

Abstract
Considerable evidence suggests that environmental factors, including diet and cigarette smoke, are involved in the pathogenesis of colon cancer. Carcinogenic nitroso compounds (NOC), such as N-nitrosodimethylamine (NDMA), are present in tobacco and processed red meat, and NOC have been implicated in colon cancer. Azoxymethane (AOM), commonly used for experimental colon carcinogenesis, is an isomer of NDMA, and it produces the same DNA adducts as does NDMA. Heterocyclic aromatic amines (HAAs) formed during the combustion of tobacco and high-temperature cooking of meats are also associated with an elevated risk of colon cancer. The most abundant carcinogenic HAA formed in tobacco smoke is 2-amino-9H-pyrido[2,3-b]indole (AαC), whereas 2-amino-3,4-dimethylimidazo[4,5-f]quinoline (MeIQ) is the most potent carcinogenic HAA formed during the cooking of meat and fish. However, the comparative tumor-initiating potential of AαC, MeIQ, and AOM is unknown. In this report, we evaluate the formation of DNA adducts as a measure of genotoxicity, and the induction of colonic aberrant crypt foci (ACF) and dysplastic ACF, as an early measure of carcinogenic potency of these compounds in the colon of male A/J mice. Both AαC and AOM induced a greater number of DNA adducts than MeIQ in the liver and colon. AOM induced a greater number of ACF and dysplastic ACF than either AαC or MeIQ. Conversely, based on adduct levels, MeIQ-DNA adducts were more potent than AαC- and AOM-DNA adducts at inducing ACF. Long-term feeding studies are required to relate levels of DNA adducts, induction of ACF, and colon cancer by these colon genotoxicants.
Keywords: tobacco smoke, heterocyclic aromatic amines, azoxymethane, DNA adducts, aberrant crypt foci (ACF), sialomucin-expressing ACF, mucin-depleted foci (MDF)
Introduction
Colon cancer is the third most common cancer and the second leading cause of cancer-related deaths in the United States [Siegel et al. 2013]. However, there is great international variation in colon cancer incidence, with industrialized countries in general having a much greater incidence than developing countries [Boyle et al. 1985]. These international differences in incidence are largely attributed to environmental factors, of which diet is one of the most important. Several foods have been implicated in increasing colon cancer risk, including red meat and processed meat [Norat et al. 2005]. Indeed, in 2007 the WCRF/AICR stated that the evidence is sufficiently strong to conclude that consumption of red meat and processed meat is a risk factor for colon cancer [WCRF/AICR 2007]. Moreover, The Working Group of IARC recently classified consumption of processed meat as “carcinogenic to humans” (Group 1) on the basis of sufficient evidence for colorectal cancer, and concluded additionally that a positive association with the consumption of processed meat was found for stomach cancer. The Working Group classified consumption of red meat as “probably carcinogenic to humans” (Group 2A) based on substantial epidemiological data showing a positive association between consumption of red meat and colorectal cancer and the strong mechanistic evidence [Bouvard et al. 2015].
The etiologic agent(s) in red and processed meats responsible for the increased colon cancer risk are uncertain, but N-nitroso compounds (NOC) have received much attention as one class of possible causative agents. N-Nitrosodimethylamine (NDMA) is a prototypical NOC, which occurs in smoked and salted fish, cured meat, and tobacco smoke [IARC 2002; Chowdhury 2013]. Several epidemiological studies have reported associations between consumption of processed meats (e.g., bacon, sausage, hot dogs) containing NOC and increased risk of colon or rectal cancer [Knekt et al. 1999; Norat et al. 2005; Loh et al. 2011; Chowdhury 2013; Zhu et al. 2014]. Further, in rats given the colon carcinogen 1,2-dimethylhydrazine (a precursor of azoxymethane, AOM) and fed meat-containing diets processed by different methods, only the group showing a greatly elevated concentration of fecal NOC had a significantly greater number of mucin-depleted foci, a preneoplastic marker of colon cancer [Santarelli et al. 2010]. To become genotoxic, NOCs, such as NDMA, must be metabolically activated by the cytochrome P450 enzymes, leading to production of α-hydroxynitrosamines, which rearrange to reactive intermediates that alkylate DNA bases. In the case of NDMA, methylation occurs at N-7 (N7-me-dG) and O-6 atoms of guanine (O6-me-dG), and the O-4 position of thymine [Mirvish 1995]. O6-Me-dG is a highly mutagenic DNA adduct, primarily inducing G-to-A transitions [Loechler et al. 1984; Belinsky et al. 1989].
The commonly used colon-specific carcinogen AOM is an isomer of NDMA. Similar to NDMA, AOM is metabolically activated by CYP2E1 [Sohn et al. 2001]. The reactive AOM intermediate, a methyldiazonium ion, methylates guanine to produce the DNA adduct, O6-me-dG [Sohn et al. 2001], the accumulation of which leads to G-to-A transitions, mutations in tumor suppressor genes (Apc and p53) and oncogenes (β-catenin and K-ras), and eventually initiates carcinogenesis [Loechler et al. 1984; Perse and Cerar 2011]. AOM is also a strong inducer of precancerous lesions (aberrant crypt foci, ACF) and dysplastic ACF in rodent colons [Perse and Cerar 2011].
The charring of meats and fish at high temperature produces heterocyclic aromatic amines (HAAs), which induce tumors at multiple sites, including the colon [IARC 2002; Sugimura et al. 2004]. The HAA 2-amino-3,4-dimethylimidazo[4,5-f]quinoline (MeIQ) is produced by a complex reaction with creatine, sugar, and amino acids during cooking [Skog 1993]. MeIQ is the most potent carcinogen formed during the cooking of meat and fish, and it induces cancers in multiple organs in animal models, including in the forestomach, liver, and colon in CDF1 mice [Ohgaki et al. 1986] and in C57BL/6 mice [Fujita et al. 1999], and induced ACF in rats [Ochiai et al. 2005]. The major DNA adduct (N-(deoxyguanosin-8-yl)-dG-C8-MeIQ (dG-C8-MeIQ) formed in vitro by MeIQ [Tada et al. 1994] also occurs in vivo in rodents [Tada et al. 1994; Tang et al. 2013].
Epidemiologic studies have consistently shown a positive association between the smoking of tobacco and an elevated risk for colon cancer [Giovannucci 2001; IARC 2002; Vineis et al. 2004; Gram et al. 2009]. In 2009, the International Agency for Research on Cancer concluded that there is sufficient evidence for a causal relationship between smoking and colon cancer [IARC 2009]. Tobacco contains NOCs [IARC 2002], and the burning of tobacco produces some HAAs [IARC 2002; Sugimura et al. 2004], with the most abundant being 2-amino-9H-pyrido[2,3-b]indole (AαC), a pyrolysis product of amino acids, such as tryptophan [IARC 2002]. AαC is present in mainstream tobacco smoke at levels ranging from 60 to 258 ng/cigarette [Manabe et al. 1990; Zhang et al. 2011]. AαC is a hepatocarcinogen in CDF1 mice [Sugimura et al. 2004] and a strong inducer of lacI transgene mutations and ACF in the colon of C57BL/6 mice [Zhang et al. 1996; Okonogi et al. 1997]. AαC undergoes bioactivation and subsequently forms the DNA adduct N-(deoxyguanosin-8-yl)-dG-C8-AαC; (dG-C8-AαC) in C57BL/6 mice [Tang et al. 2013] and human hepatocytes [Nauwelaers et al. 2011]. A previous study reported that both AαC and MeIQ induce the formation of DNA adducts in liver and extrahepatic tissues of mice [Tang et al. 2013]. However, DNA adduct formation does not necessarily indicate carcinogenicity [Nagao et al. 2001].
