

SUMMARY
Bacterial cells are fortified against osmotic lysis by a cell wall made of peptidoglycan (PG). Synthases called penicillin-binding proteins (PBPs), the targets of penicillin and related antibiotics, polymerize the glycan strands of PG and crosslink them into the cell wall meshwork via attached peptides. The average length of glycan chains inserted into the matrix by the PBPs is thought to play an important role in bacterial morphogenesis, but polymerization termination factors controlling this process have yet to be discovered. Here, we report the identification of Escherichia coli MltG (YceG) as a potential terminase for glycan polymerization that is broadly conserved in bacteria. A clone containing mltG was initially isolated in a screen for multicopy plasmids generating a lethal phenotype in cells defective for the PG synthase PBP1b. Biochemical studies revealed that MltG is an inner membrane enzyme with endolytic transglycosylase activity capable of cleaving at internal positions within a glycan polymer. Radiolabeling experiments further demonstrated MltG-dependent nascent PG processing in vivo, and bacterial two-hybrid analysis identified an MltG-PBP1b interaction. Mutants lacking MltG were also shown to have longer glycans in their PG relative to wild-type cells. Our combined results are thus consistent with a model in which MltG associates with PG synthetic complexes to cleave nascent polymers and terminate their elongation.
Keywords: cell wall, peptidoglycan, autolysin, penicillin-binding protein
INTRODUCTION
A key component of the bacterial cell envelope is the cell wall matrix, an essential structure that fortifies the cytoplasmic membrane against osmotic rupture and confers cell shape (Höltje, 1998). The cell wall is made up of a meshwork of peptidoglycan (PG), a unique bacterial heteropolymer constructed from long glycan chains of β-1-4-linked N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc) sugars connected by short stem-peptides to form a net-like macromolecule that surrounds the cell. In E. coli and other gram-negative bacteria, the cell wall exists as a thin matrix layer sandwiched between an inner and outer membrane (Vollmer and Seligman, 2010). In contrast, gram-positive bacteria possess a thick layer of PG that surrounds the inner membrane and is exposed to the environment (Vollmer and Seligman, 2010). Any perturbations to the process of PG biogenesis can lead to a catastrophic breach in the cell wall and ultimately cause cell lysis. This sequence of events is exploited by many antibiotics used clinically to treat infections, including penicillin and vancomycin. Unfortunately, resistance to these and other drugs is gradually eroding the effectiveness of our antibiotic arsenal (Taubes, 2008; McKenna, 2013). New treatments capable of overcoming the spread of resistance are therefore sorely needed. An attractive avenue towards the development of such therapies is the identification of additional weak points in the cell wall biogenesis pathway amenable to antibiotic targeting. These novel vulnerabilities are most likely to be discovered through a greater understanding of the fundamental mechanisms underlying the process.
To build the PG layer, synthases called high-molecular weight penicillin-binding proteins (PBPs) are required (Sauvage et al., 2008). These synthases are the primary targets of penicillin and related beta-lactam antibiotics (Tipper and Strominger, 1965). They are subdivided into class A (aPBPs) and class B (bPBPs) enzymes. aPBPs are bifunctional and possess both glycosyltransferase (GT) activity for polymerizing the glycan strands and transpeptidase (TP) activity for crosslinking them. bPBPs, on the other hand, are only known to possess TP activity. In addition to the PG synthases, enzymes capable of cleaving bonds in the cell wall network known as PG hydrolases or autolysins are also important for proper PG assembly (Uehara and Bernhardt, 2011). Several autolysins have recently been implicated in the expansion of the cell wall matrix during growth (Bisicchia et al., 2007; Hashimoto et al., 2012; Singh, L, et al., 2012; Meisner et al., 2013; Domínguez-Cuevas et al., 2013). They are thought to break bonds in the existing structure to allow for the insertion of new material. Enzymes with PG hydrolase activity are also known to be required for cleaving the shared cell wall septum formed during cytokinesis to promote daughter cell separation (Heidrich et al., 2001; Uehara and Bernhardt, 2011). Outside of the handful of autolysins connected with these morphogenic processes, relatively few of the many predicted PG cleaving enzymes encoded by bacteria have a clearly defined physiological function. For example, Escherichia coli encodes seven lytic transglycosylases (LTs) (Slt, MltA-F) capable of cleaving the glycan strands of PG (van Heijenoort, 2011). It also encodes RlpA, which has been shown to have LT activity in Pseudomonas aeruginosa and likely has the same enzymatic function in E. coli (Jorgenson et al., 2014). Besides Slt and RlpA, which have recently been implicated in PG quality control (Cho et al., 2014) and cell division (Jorgenson et al., 2014), respectively, little is known about the functions of the other LT enzymes other than their involvement in general PG turnover, a minor role in daughter cell separation, and potential function in the assembly of trans-envelope structures (Dijkstra and Keck, 1996; Koraimann, 2003; Scheurwater and Burrows, 2011; van Heijenoort, 2011).
Rather than simply hydrolyzing glycan strands, LTs catalyze an intramolecular transglycosylation reaction that forms an anhydro ring between the C1 and C6 atoms of MurNAc (anhMurNAc) coincident with strand breakage (van Heijenoort, 2011) (Fig. 1). Most, if not all, glycan strands in the E. coli PG matrix are capped by an anhMurNAc sugar, and pulse-chase studies indicate that this cap is formed shortly after synthesis (Burman and Park, 1983; Glauner and Höltje, 1990; Höltje, 1998). These observations have long suggested that one or more LT enzymes may function in the processing of nascent PG to terminate strand synthesis. However, the anhMurNAc content of the PG matrix changed surprisingly little in a mutant lacking six LTs (Slt, MltA-E) (Heidrich et al., 2002). This observation has led to the suggestion that either other LT enzymes may be responsible for forming the anhMurNAc caps in the PG matrix, or that the structure is formed by the polymerase enzymes through an as yet uncharacterized termination activity (Kraft et al., 1998; Heidrich et al., 2002).
Figure 1. Lytic transglycosylase reaction.
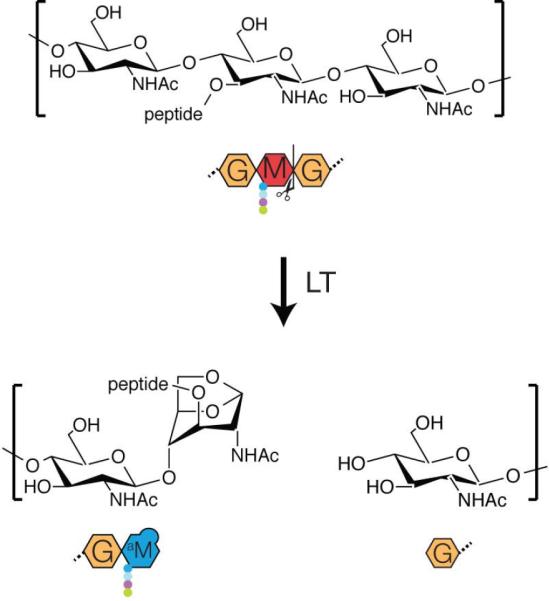
Diagram showing the lytic transglycosylase reaction. Strand cleavage results in the formation of an anhMurNAc sugar at what would normally be the reducing end of the polysaccharide. Shown below the chemical structures are cartoon representations of the substrate and products.
Because of their status as major drug targets, the structures and biochemical activities of the PBPs have been extensively characterized in vitro (Sauvage et al., 2008). However, it remains poorly understood how these proteins are regulated in vivo and what factors coordinate their activity with the PG autolysins that remodel the matrix. We therefore initiated a genetic screen designed to identify regulators of the E. coli aPBPs (Paradis-Bleau et al., 2010). The screen was carried out several years ago and was based on the synthetic lethal phenotype of mutants lacking both of the major aPBPs, PBP1a and PBP1b. We reasoned that a screen for mutations synthetically lethal with PBP1b inactivation would yield mutations in the gene for PBP1a plus mutations in any gene coding for a factor required for PBP1a function. Similarly, the complementary screen for mutations synthetically lethal with PBP1a inactivation should identify factors required for PBP1b activity. Armed with this logic and a plasmid-loss genetic assay, we successfully identified the outer membrane lipoproteins LpoA and LpoB as critical activators of PBP1a and PBP1b, respectively (Paradis-Bleau et al., 2010; Typas et al., 2010).
In this report, we revisit the screen for mutants synthetically lethal with PBP1b. However, instead of using transposon mutagenesis, we searched for genes inducing a lethal phenotype in PBP1b-defective cells when they are overexpressed from a multicopy plasmid. We reasoned that such a screen might identify negative regulators of PBP1a activity or factors causing damage to the PG network that is poorly tolerated when PG biogenesis is crippled by PBP1b inactivation. Multicopy plasmids containing the yceG gene were found to cause a severe growth defect in the absence of PBP1b. Biochemical studies indicated that YceG is a new LT enzyme with endoglycosidase activity capable of cleaving PG at internal positions along the length of glycan strands. Moreover, we found that YceG is a transmembrane protein anchored in the inner membrane and thus has a distinct subcellular localization from the other LT enzymes encoded by E. coli, which are either periplasmic (Slt) or outer membrane-anchored (MltA, B, C, D, E, F and RlpA) (van Heijenoort, 2011; Jorgenson et al., 2014). We therefore renamed this LT as MltG for membrane lytic transglycosylase G. Although loss of MltG function was not lethal or associated with a detectable morphological phenotype, its inactivation led to a significant increase in glycan strand length in the mature PG matrix. Additionally, bacterial two-hybrid analysis indicated that MltG forms a complex with PBP1b, and radiolabeling studies detected MltG-dependent processing of nascent PG in vivo. Our combined results are, therefore, consistent with a model in which MltG associates with PG synthetic complexes to cleave nascent polymers and terminate their elongation. Given its extremely broad conservation relative to other LT enzymes, we suspect that MltG performs a similar function in a wide range of bacteria. Consistent with this possibility, work in the Winkler laboratory currently in preparation for publication indicates that MltG is an essential cell wall assembly factor in Streptococcus pneumoniae (M. Winkler, personal communication).
