–1567. doi: 10.1002/art.21819. [DOI] [PubMed] [Google Scholar]
- 32.Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. 2011;186:4794–4804. doi: 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- 33.Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, Genest M, Yang H, Shaffer C, Zhao Y, Shen Y, Liu C, Liu D, Mempel TR, Rowbottom C, Chow J, Twine NC, Yu M, Gusovsky F, Ishizaka ST. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol. 2014;85:429–440. doi: 10.1124/mol.113.089821. [DOI] [PubMed] [Google Scholar]
- 34.Sacre K, Criswell LA, McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. 2012;14:R155. doi: 10.1186/ar3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costedoat-Chalumeau N, Amoura Z, Hulot JS, Hammoud HA, Aymard G, Cacoub P, Frances C, Wechsler B, Huong du LT, Ghillani P, Musset L, Lechat P, Piette JC. Low blood concentration of hydroxychloroquine is a marker for and predictor of disease exacerbations in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54:3284–3290. doi: 10.1002/art.22156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.