A main goal of our study was to understand the tumor-initiating potential of AαC, MeIQ, and AOM by comparing the formation of hepatic and colonic DNA adducts combined with the induction of ACF in the colons of mice. ACF are the first identifiable epithelial lesions to develop after administration of colon carcinogens, representing early biomarkers of colon cancer development [Bird 1998; Papanikolaou et al. 2000; Rosenberg et al. 2009]. We also enumerated dysplastic ACF, as this subclass of ACF are thought to be the ACF that progress to tumors. Finally, mouse strains vary greatly in their susceptibility to AOM. Notably, of 33 inbred mouse strains examined, the commonly used C57BL/6J mouse is one of the most resistant strains to the development of colon tumors, whereas the A/J mouse strain is highly sensitive to colon carcinogenesis, yielding about 30 times more the tumors than C57BL/6J [Liu et al. 2012]. To our knowledge, there is no study of toxicity of heterocyclic aromatic amines in the A/J mouse strain. Consequently, for this study, we employed the A/J mouse strain to compare DNA adduct formation and ACF of these 3 colon genotoxicants.
Materials and Methods
Caution
AαC, MeIQ, and AOM are carcinogenic to rodents and should be handled in a well-ventilated hood with appropriate protection.
Chemicals and materials
AOM was purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). AαC and MeIQ were purchased from Toronto Research Chemicals (Toronto, ON, Canada). [13C10]-2′-Deoxyguanosine (dG) (isotopic purity 99%) was purchased from Cambridge Isotopes (Andover, MA). 5′-[13C1015N5]-Deoxyguanosine-5′-phosphate (isotopic purity 99%), DNase I (Type IV, from bovine pancreas), alkaline phosphatase (from Escherichia coli), and nuclease P1 (from Penicillium citrinum) were purchased from Sigma-Aldrich. Phosphodiesterase I (from Crotalus adamanteus venom) was purchased from Worthington Biochemical Corp. (Lakewood, NJ). All solvents were high-purity B & J Brand from Honeywell Burdick and Jackson (Muskegon, MI). The diet ingredients were purchased from Dyets Inc. (Bethlehem, PA). N-(Deoxyguanosin-8-yl)-AαC (dG-C8-AαC) and N-(deoxyguanosin-8-yl)-MeIQ (dG-C8-MeIQ) were synthesized by reaction of their N-acetoxy-HAA derivatives with dG, [13C10]-dG, or [13C1015N5]-dG (5 mg/mL) in 100 mM potassium phosphate buffer (pH 8.0), and purified by preparative HPLC [Tang et al. 2013]. O6-me-dG and [2H3C]-O6-me-dG were synthesized as previously reported [Weng et al. 2007].
Animals and treatments
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Minnesota. Forty-four male A/J mice, 6 weeks of age, were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were housed four per cage with corn cob bedding and kept in a 12-h light/dark cycle. The mice were given water and a standard AIN-93G rodent diet containing 7% soybean oil (Dyets Inc., Bethlehem, PA) for 7 days. One week after arrival, the mice were switched to a modified AIN-93G powdered diet containing 13% Primex and 10% soybean oil. The high fat diet was employed because previous studies reported a rapid induction of ACF and tumor formation in rats given a high fat diet containing HAAs [Nakagama et al. 2002; Nakagama et al. 2005]. The carcinogen-containing diets were prepared by mixing AαC or MeIQ with a granular carrier, sucrose, for 10 min, using a kitchen blender (Kitchen Aid, St Joseph, MI, USA). The remaining diet components, except for the fat, were added and mixed for an additional 10 min. The mixture of soybean oil and Primex was added and blended for an additional 10 min. Water (30% by weight) was added to the diets to make a dough. The dough was then extruded with a sausage maker to make the pelleted diet. The diet was freeze-dried for 24 hours until the water content was less than 9% moisture by weight. The diets were kept in sealed bags at −20° C until feeding. The diet was replenished every third day. Chemical analysis, by LC/MS, showed that AαC and MeIQ were stable under these storage conditions for up to 4 weeks [carcinogen levels were within 100 ± 20% of target values, mean ± SD, n = 6 independent measurements (unpublished data, R. Turesky)]. The ingredient composition and the caloric contents of the AIN-93G and modified AIN-93G diets are provided in the supplementary data (Table S-1).
After 1 week of acclimation to the high-fat diet, mice were divided into four groups. Three groups were fed diets containing either no carcinogen (negative control) (n=8), MeIQ (300 ppm) (n=12), or AαC (400 ppm) (n=12). The fourth group was injected i.p. with AOM (5 mg/kg body weight) once a week for 6 weeks (positive control) (n=12). AOM was dissolved in 0.9% NaCl, and the injection volume was approximately 0.2 mL, depending on body weight. The doses employed were based upon previous long-term carcinogen feeding studies of AαC and MeIQ in CDF1 mice [Fujita et al. 1999; Sugimura et al. 2004], short-term (3 month) mutagenesis studies in transgenic C57BL/6 mice [Zhang et al. 1996], and our DNA adduct kinetic study in C57BL/6 mice [Tang et al. 2013]. The AOM dosing regimen is based on a standard dosing protocol used to induce ACF and colon tumors in mice [Papanikolaou et al. 2000; Rosenberg et al. 2009; Liu et al. 2012]. In this study, the dose of AOM (5 mg/kg body weight) was half of the dose (10 mg/kg body weight) commonly used for mouse model assays, because AOM dosed at 10 mg/kg body weight was found to be toxic to the A/J mice on this high-fat, with all mice dying within 24 hours of administration. In the case of AOM, O6-me-dG adduct levels peaked in colonic epithelial cells at 6 hour post-injection of AOM [Tan et al. 2011]. Thus, for the present study, four mice from the negative control, MeIQ- and AαC-treated groups were sacrificed, by CO2 asphyxiation, at the end of week 4, and four mice from the AOM group were sacrificed 6 hours after the final AOM dose. The remaining mice in all groups were fed the high-fat diet without carcinogen for an additional 8 weeks to allow for development of ACF.
Liver and colon were harvested for DNA adduct measurement. Livers were rinsed with 50 mM phosphate-buffered saline (PBS; pH 7.4), and quickly frozen in liquid nitrogen. The colon was harvested from the proximal end to the rectal end, flushed with PBS, and further processed to collect colonic epithelial cells as previously reported [Tang et al. 2013]. These samples were assayed for DNA adducts as described below.
Eight weeks after mice were harvested for DNA adduct measurement, the remaining mice were sacrificed by CO2 asphyxiation. The colons were harvested, flushed with 50 mM PBS, gently slid onto a 2 mm diameter glass rod, and bathed in 10% PBS-buffered formalin for 1 minute. The colons were cut longitudinally with a scalpel blade, and further rinsed with PBS several times to eliminate the remaining fecal waste. The colons were placed between half-folded filter paper, fixed in 10% PBS-buffered formalin overnight at 4° C, then rinsed in distilled water and kept in 70% ethanol until further analysis.
Determination of ACF and mucin-expressing ACF
Colon sections were removed from 70% ethanol and rinsed in distilled water for 5 minutes. Sections of distal colon approximately 7 cm2 in area (exact area recorded) were then stained with 0.2% PBS-buffered methylene blue (Sigma-Aldrich) for 5 min, rinsed twice with 50 mM PBS solution and once with distilled water to remove excess stain, and placed on a glass slide. The mucosal surface was examined under a standard light microscope at a magnification of 40× (BX40, Olympus Optical Co., Tokyo, Japan). Aberrant crypts (AC) can be detected as single aberrantly altered crypt, and clusters of AC that form a focus, known as ACF, are characterized by abnormally shaped foci, thickened epithelial layers, and larger pericryptal areas [Bird 1998; Perse and Cerar 2011]. Colonic AC and ACF were evaluated and recorded on the bases of their morphological features. Colon sections were coded to conceal the identity of the diet treatments until completion of the analysis. After ACF determination, the colons were de-stained and stored in 70% ethanol at 4°C.