RESULTS
A multicopy lethal screen for new cell wall biogenesis factors
Although mutants lacking the cell wall synthase PBP1b are viable, they lyse at an elevated frequency (Paradis-Bleau et al., 2014) and are hypersensitive to beta-lactam antibiotics (Schmidt et al., 1981), indicating they have an attenuated capacity for PG synthesis. In such mutants, PBP1a and its activator LpoA become essential (Paradis-Bleau et al., 2010; Typas et al., 2010). We therefore reasoned that a screen for genes rendering PBP1b essential when they are overexpressed from a multicopy plasmid might identify previously uncharacterized inhibitors of PBP1a/LpoA or novel PG-cleaving enzymes that damage the PG matrix to an extent that is not tolerated when cell wall synthesis is compromised by PBP1b inactivation.
To screen for genes causing a conditional, multicopy lethal phenotype, we adapted the plasmid-loss assay used previously to identify synthetic lethal mutant combinations (Bernhardt and de Boer, 2004; Bernhardt and de Boer, 2005; Paradis-Bleau et al., 2010). Strain TU122 [ΔponB] was transformed with the unstable mini-F vector pDY1, encoding ponB and a lacZ reporter under the control of an IPTG-inducible lactose promoter (Plac). TU122/pDY1 was then transformed with a multicopy plasmid library prepared by inserting random (1–4 kb) genomic fragments of E. coli MG1655 [WT] downstream of the arabinose promoter (Para) in a derivative of pBAD33 (Guzman et al., 1995). The resulting transformants were then plated on LB medium containing IPTG and X-gal, but without antibiotic selection for pDY1. Roughly 30,000 colonies were screened in the presence of arabinose and 20,000 in its absence. The vast majority of colonies formed under these conditions were either solid-white or sectored-blue, indicating that PBP1b was not essential and the pDY1 [Plac∷ponB lacZ] plasmid could easily be lost (Fig. 2A–C). However, rare solid-blue colonies were also formed on media with or without arabinose supplementation (Fig. 2A and 2D), suggesting that these cells harbored a multicopy clone rendering PBP1b essential such that pDY1 could not be lost. A subset of these isolates (1/6 without and 5/6 with arabinose) became IPTG-dependent for growth, confirming their reliance on ponB expression from pDY1 for viability. The multicopy plasmids were isolated from these strains and sequenced to identify the inserted genomic region.
Figure 2. MltG overproduction is lethal in cells defective for PBP1b.

A–D. Cells of TU122/pDY1 were transformed with the multicopy E. coli library and plated on LB agar supplemented with IPTG and Xgal. A typical screening plate is shown in panel A, and close-up images highlighting the different colony phenotypes are shown in B–D. See text for details. E. Diagram of the genomic locus resulting in a multicopy lethal phenotype in ΔponB cells. F. Cells of MG1655 or its ΔponB derivative containing plasmid pRY53 [Para::mltG] were grown overnight in M9 minimal maltose medium supplemented with chloramphenicol. Cells in the resulting cultures were harvested, washed twice in an equal volume of M9 salts, and resuspended in M9 salts at an OD600 of 1.0. The resulting cell suspensions were subjected to serial dilution, and 5μL of each dilution were spotted onto M9 agar plates containing 0.2% glucose or arabinose as indicated. Plates were incubated at 30°C for 2 days prior to imaging. G. Overnight cultures of the strains in F as well as the same strains carrying an empty vector control were diluted 1:100 in M9 maltose medium with chloramphenicol and grown to mid-log with shaking at 37°C. The cells were harvested, washed as above, and resuspended to an OD600 of 0.02 in M9 arabinose medium. Growth at 37°C was then monitored by following culture optical density (OD600). H–I. ΔponB cells carrying either an empty vector pCM6 [Para::empty] or plasmid pRY53 [Para::mltG] were grown on LB glucose plates, and then scraped and resuspended in M9 salts for imaging on agarose pads using DIC optics. Bars equal 4 microns.
Plasmids containing a variety of inserts were identified in the screen, which was unlikely to be saturated. The isolate we focused on for this study encoded the gene of unknown function yceG. The yceG has been implicated as potentially playing a role in cell envelope biogenesis in high-throughput studies (Nichols et al., 2011; Babu et al., 2011) (Fig. 2E). Inactivation of yceG leads to a mild hypersensitivity to the beta-lactam mecillinam (Nichols et al., 2011), which we have confirmed (data not shown), and has been reported to suppress a the phenotypes of deletions in some PG hydrolase encoding genes like yebA (Babu et al., 2011). The significance of the latter result is unclear given that growth phenotypes for single mutants of yebA were not reported or observed in more focused studies of its function (Uehara et al., 2009; Singh, SaiSree, et al., 2012). Based on the results presented below we have renamed the yceG gene mltG (membrane lytic transglycosylase G). To confirm that mltG overexpression was responsible for rendering PBP1b essential in the screen isolate, we constructed an expression vector pRY53 producing mltG under Para control and transformed it into WT and ΔponB cells. WT cells harboring pRY53 were viable on solid agar with or without arabinose supplementation and grew to saturation in liquid M9-arabinose medium (Fig. 2F–G). Conversely, plating efficiency of ΔponB cells containing pRY53 was severely compromised on arabinose agar, and these cells became extremely misshapen and lysed following plasmid induction (Fig. 2F–I). Similar phenotypes were observed in WT cells upon higher levels of MltG overproduction (data not shown). We conclude that MltG overproduction results in damage to the PG matrix or a defect in PG biogenesis thus rendering PBP1b essential for growth.
MltG degrades PG
MltG belongs to the YceG family of proteins in the Pfam database, which do not share sequence homology with proteins of known function, and are often misannotated as aminodeoxychorismate lyases (Finn et al., 2013). The structure of E. coli MltG was solved several years ago as part of the structural genomics effort (PDB: 2r1f). It has an extended N-terminus topped by a globular head domain with an observable cleft where residues with the highest level of conservation are found (Fig. 3A). To gain more insight into the potential function of MltG, we searched the Dali protein structure database (Holm and Rosenström, 2010) for structural homologs. MltG was found to share structural features with the catalytic domain of SleB from Bacillus species (PDB: 4fet and 4f55) (Li et al., 2012; Jing et al., 2012). SleB is a lytic transglycosylase important for degrading the PG cortex of Bacillus spores during germination (Giebel et al., 2009; Heffron et al., 2010; Heffron et al., 2011). Although SleB has a unique active site topology compared to other LTs, its catalytic domain displays a similar overall fold and shares similar catalytic residues with other LTs (Li et al., 2012; Jing et al., 2012). The SleB active site is located in a binding cleft at its C-terminus, within which lies the catalytic glutamate (E151) essential for SleB activity (Li et al., 2012). As previously reported by Jing and co-workers (Jing et al., 2012), alignment of MltG with SleB showed a striking structural similarity in the catalytic cleft, including the placement of the likely catalytic glutamate residue (E218 in MltG) (Fig. 3B). Thus, the available structural data suggested that MltG is likely to be a PG-processing enzyme with LT activity.
Figure 3. MltG is structurally similar to SleB.

A. Shown is a space-filling model of the MltG structure (PDB 2r1f) colored to indicate amino acid conservation using ConSurf (Ashkenazy et al., 2010). Note the high conservation of residues in the cleft-forming region. B. Ribbon diagrams showing the structural alignment of the proposed catalytic region of MltG (boxed in A) (burgundy) with the catalytic domain of the SleB lytic transglycosylase (grey) (Li et al., 2012; Jing et al., 2012). The catalytic glutamate is shown in stick form and highlighted with an asterisk. This structural homology was also reported previously by Jing and co-workers (Jing et al., 2012).
To test whether MltG degrades PG, a soluble form of the protein (sMltG) lacking its hydrophobic N-terminal domain was purified along with a control variant of sMltG with an E218Q substitution predicted to disrupt its catalytic activity. PG degradation was assessed using a dye-release assay for PG cleavage (Zhou et al., 1988). Purified E. coli PG sacculi were covalently labelled with the dye remazol brilliant blue (RBB) and incubated with the sMltG proteins (4μM) in addition to buffer and lysozyme controls. The reactions were terminated at different time points by heat inactivation (95°C for 5 min), and any remaining intact PG was pelleted by centrifugation. The amount of dye released into the supernatant was then measured as a proxy for PG cleavage by determining the absorbance at 595 nm. As expected, very little dye was released in the buffer control reaction at all incubation times, and lysozyme promoted a robust level of dye release after only 5 minutes of incubation with the RBB-labeled PG (Fig. 4). Significant dye release by sMltG(WT) was observed after 18 hours of incubation (Fig. 4). This weak PG degradation activity was unlikely to be due to a contaminating PG hydrolase because similarly purified sMltG(E218Q) was equivalent to the buffer control and did not display detectible dye-release activity (Fig. 4). Based on these results, we conclude that MltG is capable of cleaving PG and that the E218 residue is required for catalysis.
Figure 4. MltG degrades cell wall in vitro.

Dye-labeled PG sacculi were incubated with the indicated proteins (4 μM) or buffer alone for the indicated times. Reactions were terminated, undigested PG was pelleted by centrifugation, and the absorbance of the supernatant at 595 nm was measured. Shown are the results from reactions performed in triplicate, with the error bars representing the 95% confidence interval of the measurements.
MltG is a lytic transglycosylase
The structural relatedness of MltG to SleB suggested that it functions as an LT enzyme that cleaves glycan strands of PG with the concomitant formation of anhMurNAc-containing products (Fig. 1). To determine the enzymatic specificity of sMltG, we used high performance liquid chromatography coupled with high resolution mass spectrometry (LC/MS) to analyze the digestion products of unlabeled E. coli PG treated with sMltG. The enzyme generated three main products, each with the same mass to charge ratio (m/z) of 922.389, but increasing retention times and charge states of +1, +2, and +3 (Fig. 5A and 5C). An m/z of 922.389 with a charge state of +1 is consistent with a GlcNAc-anhMurNAc-tetrapeptide product formed via an LT cleavage reaction. Importantly, the same species was produced by the confirmed LT enzyme Slt (Holtje:1975vu; Mett:1980tf; Engel et al., 1991) (Fig. 5B and 5C). The additional products unique to the sMltG reaction with charged states of +2 and +3 are consistent with two possible anhMurNAc-containing products: (i) linear tetra- and hexasaccharide oligomers terminating with an anhMurNAc end, or (ii) GlcNAc-anhMurNAc-tetrapeptide fragments engaged in a peptide crosslink with one or two normal (non-anhydro containing) GlcNAc-MurNAc-tetrapeptide moieties. To distinguish between these possibilities, sMltG reaction products were compared by LC/MS with and without prior reduction by sodium borohydride (Fig. S1). Linear oligomers capped by anhMurNAc ends are nonreducing, and thus should be unaffected by sodium borohydride treatment. However, products terminating with normal MurNAc, as in the possible crosslinked products, are subject to reduction that is detectable as a small shift in the LC/MS profile and change in m/z value. The sMltG products were unaffected by sodium borohydride treatment, indicating that they are likely to be linear oligomers capped by anhMurNAc ends (Fig. S1). Such products indicate that MltG is likely to be an endo-LT that cuts within glycan strands rather than an exo-enzyme like most other characterized LTs that process the glycan chain ends and only liberate monomeric or crosslinked GlcNAc-anhMurNAc-tetrapeptide products. We therefore conclude that MltG is an endolytic LT enzyme that cleaves within glycan strands. Furthermore, MltG did not appear to release cross-linked products from purified PG, suggesting that it favors uncrosslinked strands as a substrate.
Figure 5. MltG is a lytic transglycosylase with endoglycosidase activity.

A–B. Extractedion chromatograms resulting from the digestion of purified, unlabeled PG with MltG (A) or Slt (B). Traces are for ions with an m/z of 922.38 corresponding to LT degradation products. C. Shows the identity of the peaks in A–B along with relevant mass data, including the experimental mass to charge ratio, charge (z), peak identity, and cartoon structure of product molecules.
MltG is part of a widely conserved LT enzyme family
According to the Pfam database, about 70% of bacterial genomes (2167 species out of 3060 sequenced bacterial genomes), including many species of firmicutes, proteobacteria, and actinobacteria, produce proteins that contain MltG(YceG)-like domains (Finn et al., 2013). To better assess the conservation of MltG-like proteins relative to previously characterized LTs, the presence or absence of all LT families identified to date was assessed across a diverse set of 150 representative bacterial species (Ciccarelli et al., 2006) (Fig. 6). MltG-like proteins showed a high degree of conservation, being present in 113/150 (75%) bacterial genomes analyzed (Fig. S2). The only other family with a similar degree of conservation was the Slt superfamily, including Slt, MltC, MltD, MltE, and MltF in E. coli (Fig. S2). With the exception of the RlpA family, all other LT families were present in less than half of the species analyzed. MltG and Slt family members as well as the less well conserved SleB proteins are unique in that they are more evenly distributed between gram-negative and gram-positive bacterial genomes relative to the other families, which are largely skewed towards the gram-negatives (Fig. S2). The conservation profile of MltG-like proteins suggests that these LTs likely play a fundamental role in cell wall processing in a large fraction of bacteria (see Discussion).
Figure 6. MltG is widely conserved in the bacterial domain.

Bacterial phylogenetic tree constructed using iTOL (Letunic and Bork, 2011) and a diversity set of 150 strains. The presence of MltG or other LT family members in a given species is indicated by the colored regions at the outer edge of the tree. A color-coded legend is given in the lower right portion of the panel.
MltG localizes to the inner membrane of E. coli
All of the previously characterized LTs in E. coli as well as RlpA are reported to either be soluble periplasmic proteins (Slt) or outer membrane-anchored proteins (MltA-F and RlpA) (van Heijenoort, 2011; Jorgenson et al., 2014). Depending on the algorithm used, TMHMM (Krogh et al., 2001) or Phobius (Käll et al., 2007), the N-terminal 20–25 amino acids of MltG are predicted to either encode a transmembrane helix or a signal sequence for export, respectively. A cytological assay (Lewenza et al., 2006) was therefore used to experimentally determine whether MltG resides in the inner membrane or periplasm. Cells expressing an MltG-mCherry fusion were hyperosmotically shocked to induce plasmolysis and visualized using fluorescence microscopy. Upon plasmolysis, the cytoplasm shrinks and the inner membrane pulls away from the rest of the envelope. The pattern of MltG-mCherry fluorescence followed the retracted inner membrane and closely resembled that of a control mCherry fusion to a lipoprotein signal sequence from Pal modified for inner membrane retention (Fig. 7A–D). In contrast, mCherry fused to the native Pal signal sequence for outer membrane (OM) targeting displayed a smooth peripheral fluorescence signal in the plasmolyzed cells consistent with an OM localization (Fig. 7A–D). The fluorescent signal of the periplasmic Slt-mCherry control, on the other hand, accumulated in the enlarged periplasmic compartments of the plasmolysis bays, which appear phase-light in the bright field images (Fig. 7A–D). Based on the cytological results, we conclude that MltG is localized to the inner membrane.
Figure 7. MltG is an inner membrane protein.

A–D. Cytological assay for determining the localization of MltG in the cell envelope. Cells expressing mCherry fusions to MltG (A), Slt (B), the wildtype signal-sequence of Pal for outer membrane targeting [ssPal(OM)] (C), or a modified version of ssPal retained in the inner membrane [ssPal(IM)] (D) were grown to an OD600 of 0.3–0.4, osmotically shocked by resuspension in plasmolysis buffer (30% sucrose, 50 mM HEPES (pH 7.4), 40 mM sodium azide), and imaged by mCherry (panel 1) or phase contrast (panel 2) optics. Slt-mCherry serves as a periplasmic control. ssPal(OM)-mCherry and ssPal(IM)-mCherry serve as controls for outer and inner membrane localized proteins, respectively. Arrows highlight signals that track with the inner membrane in plasmolysis bays. Bar equals 4 microns. E. Cells expressing the indicated variants of MltG were harvested 1 hr post induction with 100μM IPTG. The whole cells were left either untreated or treated with 10mM MTSES. After washing out excess MTSES, treated and untreated cells were lysed, their proteins were denatured, and the lysates were exposed to 5mM PEG-malemide (mPEG) (2kDa). Following TCA precipitation, equivalent amounts of total protein were separated by SDS-PAGE and blotted to membranes. MltG variants were detected with affinity purified anti-MltG antibodies. A cartoon diagram of the MltG membrane topology suggested by the SCAM analysis is shown below the blot image with the location of the Cys residue in each variant indicated. Asterisk marks the bands of MltG shifted to a higher molecular weight following reaction with mPEG.
TMHMM predicts that residues 1–4 of MltG reside in the cytoplasm followed by a transmembrane helix (a.a. 5–24) and a large periplasmic domain with the LT active site (a.a. 25–340). To experimentally validate this topology, the scanning cysteine accessibility method (SCAM) was employed (Zhu and Casey, 2007; Pang et al., 2009). Cells expressing MltG variants with single cysteine substitutions at positions 3, 19, and 138 were treated with the (inner) membrane-impermeable sulfhydryl-reactive reagent (2-sulfonatoethyl) methanethiosulfonate (MTSES). MTSES reacts with cysteines exposed to the periplasm or cell surface, but not those in the cytoplasm or buried in the membrane. MTSES accessibility was then determined by lysing the cells and treating with a second sulfhydryl-reactive reagent, a 2 kDa malemide-PEG derivative (mal-PEG). This compound reacts with cysteines that have not already been “protected” by MTSES, producing a 2 kDa shift in molecular weight that can be detected on an immunoblot. A fraction of both MltG(K3C) and MltG(A19C) reacted with mal-PEG and were shifted in the immunoblot whether or not cells were treated with MTSES before lysis (Fig. 7E). Conversely, MltG(S138C) reacted robustly with mal-PEG, but this reaction was almost completely blocked by MTSES pre-treatment (Fig. 7E). The SCAM results are therefore consistent with MltG being a single pass transmembrane protein with an N-in/C-out topology. Thus, MltG is unique among the characterized LTs of E. coli and other gram-negative bacteria in that it is anchored in the inner membrane.
MltG is required for normal glycan strand length distribution
As for all other E. coli LTs, inactivation of mltG alone does not result in a significant growth or morphological phenotype (data not shown). However, given the unique location of MltG in the inner membrane along with its endoglycosidase activity, we suspected that the loss of MltG function might alter the chemical composition of the PG matrix, particularly its anhMurNAc content. PG sacculi were therefore purified from WT and ΔmltG cells, digested with the muramidase mutanolysin, and the resulting muropeptide fragments were analyzed by LC/MS. Although the muropeptide composition of the PG from the two strains was largely similar, significant differences in a subset of muropeptides were observed (Fig. 8, S3, and Table S1). The largest percentage change was observed for uncrosslinked muropeptides with a pentapeptide stem, rising 252% in ΔmltG cells (Fig. 8A, S3, and Table S1). However, due to the extremely low pentapeptide content of WT PG, this change is small in absolute terms and of unclear significance. A more informative change was observed for anhydromuropeptides, which decreased in total by 20% in ΔmltG cells relative to WT, with a 16% reduction in monomeric anhdyromuropeptides, a 26% reduction in dimeric (crosslinked) muropeptides with a single anhMurNAc, and 43% reduction in trimeric (multiply crosslinked) anhydromuropeptides (Fig. 8A, S3, and Table S1). Given that anhydromuropeptides cap the strands, the reduction in anhydromuropeptide content of PG from ΔmltG cells suggests an overall increase in glycan strand length. Consistent with this interpretation, when glycan strand length was directly measured in WT and ΔmltG cell walls, a reproducible shift towards longer glycan strands was observed in strains lacking MltG (Fig. 8B and S4).
Figure 8. Alteration of muropeptide composition and glycan strand length in the PG of MltG defective cells.

A. The abundance of each muropeptide species in WT and ΔmltG PG preparations was determined. Shown are the ratios of selected groupings of muropeptide species in ΔmltG versus WT PG. Pentapeptide, Tetrapeptide, and Tripeptide indicate all muropeptide species (crosslinked and uncrosslinked) containing the indicated type of peptide. Similarly, Anhydo indicates all anhMurNAc containing species, and Tri-Lys-Arg indicates species that were linked to the outer membrane lipoprotein Lpp. Tabulated relative abundances for the complete set of known muropeptide species for each strain are presented in Table S1. B. Detailed comparison of pentapeptide containing muropeptides between the indicated strains showing the differential change between crosslinked and uncrosslinked species. C. PG preparations from the indicated strains were treated with the amidase AmiD to remove the peptide stems. The resulting peptide-free glycan strands were then separated according to length and quantified using a previously described HPLC protocol (Harz et al., 1990). Quantified peak areas for representative samples are shown. Chromatograms are presented in Figure S4.