The methylene blue de-stained colons were subsequently used to determine the mucin staining pattern, as a measure of ACF dysplasia [Caderni et al. 1995; Jenab et al. 2001]. Colon sections were removed from 70% ethanol, washed in distilled water for 5 min, and stained with freshly prepared high iron-diamine solution, which was prepared by adding 20 mg N-N′-dimethyl-p-phenylene diamine and 120 mg N-N′-dimethyl-m-phenylene diamine to 50 mL H2O, followed by the addition of 1.4 mL 60% ferric chloride, and allowing the solution to set for 8 hours in the dark at room temperature prior to use [Caderni et al. 1995]. The colon was rinsed two times in distilled water to remove excess stain. The colon was counterstained with 1% alcian blue (Sigma-Aldrich) in 3% acetic acid solution for 20 minutes at room temperature. The tissue was washed twice in 80% ethanol followed by rinsing in distilled H2O. The high iron diamine alcian blue (HID-AB) staining distinguishes the type of mucin expressed by the color or lack of color. Hyperplastic ACF stain brown, indicating sulfomucin expressing. Sialomucin-expressing (SiM) ACF, indicative of dysplastic ACF, were identified as crypts exhibiting dark or bright blue staining [Caderni et al. 1995; Jenab et al. 2001], whereas mucin-depleted ACF (MDF), indicating the greatest degree of dysplasia, were defined as having very little or no staining, a distorted lumen, and containing more than 3 crypts per foci [Caderni et al. 2003]. The colon tissue was examined under a standard light microscope at a magnification of 40×.
Tissue homogenization and DNA isolation
Liver and ~50 mg wet weight of colonic epithelial cells were homogenized in 3 vol of 50 mM Tris-HCl, 10 mM EDTA (pH 8.0) containing freshly added β-mercaptoethanol (10 mM). The homogenates were enzymatically digested with RNase A and RNAse T1, followed by proteinase K, and DNA was isolated by chloroform/phenol extraction as reported previously [Tang et al. 2013]. DNA from liver and colon (10 or 20 μg, respectively) was digested with a cocktail of nucleases as previously described [Tang et al. 2013]. The internal standards for dG-C8-AαC and dG-C8-MeIQ were added to DNA (10 μg) at a level of 5 adducts per 107 DNA bases, and [2H3C]-O6-me-dG was added to DNA (20 μg) at a level of 3.4 adducts per 106 bases.
UPLC-Electrospray Ionization/Mass Spectrometry analyses of DNA adducts
DNA adducts were determined with a NanoAcquity ultraperformance liquid chromatography (UPLC) system (Waters Corporation, Milford, MA) equipped with a linear quadrupole ion-trap mass spectrometer (Velos Pro, Thermo Fisher, San Jose, CA) and a Michrom Advance CaptiveSpray source (Auburn, CA). A Waters Symmetry trap column (180 μm × 20 mm, 5 μm particle size) was used for online solid-phase enrichment of dG-C8-AαC and dG-C8-MeIQ. The DNA digests of AαC- and MeIQ-fed animals were vacuum centrifuged to dryness and resuspended in 1:1 H2O:DMSO (30 μL). A volume of 5 μL was loaded onto the trap column, and washed with 10% CH3CN containing 0.2% HCO2H at 12 μL/min for 5 minutes, then back-flushed onto an analytical AQ C18 column (0.3 × 150 mm, 3 μm particle size, Michrom Bioresources, Auburn, CA). The analyte separation was performed with a linear gradient, starting at 99% A (mobile phase A: 0.01% HCO2H and 5% CH3CN), arriving at 95% B (mobile phase B: 0.01% HCO2H in 95% CH3CN) at 15 min, staying at 95% B for 2 min, then returning to the initial condition at 17 min. The flow rate was set to 5 μL/min.
The analysis of O6-me-dG was done following solid phase extraction (SPE). The DNA digests were loaded onto a Strata X SPE cartridge (30 mg, Phenomenex) preconditioned with CH3OH (1 mL), followed by H2O (1 mL). The non-modified deoxynucleosides were removed with H2O (1 mL), followed by 25%/75% CH3OH/H2O (1 mL), and O6-me-dG was eluted with CH3OH (1 mL). The eluents were evaporated to dryness by vacuum centrifugation and resuspended in the A mobile phase solvent (30 μL). O6-me-dG was chromatographed with the same AQ C18 column described above, employing 0.05% HCO2H and 2.5% CH3CN as the A solvent with 0.05% HCO2H in 95% CH3N as the B solvent. Chromatography commenced at 1% solvent B and arrived at 95% B at 13 min. The flow rate was set to 5 μL/min.
Xcalibur version 2.1.0 software (Thermo Fisher, San Jose, CA) was used for data acquisition and analysis. The MS3 scan mode was applied to measure the DNA adducts in the positive ionization mode following the loss of deoxyribose from the protonated DNA adducts ([M+H-116]+) at the MS2 scan stage: dG-C8-AαC (m/z 449.1 → 333.1 → 209.1, 291.1, 316.1); [13C10]-dG-C8-AαC (m/z 459.1 → 338.1 → 210.1, 295.1, 321.2); dG-C8-MeIQ (m/z 478.1 → 362.1 → 292.1, 317.2, 345.1); and [13C10 15N5]-dG-C8-MeIQ (m/z 493.1 → 372.1 → 298.1, 325.1, 354.1); O6-me-dG (m/z 282.2 → 166.1 → 134.0, 135.0, 149.1) and [2H3C]-O6-me-dG (m/z 285.2 → 169.1 → 134.0, 135.0, 152.1). Optimized instrument tuning parameters for dG-C8-AαC and dG-C8-MeIQ were set as previously reported [Nauwelaers et al. 2013; Tang et al. 2013]. For O6-me-dG, the capillary temperature was set at 275 °C and the spray voltage was set at 1.5 kV, the activation Q was set at 0.35 and the activation time of 10 ms. The normalized collision energy was set at 34 and 40, and the isolation widths were set at 3 and 1 m/z, respectively, for the MS2 and MS3 stage scan events. One μscan was used for data acquisition.
A six level calibration curve was constructed for dG-C8-AαC and dG-C8-MeIQ using DNA digest (10 μg) spiked with the internal standards at a level of 5 adducts per 107 DNA bases with the unlabeled adducts added at levels ranging from 5 to 500 adducts per 107 bases. To generate the calibration curve for O6-me-dG, [2H3C]-O6-me-dG was added at a level of 3.4 adducts per 106 bases for 20 μg of DNA digest (Calf thymus DNA matrix) following SPE (vide supra), and the unlabeled adduct was spiked at 6 levels ranging from 0.8 up to 34 adducts per 106 bases. The calibration curves were conducted in duplicate at each calibrant level, and the data were fitted to a straight line, using ordinary least squares with equal weightings for linear regression. The coefficient of determination (r2) values of the slopes equaled or exceeded 0.98.
Statistical analysis
Data were analyzed by one-way analysis of variance. When a statistically significant difference (P ≤ 0.05) was found, differences among groups were inspected using Duncan’s multiple range test. All statistical analyses were carried out using the SAS System for Windows release 9.3 (SAS Institute, Cary, NC).
Results
The structures of AαC, MeIQ, AOM and their major, stable DNA adducts are depicted in Figure 1. The DNA adducts of these carcinogens were employed as measures of bioactivation and genotoxicity, and colonic ACF were characterized as early biomarkers of colon neoplasia.
FIG. 1.

Chemical structures of AαC, MeIQ, and AOM and their stable covalent DNA adducts.