MltG interacts with PBP1b
The localization of MltG in the inner membrane places it in a position to interact with the similarly localized PG synthases as a potential means to coordinate PG synthesis with MltG-catalyzed processing of glycan strands. To probe for potential interactions between MltG and the aPBP synthases, we performed bacterial two hybrid (BACTH) analysis based on the split-adenylate cyclase fragments T18 and T25 (Karimova et al., 1998). In this assay, if two proteins interact, the T18 and T25 fragments are brought together to form a functional adenylate cyclase that can correct an E. coli cyaA defect by promoting cyclic-AMP (cAMP) production. This complementation is detectable via lacZ induction and the formation of blue color when cells are grown on agar containing X-gal. Production of T18-MltG led to a positive interaction signal in combination with T25-PBP1b and T25-MltG, but not with T25-PBP1a or the T25-Zip control (Fig. 9). Similar results were obtained with the T25-MltG fusion; it showed a positive signal with T18-PBP1b, but not with T18-PBP1a or T18-Zip (Fig. 9). Based on these results we conclude that MltG interacts, directly or indirectly, with the PG synthase PBP1b and also self-interacts. The self-interaction is consistent with the dimeric form observed in the MltG crystal structure (PDB: 2r1f). Whether MltG also interacts with PBP1a is not clear as a negative result in the BACTH assay is not interpretable without additional experimentation. Nevertheless, the BACTH results connect MltG with at least one PG synthase in the inner membrane.
Figure 9. Bacterial two-hybrid interaction of MltG with PBP1b.
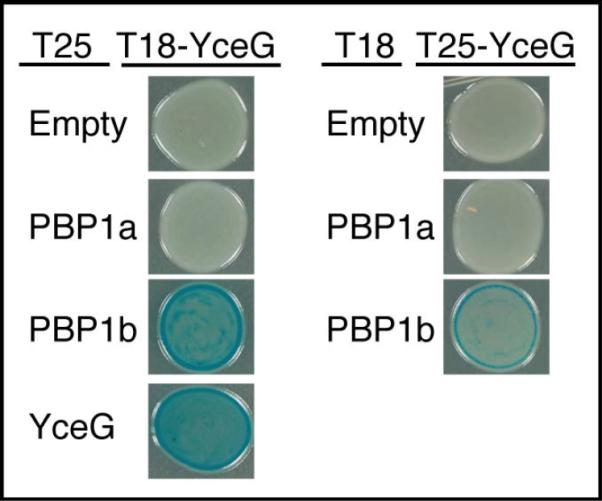
BTH101 cells containing plasmids producing the indicated T25 and T18 fusions were grown to saturation in LB with 50μg/ml amp, 25 μg/ml kan, and 500 μM IPTG and 5 μl of each culture was spotted on LB agar containing 40 μg/ml Amp, 25 μg/ml kan, 500μM IPTG, and 40 μg/ml X-gal.
MltG processes nascent PG in vivo
Our results are consistent with a model in which MltG interacts with PBP1b, and possibly other PG synthases, to process nascent PG strands. To investigate the ability of MltG to cleave nascent PG, we used an in vivo labeling method that detects the turnover of nascent material following beta-lactam treatment. Beta-lactams uncouple glycan chain synthesis from crosslinking by blocking the TP activity of their target PBPs. This uncoupling leads to the degradation of nascent uncrosslinked PG produced by the crippled synthetic machinery (Uehara and Park, 2008; Cho et al., 2014). Following specific inhibition of the bPBPs PBP2 or PBP3 with the drugs mecillinam or cephalexin, respectively, we found that the periplasmic LT Slt was the main enzyme responsible for the destruction of uncrosslinked PG (Cho et al., 2014). However, for the beta-lactam cefsulodin, which specifically targets the aPBPs PBP1a and PBP1b (Curtis et al., 1979), nascent PG degradation was only reduced by 50% upon Slt inactivation. This observation suggested that at least one other LT contributes to the cefsulodin-induced degradation of nascent, uncrosslinked PG. We suspected that this LT might be MltG, and therefore monitored cell wall synthesis and turnover in cells inactivated for Slt, MltG, or both enzymes with or without prior cefsulodin treatment.
PG synthesis and turnover can be monitored by labeling cells with [3H]-meso-diaminopimelic acid (mDAP), an amino acid unique to the peptide moiety of PG. Following [3H]-mDAP addition to growing cells, the progress of PG synthesis is followed by measuring the incorporation of new PG polymers into the matrix. The production of turnover products by LT enzymes can also be monitored following [3H]-mDAP labeling. The labeled GlcNAc-anhMurNAc-peptide turnover products produced are normally imported and processed by a number of enzymes in the cytoplasm for recycling (Park and Uehara, 2008). For our experiments, we used a strain lacking the recycling amidase AmpD to trap PG turnover products as cytoplasmic anhMurNAc-peptides.
As in our previous experiments (Cho et al., 2014), we focused our measurements on the cell elongation phase of PG synthesis by blocking cytokinetic ring assembly via induction of the FtsZ antagonist SulA thirty minutes prior to the addition of antibiotic. After five additional minutes of growth in the presence or absence of cefsulodin, [3H]-mDAP was added to the medium and growth was continued for 10 minutes (1/10th of a mass doubling). Cells were then harvested by centrifugation and extracted with hot (90°C) water. Following centrifugation of the extract, soluble compounds (PG precursors and degradation products) remained in the supernatant and were separated and quantified by HPLC and radiodetection (Cho et al., 2014). Label incorporated into the PG matrix was quantified as radioactivity released from the pellet by lysozyme. Importantly, because of the short labeling period, our measurements monitored the synthesis and degradation of nascent PG as opposed to that of bulk material in the mature matrix.
As observed previously (Cho et al., 2014), cells with a full complement of LTs were capable of incorporating label into PG with or without cefsulodin treatment (Fig. 10). However, cefsulodin induced a substantial increase in the level of turnover products detected. Inactivation of Slt did not affect the total level of label incorporation into PG, but turnover in the presence and absence of cefsulodin was reduced by approximately 50% (Fig. 10). Conversely, inactivation of MltG had little effect on turnover, but total label incorporation into PG increased approximately 20% with or without cefsulodin treatment. The most profound effects on PG synthesis and turnover were observed in cells lacking both Slt and MltG (Fig. 10). Total label incorporation into PG was increased by 60% and turnover was virtually undetectable whether or not cells were treated with cefsulodin. We infer from these results that MltG is capable of processing nascent PG in vivo, and that its activity along with that of Slt can somehow antagonize or slow the incorporation of new material into the PG matrix (see Discussion).
Figure 10. Nascent PG degradation by Slt and MltG following cefsulodin treatment.

Cells of TU278 [ΔlysA ΔampD] or its indicated derivatives were pulse labeled with [3H]-mDAP with or without prior treatment with cefsulodin (100 μg/ml). Soluble metabolites were then extracted with hot water, separated by HPLC, and turnover products were quantified. Radiolabel incorporation into PG was determined by digesting the pellets resulting from the hot water extraction with lysozyme and quantifying the amount of label released into the supernatant by scintillation counting. Results are the average of three independent experiments with the error bars representing the standard deviation. See text and Material and Methods for experimental details.
DISCUSSION
PG glycan strand length and morphogenesis
Bacterial cells come in a wide variety of shapes and sizes, and many bacteria also change their morphology in response to environmental conditions (Young, 2006). Because the PG matrix is the major determinant of bacterial cell shape, it is the collective activities of the various PG biogenesis factors that ultimately define the morphology of a given organism. Alterations of these activities are also undoubtably at the heart of any observed morphological transition. However, despite many years of study, the molecular mechanisms driving cell morphogenesis and morphogenic changes are still relatively poorly understood. One potentially important parameter for defining the size and shape of a bacterium is the length of glycan polymers inserted into the PG matrix by the PG synthases. In support of this possibility, average length of glycan strands is known to vary among different bacterial species and within a given organism upon changes in growth conditions (Glauner et al., 1988; Vollmer et al., 2008). Moreover, modeling studies predict that glycan strand length is a major variable affecting cell width (Furchtgott et al., 2011). Although PG polymerases from different organisms synthesize glycan chains of distinct lengths in vitro (Wang et al., 2008), it has remained unclear how average polymer length is controlled in vivo. Furthermore, without knowing what factors govern strand length in growing cells, perturbations to the length control process to gain an understanding of its role in bacterial growth and morphogenesis have not been possible.
A potential role for MltG as a PG polymerization terminase
In this report, we identify MltG as a novel LT in E. coli and present evidence that it plays a key role in glycan strand length determination (Fig. 11). For many years, it has been known that glycan strands in the E. coli PG matrix are “capped” by anhMurNAc sugars, and that these ends are formed early during synthesis (Höltje, 1998). These observations have led to the proposal that one or more LT enzymes may cleave nascent PG strands to terminate their synthesis (Kraft et al., 1998; Heidrich et al., 2002). Because PG strands undergoing active polymerization by the PG synthases are elongated from the reducing (MurNAc) end at the inner membrane and crosslinked into the matrix at the non-reducing (GlcNAc) end (Perlstein et al., 2007), a putative LT terminase must be able to cleave the glycan backbone within the growing chain (Fig. 11). However, most LTs studied to date are exoglycosidases that cleave glycan strands from a terminus to release soluble disaccharide products containing an anhMurNAc sugar (van Heijenoort, 2011; Lee et al., 2013). MltG, on the other hand, released anhMurNAc-containing oligomeric products from purified E. coli PG, indicating that it has the endoglycosidase activity necessary to function as a terminase. The only other E. coli LT with robust endoglycosidase activity is MltE(EmtA), which can cleave both peptide-free and peptide-containing PG strands (Kraft et al., 1998; van Heijenoort, 2011; Lee et al., 2013). RlpA from P. aeruginosa also has endoglycosidase activity, but is specific for peptide-free glycan strands (Jorgenson et al., 2014). The E. coli RlpA protein is likely to display a similar activity and substrate preference. Both MltE and RlpA, along with all other LTs previously characterized in E. coli, are reported to be localized to the outer membrane (van Heijenoort, 2011; Jorgenson et al., 2014). Although an outer membrane localization for these enzymes does not necessarily preclude them from processing nascent PG to affect glycan strand length, MltG is more likely to do so given its unique location in the inner membrane where glycan polymerization is taking place. Accordingly, a mutant lacking five outer membrane LTs (MltA, MltB, MltC, MltD, and MltE) along with periplasmic Slt did not show a large decrease in the anhMurNAc content of its PG (Heidrich et al., 2002). Conversely, the loss of MltG function alone resulted in a significant (ca. 25%) drop in these chain ends and a corresponding increase in average glycan strand length, suggesting a function for MltG in chain length determination. Notably, this change in polymer length did not result in an observable change in cell width or length, indicating that if glycan chain length plays a significant role in cell morphology, more drastic changes are needed to reveal its importance.
Figure 11. Model for MltG functioning as a PG terminase.

Shown is a model detailing the potential role of MltG as a terminase for PG polymerization. The PG matrix is drawn as in Figure 1 with GlcNAc in orange, MurNAc in red, and anhMurNAc in blue. Small circles attached to MurNAc and anhMurNAc represent the peptide side chains. On the left, an aPBP (green) is polymerizing a nascent glycan strand using its GT domain (circle closest to membrane). Although not shown, the polymers are extended by adding new dissacharides from the precursor lipid II to the growing chain at the membrane-proximal (MurNAc) end. We propose that MltG is recruited to the active polymerase via direct or indirect interactions. There, it cleaves the growing strand endolytically to terminate its elongation. It is not clear where along the strand MltG might cut. The site shown is merely to present the concept of a termination reaction. How much of the polymer remains associated with the polymerase post MltG cleavage and whether this portion of the chain can continue being elongated following cleavage also requires further investigation.
Beyond its mere presence in the inner membrane, structural data provide additional support for nascent PG being the preferred substrate of MltG. The structure of an MltG-like protein from Listeria monocytogenes was recently deposited in the PDB (PDB: 4IIW) (Fig. S5). This structure is closer to the full-length protein than the E. coli structure and includes more of the membrane-proximal domain (Fig. S5). It reveals that this region has a LysM-like fold (Fig. S5), a domain implicated in PG-binding (Buist et al., 2008). MltG thus possesses a predicted PG binding domain quite close to the membrane surface where PG precursors are being polymerized into nascent material. Although the importance of this domain for MltG function is not known, it further implies that newly synthesized PG is the target of MltG cleavage activity. Moreover, BACTH results indicated that MltG forms a complex with the PBP1b polymerase, and radiolabeling analysis demonstrated that MltG is required for the turnover of nascent PG following cefsulodin treatment. Although other models remain possible, the sum of the data presented here makes a compelling case for MltG functioning to endolytically process nascent glycan strands undergoing polymerization to terminate their synthesis (Fig. 11).
MltG-like proteins are found in roughly 70% of sequenced bacteria. Notably, a major fraction of the species lacking MltG homologs are either coccal species or bacteria that lack cell walls, such as Mycoplasma. In contrast, almost all rod-shaped and ovococcal bacteria assessed encode MltG-like proteins, implying that MltG function may be particularly important to promote proper cell elongation. Consistent with this idea, work from Winkler and colleagues in preparation demonstrates that MltG is an essential component of the elongation (peripheral synthesis) machinery in S. pneumoniae (M. Winkler, personal communication). Additionally, although MltG inactivation does not result in a significant growth or morphological phenotype in E. coli, its homologs are reported to be essential in Helicobacter pylori and Mycobacterium tuberculosis (Salama et al., 2004; Zhang et al., 2012). Thus, the processing of PG by MltG-like factors may play a fundamental role in the morphogenesis of a major fraction of rod- and ovoid-shaped bacteria. Further work will be required to test this possibility and to determine why MltG-like factors are essential in some bacteria but not in others. Given the large number of LTs encoded by E. coli, it is likely that one or more of these additional enzymes can substitute for MltG to process nascent PG, but we have yet to identify the (partially) redundant factor(s).
The MltG-PBP1b connection
Our studies have connected MltG with PBP1b in two different genetic assays: the multicopy lethal screen and the BACTH analysis. The reason why MltG overproduction is lethal in a strain defective for PBP1b is not known. Cells inactivated for PBP1b display defects in cell wall biogenesis and envelope integrity (Paradis-Bleau et al., 2010; Typas et al., 2010; Paradis-Bleau et al., 2014), suggesting that PBP1b is one of the main PG synthases in the cell. Thus, the multicopy lethal phenotype of mltG in the absence of PBP1b may simply be due to the artificial elevation of PG cleavage activity in cells that are already experiencing difficulties building their wall. Alternatively, a more intimate regulatory connection between the two proteins is possible. Although it remains unclear whether the PBP1b-MltG association is mediated via direct protein-protein contacts or through their association with a common partner, the BACTH results suggest that the two proteins are in close physical proximity. Thus, it is possible that PBP1b associates with MltG to control its activity such that when PBP1b is inactivated, MltG becomes hyperactive and toxic to the cell when overproduced. If true, such a scenario would represent an interesting case of reciprocal control between a PG synthase and cleavage enzyme, with MltG determining the length of polymers produced by PBP1b, and PBP1b controlling the processing activity of MltG. Additional studies of MltG activity and its interaction partners should reveal the extent of cooperation between MltG and PBP1b in PG biogenesis and whether this interaction is specific or if MltG also associates with other PG polymerases in the cell.
Degradation of nascent PG following cefsulodin treatment
Cefsulodin specifically blocks the TP active sites of PBP1a and PBP1b (Curtis et al., 1979). Thus, in the presence of cefsulodin, the PG produced by these polymerases is likely to be uncrosslinked or at least poorly crosslinked. The increased turnover of nascent PG following cefsulodin treatment indicates that a significant portion of this poorly crosslinked material is targeted for degradation (Cho et al., 2014). Furthermore, the increase in total radiolabel in the combined PG and turnover fractions indicates that PG polymerization is activated by TP inhibition, a phenomenon that has also been detected in vitro (Banzhaf et al., 2012). As observed previously (Cho et al., 2014), inactivation of Slt led to a 50% drop in nascent PG turnover with or without cefsulodin treatment. MltG inactivation, on the other hand, had little effect on nascent PG turnover, but degradation was almost completely blocked in a strain lacking both Slt and MltG. From these results, we infer that Slt is the major LT that intervenes when aPBPs have problems coupling PG polymerization with crosslinking. This activity is consistent with the previously proposed quality control function for Slt (Cho et al., 2014). Results from the Δslt ΔmltG mutant indicate that in the absence of Slt, MltG can step in and process the uncrosslinked or poorly crosslinked PG produced by the targeted aPBPs. This observation suggests the possibility that MltG may also function as a quality control or repair enzyme that can intervene to “rescue” damaged or malfunctioning PG polymerase complexes. However, unlike the Δslt mutant, nascent PG processing following cefsulodin treatment is unaffected by MltG inactivation alone. Additionally, MltG is essential for the activity of the peripheral PG synthesis machinery of S. pneumoniae (Tsui et al., 2015). We therefore favor the idea that the primary function of MltG is to process nascent PG during the course of normal synthesis, but that it is also poised to cleave uncrosslinked strands produced by polymerases experiencing problems coupling GT and TP activities. Indeed, the proposed terminase activity of MltG may be in part controlled by the crosslinking activity of the PBPs. For example, MltG may be poised to cleave glycan strands when the synthesis machinery reaches a region of the PG matrix where acceptor peptides for crosslinking are sparse and uncrosslinked material begins to be produced.
In addition to their effects on nascent PG turnover, MltG and Slt also appear to slow or antagonize PG synthesis. Total PG synthesis during the pulse labeling increased in the absence of MltG and was further enhanced when Slt was also inactivated. The reason for the synthesis increase is not clear, but it suggests the possibility that LT cleavage of nascent PG strands by MltG may force the polymerases to “restart” PG polymerization, which may be rate-limiting relative to chain elongation. In the absence of MltG, the weak endo-LT activity detected for Slt (Lee et al., 2013) may support some termination function in place of MltG. However, in the absence of both enzymes, termination activity may be greatly impaired such that the polymerases restart less often and therefore produce more material during the labeling pulse. Additional studies are required to test this and other possible models related to the interplay between PG cleavage by LT enzymes and synthesis by the polymerases. Nevertheless, the work reported here and in our previous report (Cho et al., 2014) reveals that these two processes are more intimately connected than previously anticipated and may represent an additional control point in PG biogenesis amenable to antibiotic targeting.
EXPERIMENTAL PROCEDURES
Media, bacterial strains and plasmids
Cells were grown in LB [1% tryptone, 0.5% yeast extract, 0.5% NaCl], 0.5xLB-0N [0.5% tryptone, 0.25% yeast extract] or minimal M9 medium (Miller, 1972) supplemented with 0.2% casamino acids and 0.2% maltose. Unless otherwise indicated, antibiotics were used at 25 (chloramphenicol; Cm), 25 (kanamycin; Kan), or 15 (ampicillin; Amp) μg/ml.
The bacterial strains used in this study are listed in Table S3. All E. coli strains used in the reported experiments are derivatives of MG1655 (Guyer et al., 1981). Plasmids used in this study are listed in Table S4. PCR was performed using KOD polymerase (Novagen) for cloning purposes and Taq DNA polymerase (NEB) for diagnostic purposes, both according to the manufacturer's instructions. Unless otherwise indicated, MG1655 chromosomal DNA was used as the template. Plasmid DNA and PCR fragments were purified using the Zyppy plasmid miniprep kit (Zymo Research) or the Qiaquick PCR purification kit (Qiagen), respectively.