Subchronic treatment of HAAs and AOM and body weight
The body weight gains of the mice during the course of this study are reported in the supplementary figure (Figure S-1). During the 4-week feeding phase, both AαC- and MeIQ-treated groups showed a loss of body weight of 0.49 g and 1.50 g, respectively, compared to a body weight gain of 2.36 g for the no carcinogen group. The decline in body weights of the two HAA-fed groups did not differ significantly from one another, but both differed from the gain in the no carcinogen group (p<0.05). During the 6 week treatment of AOM, the AOM-treated mice experienced a slight decline in body weight of 0.18 g, which was significantly different than the gain of 3.80 g in the no carcinogen group over the same period (p<0.05). However, in the post carcinogen period, food intake did not differ significantly among the groups, yet the AαC- and MeIQ-treated groups gained significantly more body weight than did the no carcinogen group (7.50 g and 6.84 g, respectively vs 4.96 g; p<0.05). Similarly, post carcinogen treatment, the AOM-treated group gained significantly more weight than the no carcinogen group (5.07 g vs. 2.94, p<0.05), and consequently body weights of the HAA-fed and AOM-treated groups were within 10% of the body weights of the untreated control animals at the time of sacrifice.
Quantitation of HAA- and AOM-DNA adducts in liver and colon
The levels of dG-C8-AαC, dG-C8-MeIQ, and O6-me-dG adducts, the major DNA adducts formed by these compounds [Pfau et al. 1997; Tang et al. 2013; Megaraj et al. 2014], were measured by the stable isotope dilution method. Representative mass chromatograms of these DNA adducts formed in the liver and colon of the treated and control mice are shown in Figures 2 and 3. The product ion mass spectra of the HAA adducts at the MS3 stage are provided as supplementary data (Figure S-2). The product ion spectra of O6-me-dG and [2H3C]-O6-me-dG at the MS3 scan stage are depicted in Figure 3B.
FIG. 2.

Representative UPLC-ESI/MS3 chromatograms of AαC and MeIQ DNA adducts and internal standards (ISTDs) in liver and colon of (A) untreated mice and (B) mice treated with carcinogens for 4 weeks. In the negative control sample (a, c, k), the intensities of the signals were normalized to the same scale as the ISTD (b, d, l). The area (A) and retention time (tR) are reported.
FIG. 3.

Reconstructed ion mass chromatogram at the MS3 scan stage of DNA adducts of mice control (A) and AOM-treated (B). The intensities of the ion signals of the untreated mice were normalized to the signals of the internal standards. The product ion spectra of O6-me-dG formed in liver and [2H3C]-O6-me-dG are depicted in the lower panel.
The levels of DNA adduct formation by the three carcinogens are summarized in Table 1. Both HAAs and AOM are efficiently bioactivated in rodent liver [Tang et al. 2013; Megaraj et al. 2014], and higher levels of DNA adducts were formed in liver than colon for all three carcinogens. The levels of DNA adducts in liver were greatest for AαC, followed by AOM, and then MeIQ. The levels of adducts were 5 to 9-fold lower in the colon. AαC and AOM had comparable levels of colonic adducts, both of which were significantly greater than the levels of dG-C8-MeIQ. O6-me-dG was also measured following acid hydrolysis (1N HCl, 80 °C for 60 min), and the estimates of the mean adduct levels of O6-me-Guanine were within 20% of the adduct values obtained by enzymatic digestion (unpublished data, J. Guo). The levels of dG-C8-AαC and dG-C8-MeIQ in liver of A/J mice are about ~4-fold higher than adduct levels in liver of male C57BL/6 mice fed AαC and MeIQ for 4 weeks [Tang et al. 2013].
Table 1.
DNA adducts of AαC, MeIQ, and AOM in liver and colon tissues of A/J mice
| O6-me-dG (per 107 bases) | dG-C8-MeIQ (per 107 bases) | 4dG-C8-AαC (per 107 bases) | |
|---|---|---|---|
| Liver | 290.2 ± 19.3b | 70.31 ± 8.73c | 470.11 ± 45.83a |
| Colon | 62.5 ± 4.0a | 8.24 ± 2.72b | 50.93 ± 13.43a |
Values represent the means ± SD of four animals. Values within a row not sharing a common superscript are significantly different (p<0.05).
AC and ACF number
AC and ACF were enumerated after methylene blue staining. A representative image of an ACF induced in an AαC-fed mouse colon is shown in Figure 4A. ACF were found throughout the distal colons, but the greatest number of ACF was found near the rectum in all carcinogen-treated mice. AOM treatment induced the greatest number of ACF and at significantly greater numbers than found in the AαC- and MeIQ-fed mice (P < 0.05), which were similar to each other (Table 2). Only one mouse from the negative control group (n=4 per group) developed a single aberrant crypt (AC).
FIG. 4.
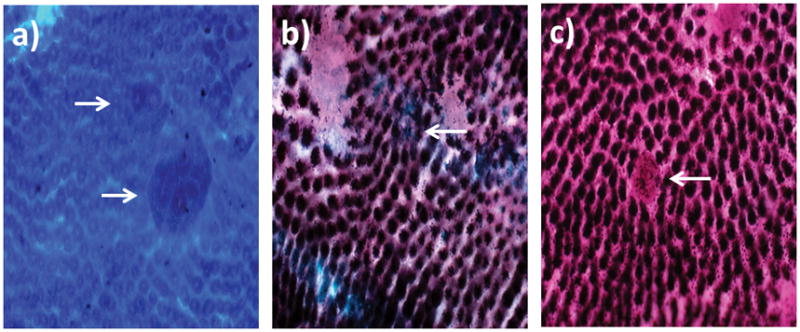
Representative images of (A) methylene-blue stained ACF, (B) sialomucin-expressing ACF, and (C) MDF observed in colons of AαC-fed mice (magnification 40×).
Table 2.
Aberrant crypt (AC) foci (ACF), ACF multiplicity (AC/ACF), large ACF (>4 AC/ACF), SiM, MDF, and ACF per colonic DNA adducts in rats fed AaC or MeIQ or treated with AOM1
| Number/cm2 of distal colon | AOM | MeIQ | AαC |
|---|---|---|---|
| AC | 7.94 ± 1.01a | 1.61 ± 0.22b | 1.81 ± 0.30b |
| ACF | 3.90 ± 0.29a | 1.12 ± 0.15b | 1.30 ± 0.19b |
| AC/ACF | 1.99 ± 0.12a | 1.39 ± 0.07b | 1.37 ± 0.09b |
| >4 AC/ACF | 0.15 ± 0.07 | 0 | 0 |
| SiM | 0.31 ± 0.09a | 0.03 ± 0.02b | 0.08 ± 0.04b |
| MDF | 0.76 ± 0.17a | 0.11 ± 0.05b | 0.13 ± 0.05b |
| ACF/107 colonic DNA adducts | 0.0624 | 0.1359 | 0.0255 |
| ACF with dysplasia (SiM + MDF)/ 107 colonic DNA adducts | 0.0171 | 0.0171 | 0.00412 |
Values represent means ± SEM, n=8. Values within a row that do not share a superscript are significantly different (P < 0.05).
Mucin expression pattern
Normal crypts and hyperplastic ACF stain for sulfomucin, whereas dysplastic ACF stain for sialomucin (SiM), with the most dysplastic crypts showing no mucin staining (MDF) [Perse and Cerar 2011]. SiM and MDF were observed in the AαC, MeIQ, and AOM groups but not in the negative control animals. Representative images of dysplastic ACF, characterized by SiM and MDF, in the colon of AαC-fed mice are shown in Figures 4A–C. The number of SiM and MDF was similar in the AαC- and MeIQ-fed mice, whereas a significantly greater number of both markers were induced in AOM-treated mice (P < 0.05) (Table 2). The SiM or MDF appeared to be largely or entirely coincident with ACF detected by methylene blue staining. Representative images of the coincident lesions, recognized as ACF and SiM, with the different staining methods, is shown in Figure S-3.