Multicopy Library Preparation
MG1655 genomic DNA was partially digested with Sau3AI. Digested DNA fragments were separated on an agarose gel, and fragments in the 1–4kb range were excised and extracted from the matrix. The library of fragments was ligated with BamHI-digested pCM6 plasmid, a derivative of pBAD33 with only a single BamHI cleavage site in the multiple cloning region; the BamHI site in the arabinose promoter region was removed by site-directed mutagenesis. Ligation reactions were transformed into commercial electrocompetent DH5-alpha cells (New England Biolabs) yielding a library of approximately 30,000 transformants. The transformant colonies were then slurried in LB broth. A portion of the slurry was used to prepare plasmid DNA using Qiagen miniprep kit, and the remainder was stored frozen at −80°C.
Multicopy lethal screen
Electrocompetent TU122/pDY1 [ΔlacIZYA ΔponB/Plac∷ponB lacZ] cells were transformed with miniprepped DNA from the multicopy plasmid library. Transformants (>106 to retain library complexity) were selected on LB media containing ampicillin, chloramphenicol, and IPTG (250μM). Amp was required to ensure that cells retained the mini-F plasmid whereas Cm selected for transformants that received a multicopy plasmid, respectively. Induction of ponB expression was controlled with IPTG (250μM). The library was collected and stored in aliquots at −80°C in LB containing 15% glycerol.
For the screen, the TU122/pDY1 multicopy library was plated on LB plates supplemented with IPTG (250mM) and X-gal (40 μg/ml) to identify multicopy plasmids causing a lethal defect in the ΔponB background. For our screen, chloramphenicol (10 μg/mL) was also added to the screening plates to ensure retention of the multicopy plasmid. A total of 30,000 colonies were screened on media without arabinose, and 20,000 colonies were screened on media with 0.2% arabinose to induce Para on the pBAD33-derived vectors, yielding 6 solid-blue isolates that displayed varying degrees of IPTG-dependent growth. Multicopy plasmids were isolated from candidates of interest and their plasmid-insert junctions were sequenced to identify the chromosomal fragment contained within each plasmid.
ΔmltG∷CmR strain construction
E. coli mltG resides upstream of essential genes (holB and tmk). To avoid potential effects on the holB and tmk genes when inactivating mltG, a ΔmltG∷CmR allele was constructed using lambda red recombineering to replace the region between the 2nd codon and the 25th codon of mltG with the CmR cassette as described previously (Yu et al., 2000). This effectively eliminates the transmembrane region of MltG encoded by the gene. Also, upon removing the Cm cassette with FLP recombinase, a frame-shift in mltG is created to disrupt its translation. The CmR cassette was amplified from pKD3 (Datsenko and Wanner, 2000) using the primers 5′-AATATTTAGCCCCACTTTGTGAGCGCCCGAATTAGTCATGAATATCCTCCTTAGTTCCTA-3′ and 5′-GTCTCTTCTTTGATAAGCAATTTGCTGTCGGCAAGATGGCGTGTAGGCTGGAGCTGCTTC-3′ (underlined regions correspond to homology to pKD3 necessary for PCR amplification of the Cm cassette, whereas the non-underlined regions correspond to homology to mltG necessary for recombineering). The resulting PCR product was purified and electroporated into strain TB10 as described previously (Bernhardt and de Boer, 2004), and the recombinants were selected at 30°C on a LB plate containing 25 μg/ml chloramphenicol.
Protein Purification
MltG(27–340), referred to as sMltG, and sMltG(E218Q) with a 6xHis-SUMO (H-SUMO) tag fused to their N-termini were purified from Rosetta 2(λDE3)/pRY54 and Rosetta 2(λDE3)/pRY62, respectively, closely following procedures described previously (Uehara et al., 2010). The sequence of the affinity tag in all cases was MRGSHHHHHHMASG. After purification of the H-SUMO fusion protein by metal-affinity chromatography, the H-SUMO tag was removed using 6xHis-tagged SUMO protease (H-SP) (Bendezú et al., 2009). Cleavage reactions were passed through Ni-NTA resin to remove free H-SUMO and H-SP, yielding a pure preparation of the desired protein. As a result of fusion construction, all proteins have an additional Ser residue at their N-termini.
Overnight cultures were grown at 37°C in LB supplemented with ampicillin (15 μg/ml) and glucose (0.2%). The cultures were diluted 1:200 into 1L of LB supplemented with ampicillin (15 μg/ml) and glucose (0.04%), and cells were grown at 37°C to an OD600 of 0.5. IPTG was added to 0.5 mM, and the cultures were grown for an additional 3 hrs at 37°C. Cells were harvested by centrifugation, and the cell pellets were resuspended in 20 ml of buffer A (50 mM Tris-HCl pH 8.0, 300 mM NaCl) with 20 mM imidazole and stored at −80°C prior to use for protein purification. Cells were thawed and disrupted by passing them through a french pressure cell two times at 15,000 psi. Cell debris was pelleted by centrifugation at 17,000 × g for 30 min at 4°C.
The H-SUMO fusions were purified using 1mL Ni-NTA agarose resin (Qiagen) according to the manufacturer's instructions. Resin was equilibrated in buffer A with 20 mM imidazole. Thawed cell extract (20 mL) was combined with the equilibrated resin and incubated at 4°C with agitation to promote binding of the H-SUMO fusions. The resin was collected by centrifugation at 1000 × g for 3 min at 4°C to remove the non-conjugated material (wash 1), resuspended in buffer A with 20mM imidazole, and loaded onto a Poly-prep Chromatography Column (Biorad). After draining the supernatant, the column was washed with 4 ml buffer A with 20 mM imidazole (wash 2). The fusion proteins were then eluted with buffer A containing 300 mM imidazole and peak fractions were collected. The purified H-SUMO fusions were incubated with a 1:1000 dilution of 6xHis-tagged SUMO protease (H-SP) and dialyzed overnight in buffer D (50mM Tris-HCl pH 8.0, 300mM NaCl, glycerol 10%) at 4°C (Uehara et al., 2010). The following morning, the protein preparation was passed over 1 ml Ni-NTA agarose resin (Qiagen). Untagged proteins eluted from the resin were dialyzed in 1 L of buffer D with 10% glycerol and stored at −80°C.
Dye-release assay for PG hydrolysis
Sacculi were prepared from strain TU163 (Δlpp) and labeled with Remazol-Brilliant Blue as previously described (Uehara et al., 2010). RBB-labelled sacculi (8 μl) were incubated at 37°C with purified sMltG (4μM), sMltG(E218Q) (4μM), lysozyme (4μM) or buffer D at 37°C in 40 μl of PBS buffer (10 mM Na2HPO4, 2 mM KH2PO4, 137 mM NaCl and 2.7 mM KCl, pH 7.4) for the indicated time points. After the indicated time periods, reactions were terminated by incubating them at 95°C for 5 min. Following termination, all reactions were centrifuged at 16,000 × g for 20 min at room temperature. Supernatants were removed, and their absorbance was measured at 595 nm.
Cytological Assay for Membrane Localization
Fluorescence microscopy was performed essentially as described previously (Uehara et al., 2009). The cytological assay for membrane localization of lipoproteins was performed as described previously (Lewenza et al., 2006). Specific growth conditions used for each experiment are given in figure legends.
Overnight cultures of the strains used in Figure 4 were back-diluted to an OD600 of 0.02 in 5 mL M9-maltose supplemented with 100 μM IPTG. Cells were grown at 37°C until they reached OD600 of 0.3, at which point they were harvested by centrifugation (5 min at 5000 × g) and washed in M9-Maltose twice. The washed cells were resuspended in a final volume of 100μl M9-Maltose. 10μl of the cell resuspensions were transferred to a separate tube containing 10μl of 2x plasmolysis buffer [30% sucrose, 50 mM HEPES (pH 7.4), 40 mM sodium azide], and immediately imaged using phase contrast and mCherry optics.
Purification of PG Sacculi
Exponential phase cultures of TB28 and its ΔmltG derivative, RY36, were diluted 1:200 in 300 ml LB and grown at 37°C to OD600 ~0.6. Cells in the resulting cultures were pelleted, resuspended in 4 ml PBS, added slowly to 4 ml boiling 8% SDS (final [SDS] = 4%), and boiled for 30 min. Insoluble material after the boiling was collected by centrifugation (125,000 × g, 1 hr, 25°C). Following several washes with water, the PG pellets were resuspended in 1 ml PBS and incubated with 200 μg/ml α-amylase (Sigma) for 2 hr at 37°C. The sacculi were treated again with pronase (final concentration: 200 μg/ml) overnight at 37°C. The following morning, the sacculi were treated again with pronase (final concentration: 200 μg/ml) for 1 hr at 50°C. Then, 10% SDS was added to each sample to a final concentration of 2%, and the samples were heated at 95°C for 1 hr to inactivate pronase. The de-proteinized sacculi were pelleted by centrifugation (200,000 × g, 20 min, 25°C), washed several times with water to remove SDS, and resuspended in 200 μl water containing 0.02% sodium azide.
LC/MS Analysis of Mutanolysin-Digested Peptidoglycan
Sacculi prepared from WT (TB28) and ΔmltG (RY36) strains were digested with mutanolysin, and the resulting muropeptides were analyzed by LC/MS. Sacculi equivalent to the amount isolated from a 100 ml culture (ca. 1010 cells) were digested overnight with 10 U of mutanolysin (Sigma) in 50 mM sodium phosphate buffer (pH 6.0) at 37°C. The digests were centrifuged (21,000 × g for 30 min at 4°C), and the resulting supernatants were reduced by addition of 1 mg of sodium borohydride in sodium borate buffer (pH 9.0) for 30 min at room temperature. The residual sodium borohydride was inactivated by addition of phosphoric acid to bring the sample pH to 6. The samples were then centrifuged again as before to remove any particulate matter, lyophilized, resuspended in water, and analyzed using an Bruker Maxi Impact Q-TOF LC/MS system with Agilent 1290 HPLC outfitted with a Kinetex core shell C18 column (4.6 by 250 mm, 5 μm resin size, 100 Å pore size) with a Gemini C18 guard column. Muropeptides were eluted at a flow rate of 0.5 ml/min at a temperature of 50°C as follows: isocratic elution with 98% solvent A (water with 0.1% formic acid) and 2% solvent B (acetonitrile with 0.1% formic acid) for 5 min, a 40 min linear gradient to 18% B, a 15 min linear gradient to 90% B, a 10 min linear gradient back to 2% B, and an isocratic elution with 2% B for 5 min. For comparing the abundance of each muropeptide species among different samples, the representative ion species derived from each muropeptide was extracted and integrated from total ion chromatograms using Bruker Data Analysis software. The integrated ion counts were compared after normalizing them to the total ion counts for each sample.