ACF and dysplastic ACF per DNA adducts
We related the induction of the number of ACF and dysplastic ACF expressed per 107 DNA adducts present in the colon. The adduct measurements and ACF were obtained from different sets of animals. The adduct levels were measured at 4 weeks post-treatment with HAAs and 6 hours following the last treatment with AOM, whereas ACF were measured following an additional 8 weeks to allow for development of ACF. The number of ACF induced per 107 DNA adducts varied substantially among the carcinogens (Table 2). The number of total ACF per 107 colonic DNA adducts was greatest for the MeIQ-fed group. The number of the AOM-treated group was about half that found in the MeIQ-fed group, and the ACF per 107 colonic DNA adducts in AαC-fed group was only about 20% of the MeIQ-fed group. However, the number of dysplastic ACF (SiM+MDF) per 107 colonic DNA adducts was comparable for the MeIQ-fed and AOM-treated groups, but about 70% less in the AαC-fed group.
Discussion
Tobacco smoke is a primary cause of lung cancer, but also causes several other types of cancers, including urinary bladder, liver, larynx, pancreas, and colorectum [Giovannucci 2001; IARC 2002; Vineis et al. 2004; IARC 2009]. There are more than 60 carcinogens in cigarette smoke [Hecht 2003]; however, the causal agent(s) of tobacco-associated colon carcinogenesis is uncertain [Giovannucci 2001; Vineis et al. 2004]. Although a number of HAAs are experimental colon carcinogens in rodents [Sugimura et al. 2004], other than AαC and its methylated homologue, 2-amino-3-methy-9H-pyrido[2,3-b]indole (MeAαC), the amounts of individual HAAs present in mainstream tobacco smoke are generally <1 ng/cig and unlikely to significantly contribute to the overall DNA adduct burden load and genotoxicity [IARC 2002]. Given the high amounts of AαC present in mainstream tobacco smoke and its potential to undergo bioactivation to form DNA adducts in liver, lung, bladder, and colon of mice [Tang et al. 2013], and in human hepatocytes [Nauwelaers et al. 2013], it is likely that AαC can induce DNA damage in these organs of smokers.
AOM and its precursor, 1,2-dimethylhydrazine, are commonly used to induce ACF and to examine the morphology of different types of ACF during the progression to colon cancer [Caderni et al. 1995; Papanikolaou et al. 2000; Caderni et al. 2003; Rosenberg et al. 2009; Perse and Cerar 2011]. While AOM is not a naturally occurring environmental or dietary carcinogen, its carcinogenic isomer, NDMA is present in tobacco smoke and processed meats [IARC 2002]. Both AOM and NDMA form the same DNA adducts [IARC 2002; Megaraj et al. 2014]. In contrast to AOM, studies on the induction of ACF and tumors by other colon genotoxicants, such as HAAs, are few because of the expense of the chemicals and the relatively small number of ACF produced [Sugimura et al. 2004]. The A/J mouse strain is highly sensitive to AOM and forms large numbers of ACF and colon tumors following treatment with AOM [Papanikolaou et al. 2000]. The A/J mouse is also susceptible to tobacco-specific nitrosamines and develops lung tumors [Hecht et al. 1994].
Genotoxicity studies have not been previously conducted with HAAs in A/J mice [Sugimura et al. 2004], though MeIQ and AαC are potent inducers of lacI transgene mutations in the colon of Big Blue mice, which have a genetic background of C57BL/6N [Zhang et al. 1996; Nagao et al. 2001]. MeIQ is a colon carcinogen in the C57BL/6N strain [Fujita et al. 1999], but long-term feeding studies with AαC have not been conducted in this mouse strain. Both MeIQ and AαC are liver carcinogens in CDF1 mice [Sugimura et al. 2004]. In a preliminary study, we found that AαC and MeIQ, fed at the same dietary concentrations used in the present study, formed high levels of DNA adducts in the colon of C57BL/6N mice, but induced very few ACF, consistent with a previous study [Okonogi et al. 1997]. However, the C57BL/6J mouse is one of the most resistant strains to the development of AOM-induced colon tumors, whereas the A/J mouse strain is highly sensitive to colon carcinogenesis, yielding about 30 times more the tumors than C57BL/6J for AOM [Liu et al. 2012]. Hence, we examined the potential of AOM, MeIQ, and AαC to form DNA adducts and to induce ACF as early biomarkers of tumorigenesis in A/J mice. To our knowledge, our study is the first to relate DNA adduct formation, ACF, and dysplastic ACF from three different colon genotoxicants.
DNA adducts are believed to be a necessary event to initiate carcinogenesis by genotoxic carcinogens, but adduct formation itself is insufficient for cancer induction, as the process of carcinogenesis requires multiple steps [Schut and Snyderwine 1999]. AαC and MeIQ preferentially bind to the C8 position of dG [Pfau et al. 1997; Sugimura et al. 2004; Tang et al. 2013], whereas AOM preferential forms adducts at the endocyclic ring nitrogen (N-7 position) and exocyclic oxygen (O6 position) of guanine (Figure 1) [Boysen et al. 2010]. N7-me-dG is formed at higher levels than O6-me-dG in mice treated with AOM [Megaraj et al. 2014], but N7-me-dG undergoes rapid depurination to leave an abasic site, which can be repaired [Boysen et al. 2009]. Studies have shown that O6-me-dG stably pairs with thymidine during replication, creating an O6-methyl-G-T mismatch leading to G-to-A transition mutations [Delaney and Essigmann 2008]. This mutation has been observed in the K-ras gene isolated from lung and colon tumors of mice treated with DNA methylating agents [Perse and Cerar 2011]. MeIQ frequently induces a G-to-T transversion mutation in Ha-ras codon 13 of Zymbal gland or forestomach tumors of CDF1 mice [Nagao et al. 1997]; however, there are no reports about the genetic changes in AαC-induced tumors in rodents. Mutational hot spots induced by MeIQ and AαC occurred in distinct sites, respectively at 5′-GC-3′ and 5′-CGT-3′, in the lacI transgene of Big Blue mice, both leading to G-to-T transversion mutations [Okonogi et al. 1997]. Mutations induced by DNA adducts of genotoxicants are strongly influenced by sequence context [Delaney and Essigmann 2008]. The structures of the different types of DNA adducts formed by AαC, MeIQ, and AOM in different genes undoubtedly contribute to the differences in the rate and development of colonic ACF and tumors. Long-term studies examining the potential of these three chemicals to induce tumors and tumor multiplicity in colon are warranted. The mutational characteristics of MeIQ and AαC in tumor-related genes of the colon of A/J or other mouse strains also remain to be elucidated.
Considerable evidence suggests that ACF are preneoplastic lesions. ACF have a greater cell proliferation rate than normal crypts [Yamashita et al. 1994], several oncogenes are activated [Perse and Cerar 2011], and the number of ACF correlate with adenoma formation [Takayama et al. 1998]. Different morphological features of ACF have been proposed as indicators of the degree of colon cancer risk, including the number of aberrant crypts (AC), the number of ACF, ACF multiplicity (AC/ACF), and the number of large ACF (sometimes defined as ACF with ≥ 4 AC). Both ACF multiplicity [Perse and Cerar 2011] and the number of large ACF [Caderni et al. 1995] may be better predictors of tumor development than ACF number alone. Also, there are different sub-populations of ACF that vary in their tumorigenic potential [Papanikolaou et al. 2000; Caderni et al. 2003; Femia et al. 2008; Rosenberg et al. 2009]. The vast majority of ACF are hyperplastic, which are generally considered not likely to progress to tumors. In contrast, ACF with dysplastic characteristics are viewed as preneoplastic lesions [Perse and Cerar 2011].