LC/MS Analysis of LT-Digested Peptidoglycan
Sacculi prepared from WT (TB28) were digested with purified sMltG or purified Slt (Morlot et al., 2010) and the resulting muropeptides were analyzed by LC/MS. Sacculi equivalent to the amount isolated from a 100 ml culture (ca. 1010 cells) were digested overnight with 4μM enzyme in 1 x PBS (sMltG) or 25 mM potassium acetate buffer, pH 4.5 (Slt) at 37°C. The digests were centrifuged (21,000 × g for 30 min at 4°C), and the resulting supernatants were either reduced as described with mutanolysin-generated muropeptides or left unreduced. Analysis on LC/MS was performed as described for mutanolysin-generated muropeptides.
Glycan strand analysis
PG sacculi isolated from WT (TB28) and ΔmltG (RY36) strains of E. coli were treated with purified E. coli AmiD (Morlot et al., 2010) for 48 hours at 37°C to release the glycan strands from the PG matrix. The digestion reaction was heat inactivated by incubating at 95°C for 10 min. Uncleaved PG was pelleted, and the remaining supernatant was run directly on a Dionex HPLC system with a Nucleosil C18 column (4.6 by 250 mm, 5 μm resin size, 300 Å pore size) using the method described previously (Harz et al., 1990).
Scanning Cysteine Accessibility Mutagenesis (SCAM)
Overnight cultures of RY8 [ΔmltG] carrying plasmids encoding IPTG-inducible variants of MltG with single cysteine substitutions (pRY66 - pRY70) were grown at 37°C in LB supplemented with chloramphenicol (25 μg/ml). The cultures were diluted 1:100 into 5mL of LB supplemented with chloramphenicol (25 μg/ml), and cells were grown at 30°C to an OD600 of 0.5. IPTG was added to 100 μM to induce expression of the MltG variants and the cultures were grown for an additional 1 hr at 30°C.
An aliquot of each culture (1 ml) was normalized to OD600 of 1, washed twice with 1 ml 1xPBS, and resuspended in a final volume of 90 μl 1xPBS buffer. For the blocking step, tubes were set up containing 90 μl of cell suspension and 10 μl of either 1xPBS (untreated positive- and negative- label control tubes) or 100mM sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES, Biotium), [MTSES]final = 10mM). After rotating at room temperature for 10 min, cells were washed three times with 1 ml 1 x PBS and resuspended in 100 μl PEGylation buffer (60 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1% SDS, 10 M Urea) (Pang et al., 2009). All samples except the negative-control tube were labelled with 5 mM maleimide-polyethylene glycol (Mal-PEG, molecular weight 2 kDa, Nof Corporation Sunbright) for 30 min at 37°C. To remove excess mPEG-mal and minimize its interference on the SDS-PAGE gel, each sample was TCA-precipitated by incubating with 10% TCA on ice for 1hr, harvesting the protein pellet, and washing with 600 μl ice-cold acetone. After letting the samples sit on ice with acetone for 20 min, the protein pellet was collected and resuspended in 100μl PEGylation buffer. The labelling of each sample was assessed using SDS-PAGE followed by immunoblotting with anti-MltG antibody.
Immunoblotting
Polyclonal rabbit antiserum was raised against purified sMltG by Covance Custom Immunology according to their standard protocol. The resulting anti-MltG antibodies were affinity purified using sMltG coupled to AminoLink resin (Pierce) using the standard protocol. For immunoblots, strains were grown as described in the figure legends. At the designated times, cells were harvested and whole-cell extracts were prepared as described previously (Hale and de Boer, 1999). The protein concentration of each extract was determined using the non-interfering protein assay (Genotech) according to the manufacturer's instructions. Protein concentrations were normalized between extracts, the samples were combined with 2x Laemmeli sample buffer with 10% ß-merceptoethanol, and the indicated amount of total protein from each extract was separated on a 12% SDS-PAGE gel (with 4% stacking gel). Proteins were transferred to a PVDF membrane (Whatman) and the membrane was blocked with Rapid-Block (Amresco) for 5 minutes. The membrane was incubated with primary anti-MltG antibodies diluted in Rapid-Block (1:10,000) for 1 hour with gentile agitation at room temperature. The primary antibody solution was then removed and the membrane was quickly rinsed with TBS-T (10mM Tris-HCl pH 7.5, 100mM NaCl, 0.1% Tween-20) and thoroughly washed three times with 25ml TBS-T for 10 minutes each wash. Following the final wash, the membrane was incubated with secondary goat anti-rabbit antibodies conjugated to horseradish peroxidase (Rockland) diluted 1:35,000 in Rapid-Block for 1 hour with gentle agitation at room temperature. After this incubation period, the secondary antibody solution was discarded and the membrane was again quickly rinsed with TBS-T and then thoroughly washed an additional three times with 25ml TBS-T for 10 minutes each. The blot was developed using the Super Signal West Pico system (Pierce) according to the manufacturer's protocol. Chemiluminescence was detected using a BioRad Chemidoc system.
Bacterial two hybrid analysis
BACTH plasmids were co-transformed into BTH101 (cya-99). Transformants were grown overnight in LB supplemented with 50 μg/ml ampicillin, 25 μg/ml kanamycin, and 500 μM IPTG. Spots of 5μl of stationary-phase cultures were then spotted onto LB agar plates containing 50 μg/ml ampicillin, 25 μg/ml kanamycin, and 500 μM IPTG with 40 μg/mL X-gal. Plates were imaged after incubation at 30°C for 24–48 hours.
Measurement of PG synthesis and turnover
The synthesis and turnover of nascent PG was monitored by using ΔlysA ΔampD strains essentially as described previously (Uehara and Park, 2008; Cho et al., 2014). Cells of the relevant strains were grown overnight in M9-glycerol medium supplemented with 0.2% casamino acids. The resulting cultures were then diluted to an OD600 of 0.04 in the same medium and grown to an OD600 between 0.3–0.4 at 30°C. Cell division was then blocked by the induction of sulA from a chromosomally integrated plasmid (pHC859) for 30 min before drug treatment. The cultures were then treated with the cefsulodin (100 μg/ml) for 5 min after adjusting the culture OD600 to 0.3. Following drug treatment, [3H]-mDAP (1 μCi) was added to 1mL of each culture for 10 min (1/10 of the doubling time) to label the newly synthesized PG and its turnover products. After the labeling, cells were pelleted, resuspended in 0.7 ml water, and heated at 90°C for 30 min to extract water-soluble compounds. After the hot water extraction, insoluble material was pelleted by ultracentrifugation (200,000 × g for 20 min at 4°C). The resulting supernatant was then removed, lyophilized, and resuspended in 0.1% formic acid for HPLC analysis. To determine [3H]-mDAP incorporated into the PG matrix, the pellet fraction was washed with 0.7 mL buffer A (20 mM Tris-HCl, pH 7.4, 25 mM NaCl) and resuspended in 0.5 mL buffer A containing 0.25 mg lysozyme. The suspensions were incubated overnight at 37°C. Insoluble material was then pelleted by centrifugation (21,000 × g for 30 min at 4°C), and the resulting supernatant was mixed with 10 mL EcoLite (MP biomedicals) scintillation fluid and quantified in Microbeta Trilux 1450 liquid scintillation counter (Perkin-Elmer).
Conservation Analysis
A previously constructed phylogenic tree containing a diverse representation of bacteria was used to select the genomes for analysis (Ciccarelli et al., 2006). The genome set was assessed for the presence or absence of proteins containing signature Pfams (Finn et al., 2013) that serve as the minimal domain constituent for each major family of LTs.
Supplementary Material
ABBREVIATED SUMMARY.
The cell wall layer of bacteria is composed of long glycan chains crosslinked to one another by attached peptide moieties. It has been known for many years that glycan polymerization and crosslinking are catalyzed by the penicillin-binding proteins (PBPs). However, it has remained unclear how glycan chain length is controlled. Here, we identify MltG as a candidate terminase enzyme that cleaves nascent glycans produced by the PBPs to help determine chain length.
ACKNOWLEDGEMENTS
The authors would like to thank all members of the Bernhardt and Rudner labs for advice and helpful discussions. We would also like to thank Dana Boyd for initial help with the bioinformatic analysis of MltG conservation. We are also grateful to Suzanne Walker and her laboratory for help with LC/MS. Muropeptide analysis was performed using LC/MS at the Harvard Small Molecular Mass Spectrometry Core with the help of Sunia Trauger and Gary Byrd. Special thanks to Malcolm Winkler for communicating results prior to publication. This work was supported by the National Institute of Allergy and Infections Diseases of the National Institutes of Health (R01AI083365). R.Y. was supported in part by predoctoral fellowships from the National Science Foundation (DGE0946799, DGE0644491, DGE1144152), the ASM Robert D. Watkins Fellowship, and the National Institutes of Health (F31AI110046).