We enumerated dysplastic ACF in unsectioned whole mount colons by distinguishing the presence and type of mucin using the HID-AB staining method [Caderni et al. 1995; Caderni et al. 2003]. Normal crypts and hyperplastic ACF stain for sulfomucin, whereas dysplastic ACF stain for sialomucin, and the most dysplastic of the crypts show no mucin staining (MDF) [Perse and Cerar 2011]. The relative increase in SiM staining and reduction in SuM staining has been observed in colon cancer in both humans and rats [Dawson et al. 1978; Thorup 1997; Yoshimi et al. 2004], and MDF have been identified in the colon of rats treated with AOM and 1,2-dimethylhydrazine [Caderni et al. 2003; Yoshimi et al. 2004] and patients at high risk of colon cancer [Femia et al. 2008]. In our study, mice administered AOM had significantly more SiM and MDF compared to mice consuming either of the two HAA-containing diets, which were similar to each other in SiM and MDF number. Yet the dose of AOM administered over the course of the study (~9 μmol) was much less than the amount of HAA consumed by the mice (~123 μmol MeIQ and ~190 μmol AαC). However, it is important to note that AαC and MeIQ were given as part of the diet, whereas azoxymethane was given by i.p. injection. Hence, the bioavailability of the HAAs was probably lower than for AOM. Moreover, the differences in the route of administration could have influenced the levels of DNA adduct formation of these three compounds, which are mainly bioactivated in liver [Megaraj et al. 2014; Turesky et al. 2015]. The doses and route of administration were chosen based on previous studies which examined ACF formation by AOM [Papanikolaou et al. 2000; Rosenberg et al. 2009; Liu et al. 2012], and the induction of lacI transgene mutations in colon of mice by MeIQ and AαC [Zhang et al. 1996; Nagao et al. 2001]. The differences in ACF induction by these chemicals also may be driven by the different types of DNA adducts formed and their differing biological impact. Our findings with regards to number of ACF with AOM treatment are similar to those of Megaraj et al. [2014] who subcutaneously administered AOM to A/J mice (~ 7 μmol) at a dose comparable to that used in our study (~ 9 μmol). Both studies showed a similar number of total ACF, 29 ± 1 and 31 ± 10, respectively.
dG-C8-AαC and dG-C8-MeIQ are the primary DNA adducts formed by AαC and MeIQ in liver and colon of mice [Tang et al. 2013]. O6-me-dG is the major stable adduct formed in the colon of mice following activation by AOM [Boysen et al. 2009; Megaraj et al. 2014]. While we cannot exclude the possibility that other DNA adducts of AαC and MeIQ contribute to ACF and tumor formation, other adducts, if present, are minor [Tang et al. 2013]. In the case of AOM, the major N7-me-dG adduct is cationic and undergoes rapid depurination to produce an abasic site. N7-me-dG is generally believed to be benign since N7 does not participate in Watson-Crick base pairing [Boysen et al. 2009]. However, we cannot exclude that abasic site formation contributes to the induction of ACF. If we may assume that dG-C8-AαC, dG-C8-MeIQ, and O6-me-dG are primarily responsible for the biological effects of these genotoxicants, dG-C8-MeIQ is more potent than either dG-C8-AαC or O6-me-dG in the induction of total ACF. When only dysplastic ACF are considered (SiM plus MDF), the induction of lesions by dG-C8-MeIQ and O6-me-dG adducts is equivalent, and dG-C8-AαC has less potential to induce dysplastic ACF. However, AαC forms DNA adducts at 8-fold higher levels than MeIQ (P < 0.05), and the total number of dysplastic ACF induced by AαC and MeIQ were comparable. Long-term studies are required to determine the relationship between DNA adduct formation, different types of ACF, and colon tumorigenesis in A/J mice treated with these chemicals.
The toxicological and epidemiological data of tobacco smoking has led the IARC to declare that tobacco smoking causes colorectal cancer [IARC 2009]. Our findings show that AαC, when given as part of the diet, results in the formation of high levels of DNA adducts and precancerous colonic lesions in the A/J mouse, providing biological plausibility for a causal role of AαC in tobacco-associated colorectal carcinogenesis in smokers. Although the amounts of test chemicals employed here and in other rodent model studies investigating ACF are much greater than daily human exposures [Rosenberg et al. 2009; Perse and Cerar 2011], appreciable levels of AαC are detected in the urine of smokers [Turesky et al. 2007], and AαC DNA adducts are formed in human hepatocytes at environmental exposure levels [Nauwelaers et al. 2013], signifying that DNA damage by AαC is likely to occur in tobacco smokers. Further studies on the potential role of AαC in colon carcinogenesis are warranted.
Supplementary Material
Table S-1. Diet ingredients and caloric contents of AIN 93G (for the adjustment period) and modified AIN 93G (Dyet#102987).
Figure S-1. Average body weight of mice during the period of carcinogen administration and after carcinogen administration.
Figure S-2. MS3 product ion spectra of dG-C8-AαC, dG-C8-MeIQ from liver of mice and their isotopically-labelled internal standards.
Acknowledgments
Source of Funding
National Cancer Institute at the National Institutes of Health (R01 CA134700 to R.J.T.), and mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, supported in part by National Cancer Institute Cancer Center Support grant no. CA077598 (R.J.T.)
Footnotes
Conflict of Interest
The authors have no conflict of interests to disclose
Statement of Author Contributions
Drs. Turesky and Gallaher designed the study, with intellectual input from Dr. O’Sullivan. Dr. Turesky obtained animal use approval. S. Kim conducted the research. Dr. J. Guo established LC/MS methods to measure O6-me-dG, dG-C8-AαC, and dG-C8-MeIQ. S. Kim and Dr. Gallaher analyzed the data. S. Kim and Drs. Turesky and Gallaher prepared the manuscript. All authors approved the final manuscript.
References
- Belinsky SA, Devereux TR, Maronpot RR, Stoner GD, Anderson MW. Relationship between the Formation of Promutagenic Adducts and the Activation of the K-ras Protooncogene in Lung Tumors from A/J Mice Treated with Nitrosamines. Cancer Research. 1989;49:5305–5311. [PubMed] [Google Scholar]
- Bird RP. Aberrant Crypt Foci System to Study Cancer Preventive Agents in the Colon. In: Hanausek M, Walaszek Z, editors. Tumor Marker Protocols. Springer; New York: 1998. pp. 465–474. [Google Scholar]
- Bouvard V, Loomis D, Guyton KZ, Grosse Y, Ghissassi FE, Benbrahim-Tallaa L, Guha N, Mattock H, Straif K. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015 doi: 10.1016/S1470-2045(15)00444-1. [DOI] [PubMed] [Google Scholar]
- Boyle P, Zaridze DG, Smans M. Descriptive epidemiology of colorectal cancer. Int J Cancer. 1985;36:9–18. doi: 10.1002/ijc.2910360103. [DOI] [PubMed] [Google Scholar]
- Boysen G, Collins LB, Liao S, Luke AM, Pachkowski BF, Watters JL, Swenberg JA. Analysis of 8-oxo-7,8-dihydro-2′-deoxyguanosine by ultra high pressure liquid chromatography-heat assisted electrospray ionization-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:375–380. doi: 10.1016/j.jchromb.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. The formation and biological significance of N7-guanine adducts. Mutat Res. 2009;678:76–94. doi: 10.1016/j.mrgentox.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caderni G, Femia AP, Giannini A, Favuzza A, Luceri C, Salvadori M, Dolara P. Identification of mucin-depleted foci in the unsectioned colon of azoxymethane-treated rats: correlation with carcinogenesis. Cancer Res. 2003;63:2388–2392. [PubMed] [Google Scholar]
- Caderni G, Giannini A, Lancioni L, Luceri C, Biggeri A, Dolara P. Characterisation of aberrant crypt foci in carcinogen-treated rats: association with intestinal carcinogenesis. Br J Cancer. 1995;71:763–769. doi: 10.1038/bjc.1995.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S. N-Nitrosodimethylamine (NDMA) in Food and Beverages: A Comparison in Context to Drinking Water. Human and Ecological Risk Assessment: An International Journal. 2013;20:1291–1312. [Google Scholar]
- Dawson PA, Patel J, Filipe MI. Variations in sialomucins in the mucosa of the large intestine in malignancy: a quantimet and statistical analysis. Histochem J. 1978;10:559–572. doi: 10.1007/BF01003137. [DOI] [PubMed] [Google Scholar]
- Delaney JC, Essigmann JM. Biological properties of single chemical-DNA adducts: a twenty year perspective. Chem Res Toxicol. 2008;21:232–252. doi: 10.1021/tx700292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femia AP, Giannini A, Fazi M, Tarquini E, Salvadori M, Roncucci L, Tonelli F, Dolara P, Caderni G. Identification of mucin depleted foci in the human colon. Cancer Prev Res. 2008;1:562–567. doi: 10.1158/1940-6207.CAPR-08-0125. [DOI] [PubMed] [Google Scholar]
- Fujita H, Nagano K, Ochiai M, Ushijima T, Sugimura T, Nagao M, Matsushima T. Difference in target organs in carcinogenesis with a heterocyclic amine, 2-amino-3,4-dimethylimidazo[4,5-f]quinoline, in different strains of mice. Jpn J Cancer Res. 1999;90:1203–1206. doi: 10.1111/j.1349-7006.1999.tb00696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci E. An updated review of the epidemiological evidence that cigarette smoking increases risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2001;10:725–731. [PubMed] [Google Scholar]
- Gram IT, Braaten T, Lund E, Le Marchand L, Weiderpass E. Cigarette smoking and risk of colorectal cancer among Norwegian women. Cancer Causes Control. 2009;20:895–903. doi: 10.1007/s10552-009-9327-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3:733–744. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- Hecht SS, Isaacs S, Trushin N. Lung tumor induction in A/J mice by the tobacco smoke carcinogens 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and benzo[a]pyrene: a potentially useful model for evaluation of chemopreventive agents. Carcinogenesis. 1994;15:2721–2725. doi: 10.1093/carcin/15.12.2721. [DOI] [PubMed] [Google Scholar]
- IARC. Tobacco smoke and involuntary smoking. Lyon, France: International Agency for Research on Cancer; 2002. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. [PMC free article] [PubMed] [Google Scholar]
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Lyon, France: International Agency for Research on Cancer; 2009. A review of human carcinogens. Part E: Personal Habits And Indoor Combustions. Working Group On The Evaluation Of Carcinogenic Risks To Humans. [Google Scholar]
- Jenab M, Chen J, Thompson LU. Sialomucin production in aberrant crypt foci relates to degree of dysplasia and rate of cell proliferation. Cancer Lett. 2001;165:19–25. doi: 10.1016/s0304-3835(00)00706-0. [DOI] [PubMed] [Google Scholar]
- Knekt P, Jarvinen R, Dich J, Hakulinen T. Risk of colorectal and other gastro-intestinal cancers after exposure to nitrate, nitrite and N-nitroso compounds: a follow-up study. Int J Cancer. 1999;80:852–856. doi: 10.1002/(sici)1097-0215(19990315)80:6<852::aid-ijc9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Liu P, Lu Y, Liu H, Wen W, Jia D, Wang Y, You M. Genome-wide association and fine mapping of genetic loci predisposing to colon carcinogenesis in mice. Mol Cancer Res. 2012;10:66–74. doi: 10.1158/1541-7786.MCR-10-0540. [DOI] [PubMed] [Google Scholar]
- Loechler EL, Green CL, Essigmann JM. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci U S A. 1984;81:6271–6275. doi: 10.1073/pnas.81.20.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YH, Jakszyn P, Luben RN, Mulligan AA, Mitrou PN, Khaw KT. N-Nitroso compounds and cancer incidence: the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk Study. Am J Clin Nutr. 2011;93:1053–1061. doi: 10.3945/ajcn.111.012377. [DOI] [PubMed] [Google Scholar]
- Manabe S, Wada O, Kanai Y. Simultaneous determination of amino-alpha-carbolines and amino-gamma-carbolines in cigarette smoke condensate by high-performance liquid chromatography. J Chromatogr. 1990;529:125–133. doi: 10.1016/s0378-4347(00)83813-x. [DOI] [PubMed] [Google Scholar]
- Megaraj V, Ding X, Fang C, Kovalchuk N, Zhu Y, Zhang QY. Role of hepatic and intestinal p450 enzymes in the metabolic activation of the colon carcinogen azoxymethane in mice. Chem Res Toxicol. 2014;27:656–662. doi: 10.1021/tx4004769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 1995;93:17–48. doi: 10.1016/0304-3835(95)03786-V. [DOI] [PubMed] [Google Scholar]
- Nagao M, Ochiai M, Okochi E, Ushijima T, Sugimura T. LacI transgenic animal study: relationships among DNA-adduct levels, mutant frequencies and cancer incidences. Mutat Res. 2001;477:119–124. doi: 10.1016/s0027-5107(01)00113-0. [DOI] [PubMed] [Google Scholar]
- Nagao M, Ushijima T, Toyota M, Inoue R, Sugimura T. Genetic changes induced by heterocyclic amines. Mutat Res. 1997;376:161–167. doi: 10.1016/s0027-5107(97)00039-0. [DOI] [PubMed] [Google Scholar]
- Nakagama H, Nakanishi M, Ochiai M. Modeling human colon cancer in rodents using a food-borne carcinogen, PhIP. Cancer Sci. 2005;96:627–636. doi: 10.1111/j.1349-7006.2005.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagama H, Ochiai M, Ubagai T, Tajima R, Fujiwara K, Sugimura T, Nagao M. A rat colon cancer model induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, PhIP. Mutat Res. 2002;506–507:137–144. doi: 10.1016/s0027-5107(02)00160-4. [DOI] [PubMed] [Google Scholar]
- Nauwelaers G, Bellamri M, Fessard V, Turesky RJ, Langouet S. DNA adducts of the tobacco carcinogens 2-amino-9H-pyrido[2,3-b]indole and 4-aminobiphenyl are formed at environmental exposure levels and persist in human hepatocytes. Chem Res Toxicol. 2013;26:1367–1377. doi: 10.1021/tx4002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauwelaers G, Bessette EE, Gu D, Tang Y, Rageul J, Fessard V, Yuan JM, Yu MC, Langouet S, Turesky RJ. DNA adduct formation of 4-aminobiphenyl and heterocyclic aromatic amines in human hepatocytes. Chem Res Toxicol. 2011;24:913–925. doi: 10.1021/tx200091y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norat T, Bingham S, Ferrari P, Slimani N, Jenab M, Mazuir M, Overvad K, Olsen A, Tjonneland A, Clavel F, Boutron-Ruault MC, Kesse E, Boeing H, Bergmann MM, Nieters A, Linseisen J, Trichopoulou A, Trichopoulos D, Tountas Y, Berrino F, Palli D, Panico S, Tumino R, Vineis P, Bueno-de-Mesquita HB, Peeters PH, Engeset D, Lund E, Skeie G, Ardanaz E, Gonzalez C, Navarro C, Quiros JR, Sanchez MJ, Berglund G, Mattisson I, Hallmans G, Palmqvist R, Day NE, Khaw KT, Key TJ, San Joaquin M, Hemon B, Saracci R, Kaaks R, Riboli E. Meat, fish, and colorectal cancer risk: the European Prospective Investigation into cancer and nutrition. J Natl Cancer Inst. 2005;97:906–916. doi: 10.1093/jnci/dji164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai M, Watanabe M, Nakanishi M, Taguchi A, Sugimura T, Nakagama H. Differential staining of dysplastic aberrant crypt foci in the colon facilitates prediction of carcinogenic potentials of chemicals in rats. Cancer Lett. 2005;220:67–74. doi: 10.1016/j.canlet.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Hasegawa H, Suenaga M, Kato T, Sato S, Takayama S, Sugimura T. Induction of hepatocellular carcinoma and highly metastatic squamous cell carcinomas in the forestomach of mice by feeding 2-amino-3,4-dimethylimidazo[4,5-f]quinoline. Carcinogenesis. 1986;7:1889–1893. doi: 10.1093/carcin/7.11.1889. [DOI] [PubMed] [Google Scholar]