REFERENCES
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. - PubMed - NCBI. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, Díaz-Mejía JJ, Vlasblom J, Gagarinova A, Phanse S, Graham C, et al. Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 2011;7:e1002377. doi: 10.1371/journal.pgen.1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banzhaf M, van den Berg van Saparoea B, Terrak M, Fraipont C, Egan A, Philippe J, et al. Cooperativity of peptidoglycan synthases active in bacterial cell elongation. Molecular Microbiology. 2012;85:179–194. doi: 10.1111/j.1365-2958.2012.08103.x. [DOI] [PubMed] [Google Scholar]
- Bendezú FO, Hale CA, Bernhardt TG, de Boer PAJ. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 2009;28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PAJ. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Molecular Microbiology. 2004;52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PAJ. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over Chromosomes in E. coli. Mol Cell. 2005;18:555–564. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, et al. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Molecular Microbiology. 2007;65:180–200. doi: 10.1111/j.1365-2958.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- Buist G, Steen A, Kok J, Kuipers OP. LysM, a widely distributed protein motif for binding to (peptido)glycans. Molecular Microbiology. 2008;68:838–847. doi: 10.1111/j.1365-2958.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- Burman LG, Park JT. Changes in the composition of Escherichia coli murein as it ages during exponential growth. J Bacteriol. 1983;155:447–453. doi: 10.1128/jb.155.2.447-453.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Uehara T, Bernhardt TG. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, Doerks T, Mering, von C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Curtis NA, Orr D, Ross GW, Boulton MG. Affinities of penicillins and cephalosporins for the penicillin-binding proteins of Escherichia coli K-12 and their antibacterial activity. Antimicrob Agents Chemother. 1979;16:533–539. doi: 10.1128/aac.16.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra AJ, Keck W. Peptidoglycan as a barrier to transenvelope transport. J Bacteriol. 1996;178:5555–5562. doi: 10.1128/jb.178.19.5555-5562.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez-Cuevas P, Porcelli I, Daniel RA, Errington J. Differentiated roles for MreB-actin isologues and autolytic enzymes in Bacillus subtilismorphogenesis. Molecular Microbiology. 2013;89:1084–1098. doi: 10.1111/mmi.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel H, Kazemier B, Keck W. Murein-metabolizing enzymes from Escherichia coli: sequence analysis and controlled overexpression of the slt gene, which encodes the soluble lytic transglycosylase. J Bacteriol. 1991;173:6773–6782. doi: 10.1128/jb.173.21.6773-6782.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2013;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchtgott L, Wingreen NS, Huang KC. Mechanisms for maintaining cell shape in rod-shaped Gram-negative bacteria. Molecular Microbiology. 2011;81:340–353. doi: 10.1111/j.1365-2958.2011.07616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel JD, Carr KA, Anderson EC, Hanna PC. The Germination-Specific Lytic Enzymes SleB, CwlJ1, and CwlJ2 Each Contribute to Bacillus anthracis Spore Germination and Virulence. J Bacteriol. 2009;191:5569–5576. doi: 10.1128/JB.00408-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauner B, Höltje JV. Growth pattern of the murein sacculus of Escherichia coli. J Biol Chem. 1990;265:18988–18996. [PubMed] [Google Scholar]
- Glauner B, Höltje JV, Schwarz U. The composition of the murein of Escherichia coli. J Biol Chem. 1988;263:10088–10095. [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 1981;45(Pt 1):135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PA. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ and independent of FtsA. J Bacteriol. 1999;181:167–176. doi: 10.1128/jb.181.1.167-176.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harz H, Burgdorf K, Höltje JV. Isolation and separation of the glycan strands from murein of Escherichia coli by reversed-phase high-performance liquid chromatography. Anal Biochem. 1990;190:120–128. doi: 10.1016/0003-2697(90)90144-x. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Ooiwa S, Sekiguchi J. Synthetic lethality of the lytE cwlO genotype in Bacillus subtilis is caused by lack of D,L-endopeptidase activity at the lateral cell wall. J Bacteriol. 2012;194:796–803. doi: 10.1128/JB.05569-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron JD, Lambert EA, Sherry N, Popham DL. Contributions of Four Cortex Lytic Enzymes to Germination of Bacillus anthracis Spores. J Bacteriol. 2010;192:763–770. doi: 10.1128/JB.01380-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron JD, Sherry N, Popham DL. In vitro studies of peptidoglycan binding and hydrolysis by the Bacillus anthracis germination-specific lytic enzyme SleB. J Bacteriol. 2011;193:125–131. doi: 10.1128/JB.00869-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, et al. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Molecular Microbiology. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Heidrich C, Ursinus A, Berger J, Schwarz H, Höltje J-V. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J Bacteriol. 2002;184:6093–6099. doi: 10.1128/JB.184.22.6093-6099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höltje J-V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X, Robinson HR, Heffron JD, Popham DL, Schubot FD. The catalytic domain of the germination-specific lytic transglycosylase SleB from Bacillus anthracis displays a unique active site topology. Proteins. 2012;80:2469–2475. doi: 10.1002/prot.24140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson MA, Chen Y, Yahashiri A, Popham DL, Weiss DS. The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa. Molecular Microbiology. 2014;93:113–128. doi: 10.1111/mmi.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res. 2007;35:W429–32. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koraimann G. Lytic transglycosylases in macromolecular transport systems of Gram-negative bacteria. Cell Mol Life Sci. 2003;60:2371–2388. doi: 10.1007/s00018-003-3056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AR, Templin MF, Höltje JV. Membrane-bound lytic endotransglycosylase in Escherichia coli. J Bacteriol. 1998;180:3441–3447. doi: 10.1128/jb.180.13.3441-3447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, Heijne, von G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Lee M, Hesek D, Llarrull LI, Lastochkin E, Pi H, Boggess B, Mobashery S. Reactions of all Escherichia coli lytic transglycosylases with bacterial cell wall. J Am Chem Soc. 2013;135:3311–3314. doi: 10.1021/ja309036q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–8. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewenza S, Vidal-Ingigliardi D, Pugsley AP. Direct visualization of red fluorescent lipoproteins indicates conservation of the membrane sorting rules in the family Enterobacteriaceae. J Bacteriol. 2006;188:3516–3524. doi: 10.1128/JB.188.10.3516-3524.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jin K, Setlow B, Setlow P, Hao B. Crystal structure of the catalytic domain of the Bacillus cereus SleB protein, important in cortex peptidoglycan degradation during spore germination. J Bacteriol. 2012;194:4537–4545. doi: 10.1128/JB.00877-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M. Antibiotic resistance: The last resort. Nature. 2013;499:394–396. doi: 10.1038/499394a. [DOI] [PubMed] [Google Scholar]
- Meisner J, Montero Llopis P, Sham L-T, Garner E, Bernhardt TG, Rudner DZ. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Molecular Microbiology. 2013;89:1069–1083. doi: 10.1111/mmi.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; New York: 1972. [Google Scholar]
- Morlot C, Uehara T, Marquis KA, Bernhardt TG, Rudner DZ. A highly coordinated cell wall degradation machine governs spore morphogenesis in Bacillus subtilis. Genes Dev. 2010;24:411–422. doi: 10.1101/gad.1878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, et al. Phenotypic Landscape of a Bacterial Cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang T, Savva CG, Fleming KG, Struck DK, Young R. Structure of the lethal phage pinhole. Proc Natl Acad Sci USA. 2009;106:18966–18971. doi: 10.1073/pnas.0907941106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis-Bleau C, Kritikos G, Orlova K, Typas A, Bernhardt TG. A Genome-Wide Screen for Bacterial Envelope Biogenesis Mutants Identifies a Novel Factor Involved in Cell Wall Precursor Metabolism. PLoS Genet. 2014;10:e1004056. doi: 10.1371/journal.pgen.1004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010;143:1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JT, Uehara T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan) Microbiol Mol Biol Rev. 2008;72:211–27. doi: 10.1128/MMBR.00027-07. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlstein DL, Zhang Y, Wang T-S, Kahne DE, Walker S. The direction of glycan chain elongation by peptidoglycan glycosyltransferases. J Am Chem Soc. 2007;129:12674–12675. doi: 10.1021/ja075965y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol. 2004;186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- Scheurwater EM, Burrows LL. Maintaining network security: how macromolecular structures cross the peptidoglycan layer. FEMS Microbiol Lett. 2011;318:1–9. doi: 10.1111/j.1574-6968.2011.02228.x. [DOI] [PubMed] [Google Scholar]
- Schmidt LS, Botta G, Park JT. Effects of furazlocillin, a beta-lactam antibiotic which binds selectively to penicillin-binding protein 3, on Escherichia coli mutants deficient in other penicillin-binding proteins. J Bacteriol. 1981;145:632–637. doi: 10.1128/jb.145.1.632-637.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, L S, Amrutha RN, Reddy M. Three redundant murein endopeptidases catalyze an essential cleavage step in peptidoglycan synthesis of Escherichia coli K12. Molecular Microbiology. 2012;86:1036–1051. doi: 10.1111/mmi.12058. [DOI] [PubMed] [Google Scholar]
- Singh SK, SaiSree L, Amrutha RN, Reddy M. Three redundant murein endopeptidases catalyse an essential cleavage step in peptidoglycan synthesis of Escherichia coliK12. Molecular Microbiology. 2012;86:1036–1051. doi: 10.1111/mmi.12058. [DOI] [PubMed] [Google Scholar]
- Taubes G. The bacteria fight back. Science. 2008;321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- Tipper DJ, Strominger JL. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci USA. 1965;54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui H-CT, Zheng JJ, Magallon AN, Yunck R, Rued BE, Boersma MJ, et al. Identification of a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae. Molecular Microbiology. 2015;1:1–2. doi: 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010;143:1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Bernhardt TG. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr Opin Microbiol. 2011;14:698–703. doi: 10.1016/j.mib.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Park JT. Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and septation. J Bacteriol. 2008;190:3914–3922. doi: 10.1128/JB.00207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Dinh T, Bernhardt TG. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J Bacteriol. 2009;191:5094–5107. doi: 10.1128/JB.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Parzych KR, Dinh T, Bernhardt TG. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 2010;29:1412–1422. doi: 10.1038/emboj.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heijenoort J. Peptidoglycan hydrolases of Escherichia coli. Microbiol Mol Biol Rev. 2011;75:636–663. doi: 10.1128/MMBR.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Seligman SJ. Architecture of peptidoglycan: more data and more models. Trends in Microbiology. 2010;18:59–66. doi: 10.1016/j.tim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Blanot D, de Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- Wang T-SA, Manning SA, Walker S, Kahne D. Isolated peptidoglycan glycosyltransferases from different organisms produce different glycan chain lengths. J Am Chem Soc. 2008;130:14068–14069. doi: 10.1021/ja806016y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KD. The selective value of bacterial shape. Microbiol Mol Biol Rev. 2006;70:660–703. doi: 10.1128/MMBR.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 2012;8:e1002946. doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Chen S, Recsei P. A dye release assay for determination of lysostaphin activity. Anal Biochem. 1988;171:141–144. doi: 10.1016/0003-2697(88)90134-0. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Casey JR. Topology of transmembrane proteins by scanning cysteine accessibility mutagenesis methodology. Methods. 2007;41:439–450. doi: 10.1016/j.ymeth.2006.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.