- Okonogi H, Ushijima T, Shimizu H, Sugimura T, Nagao M. Induction of aberrant crypt foci in C57BL/6N mice by 2-amino-9H-pyrido[2,3-b]indole (A alphaC) and 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) Cancer Lett. 1997;111:105–109. doi: 10.1016/s0304-3835(96)04505-3. [DOI] [PubMed] [Google Scholar]
- Papanikolaou A, Wang QS, Papanikolaou D, Whiteley HE, Rosenberg DW. Sequential and morphological analyses of aberrant crypt foci formation in mice of differing susceptibility to azoxymethane-induced colon carcinogenesis. Carcinogenesis. 2000;21:1567–1572. [PubMed] [Google Scholar]
- Perse M, Cerar A. Morphological and molecular alterations in 1,2 dimethylhydrazine and azoxymethane induced colon carcinogenesis in rats. J Biomed Biotechnol. 2011;2011:473964. doi: 10.1155/2011/473964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau W, Schulze C, Shirai T, Hasegawa R, Brockstedt U. Identification of the major hepatic DNA adduct formed by the food mutagen 2-amino-9H-pyrido[2,3-b]indole (A alpha C) Chem Res Toxicol. 1997;10:1192–1197. doi: 10.1021/tx9701182. [DOI] [PubMed] [Google Scholar]
- Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–196. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli RL, Vendeuvre JL, Naud N, Tache S, Gueraud F, Viau M, Genot C, Corpet DE, Pierre FH. Meat processing and colon carcinogenesis: cooked, nitrite-treated, and oxidized high-heme cured meat promotes mucin-depleted foci in rats. Cancer Prev Res (Phila) 2010;3:852–864. doi: 10.1158/1940-6207.CAPR-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–368. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Skog K. Cooking procedures and food mutagens: a literature review. Food Chem Toxicol. 1993;31:655–675. doi: 10.1016/0278-6915(93)90049-5. [DOI] [PubMed] [Google Scholar]
- Sohn OS, Fiala ES, Requeijo SP, Weisburger JH, Gonzalez FJ. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001;61:8435–8440. [PubMed] [Google Scholar]
- Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004;95:290–299. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada A, Ochiai M, Wakabayashi K, Nukaya H, Sugimura T, Nagao M. Identification of N-(deoxyguanosin-8-yl)-2-amino-3,4-dimethylimidazo[4,5-f]quinoline (dG-C8-MeIQ) as a major adduct formed by MeIQ with nucleotides in vitro with DNA in vivo. Carcinogenesis. 1994;15:1275–1278. doi: 10.1093/carcin/15.6.1275. [DOI] [PubMed] [Google Scholar]
- Takayama T, Katsuki S, Takahashi Y, Ohi M, Nojiri S, Sakamaki S, Kato J, Kogawa K, Miyake H, Niitsu Y. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N Engl J Med. 1998;339:1277–1284. doi: 10.1056/NEJM199810293391803. [DOI] [PubMed] [Google Scholar]
- Tan SL, Gerber JP, Cosgrove LJ, Lockett TJ, Clarke JM, Williams DB, Head RJ. Is the tissue persistence of O(6)-methyl-2′-deoxyguanosine an indicator of tumour formation in the gastrointestinal tract? Mutat Res. 2011;721:119–126. doi: 10.1016/j.mrgentox.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Tang Y, Kassie F, Qian X, Ansha B, Turesky RJ. DNA adduct formation of 2-amino-9H-pyrido[2,3-b]indole and 2-amino-3,4-dimethylimidazo[4,5-f]quinoline in mouse liver and extrahepatic tissues during a subchronic feeding study. Toxicol Sci. 2013;133:248–258. doi: 10.1093/toxsci/kft077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorup I. Histomorphological and immunohistochemical characterization of colonic aberrant crypt foci in rats: relationship to growth factor expression. Carcinogenesis. 1997;18:465–472. doi: 10.1093/carcin/18.3.465. [DOI] [PubMed] [Google Scholar]
- Turesky RJ, Konorev D, Fan X, Tang Y, Yao L, Ding X, Xie F, Zhu Y, Zhang QY. The Effect of Cytochrome P450 Reductase Deficiency on 2-Amino-9H-Pyrido[2,3-b]Indole Metabolism and DNA Adduct Formation in Liver and Extrahepatic Tissues of Mice. Chem Res Toxicol. 2015 doi: 10.1021/acs.chemrestox.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky RJ, Yuan JM, Wang R, Peterson S, Yu MC. Tobacco smoking and urinary levels of 2-amino-9H-pyrido[2,3-b]indole in men of Shanghai, China. Cancer Epidemiol Biomarkers Prev. 2007;16:1554–1560. doi: 10.1158/1055-9965.EPI-07-0132. [DOI] [PubMed] [Google Scholar]
- Vineis P, Alavanja M, Buffler P, Fontham E, Franceschi S, Gao YT, Gupta PC, Hackshaw A, Matos E, Samet J, Sitas F, Smith J, Stayner L, Straif K, Thun MJ, Wichmann HE, Wu AH, Zaridze D, Peto R, Doll R. Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst. 2004;96:99–106. doi: 10.1093/jnci/djh014. [DOI] [PubMed] [Google Scholar]
- WCRF/AICR. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. Washington, D.C: AICR; 2007. [Google Scholar]
- Weng Y, Fang C, Turesky RJ, Behr M, Kaminsky LS, Ding X. Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res. 2007;67:7825–7832. doi: 10.1158/0008-5472.CAN-07-1006. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Minamoto T, Onda M, Esumi H. Increased cell proliferation of azoxymethane-induced aberrant crypt foci of rat colon. Jpn J Cancer Res. 1994;85:692–698. doi: 10.1111/j.1349-7006.1994.tb02416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi N, Morioka T, Kinjo T, Inamine M, Kaneshiro T, Shimizu T, Suzui M, Yamada Y, Mori H. Histological and immunohistochemical observations of mucin-depleted foci (MDF) stained with Alcian blue, in rat colon carcinogenesis induced with 1,2-dimethylhydrazine dihydrochloride. Cancer Sci. 2004;95:792–797. doi: 10.1111/j.1349-7006.2004.tb02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ashley DL, Watson CH. Quantitative analysis of six heterocyclic aromatic amines in mainstream cigarette smoke condensate using isotope dilution liquid chromatography-electrospray ionization tandem mass spectrometry. Nicotine Tob Res. 2011;13:120–126. doi: 10.1093/ntr/ntq219. [DOI] [PubMed] [Google Scholar]
- Zhang XB, Felton JS, Tucker JD, Urlando C, Heddle JA. Intestinal mutagenicity of two carcinogenic food mutagens in transgenic mice: 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and amino(alpha)carboline. Carcinogenesis. 1996;17:2259–2265. doi: 10.1093/carcin/17.10.2259. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wang PP, Zhao J, Green R, Sun Z, Roebothan B, Squires J, Buehler S, Dicks E, Zhao J, Cotterchio M, Campbell PT, Jain M, Parfrey PS, McLaughlin JR. Dietary N-nitroso compounds and risk of colorectal cancer: a case-control study in Newfoundland and Labrador and Ontario, Canada. Br J Nutr. 2014;111:1109–1117. doi: 10.1017/S0007114513003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S-1. Diet ingredients and caloric contents of AIN 93G (for the adjustment period) and modified AIN 93G (Dyet#102987).
Figure S-1. Average body weight of mice during the period of carcinogen administration and after carcinogen administration.
Figure S-2. MS3 product ion spectra of dG-C8-AαC, dG-C8-MeIQ from liver of mice and their isotopically-labelled internal standards.