

Abstract
The cannabinoid 1 receptor (CB1) is an important regulator of energy metabolism. Reports of in vivo and in vitro studies give conflicting results regarding its role in insulin secretion, possibly due to circulatory factors, such as incretins. We hypothesized that this receptor may be a regulator of the entero-insular axis. We found that despite lower food consumption and lower body weight postprandial GLP-1 plasma concentrations were increased in CB1−/− mice compared to CB1+/+ mice administered a standard diet or high fat/sugar diet. Upon exogenous GLP-1 treatment, CB1−/− mice had increased glucose-stimulated insulin secretion. In mouse insulinoma cells, cannabinoids reduced GLP-1R-mediated intracellular cAMP accumulation and subsequent insulin secretion. Importantly, such effects were also evident in human islets, and were prevented by pharmacologic blockade of CB1. Collectively, these findings suggest a novel mechanism in which endocannabinoids are negative modulators of incretin-mediated insulin secretion.
Keywords: Adenylyl cyclase, cAMP, CB1, GLP-1, Incretin, Insulin secretion
Graphical Abstract
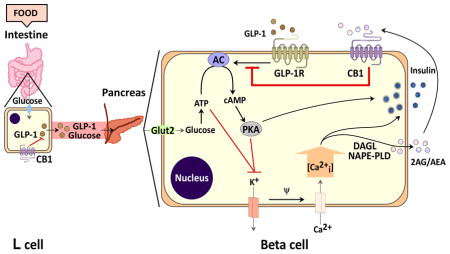
Schematic representation depicting theoretical postprandial regulation of the entero-insular axis by CB1. After consuming a meal, GLP-1 is secreted into the circulation from L-cells. Upon binding to their specific receptors on pancreatic β cells, they activate adenylyl cyclase (AC), which results in increased cAMP production and PKA activation. Additionally, elevated circulating glucose concentrations lead to increased ATP production and inhibition of the ATP/K+ channels, which initiates calcium (Ca2+) entry through voltage-dependent Ca2+ channels. The resultant rise of intracellular Ca2+, in conjunction with incretin-mediated PKA activation, leads to secretion of insulin and synthesis (by NAPE-PLD and DAGL) of ECs (2-AG/AEA) (Kim et al., 2011). Subsequently, AEA/2-AG activate CB1, thereby blocking the action of incretin-mediated AC activation, which in turn negatively impacts glucose-mediated insulin secretion. (Glut2= glucose transporter 2; PKA= protein kinase A; NAPE-PLD= N-Acyl phosphatidylethanolamine-specific phospholipase D; DAGL= Diacylglycerol lipase; 2-AG= 2-arachidonoylglycerol; AEA= anandamide. GLP-1= glucagon-like peptide-1; GIP= glucose-dependent insulinotropic peptide).
1. Introduction
In the past two decades, the endogenous cannabinoid system (ECS) has emerged as an important regulator of energy metabolism (Silvestri and Di Marzo, 2013). Up-regulation of this system, which includes endocannabinoids (ECs), their synthesizing/degrading enzymes and their receptors, is associated with dyslipidemia, obesity and type 2 diabetes mellitus (Engeli, 2008; Matias and Di Marzo, 2007). Many of the metabolic regulatory effects associated with the ECS are modulated through the cannabinoid 1 receptor (CB1), a Gi/o-protein coupled receptor found in brain and peripheral tissues. Central CB1 stimulation leads to increase food intake (Di Marzo et al., 2001; Silvestri and Di Marzo, 2013), and CB1 blockade induces weight loss (Van Gaal et al., 2005). Conversely, peripheral CB1s play an important role in glucose homeostasis by modulating lipogenesis in liver and adipose tissues (Cota et al., 2003; Matias et al., 2006; Osei-Hyiaman et al., 2005), glucose uptake in skeletal muscle (Esposito et al., 2008) and motility of the gastrointestinal tract (Izzo and Sharkey, 2010; Troy-Fioramonti et al., 2014). While most researchers agree that pancreatic beta (β) cells also contain CB1 (Bermúdez-Silva et al., 2008; Kim et al., 2011; Starowicz et al., 2008), its exact role in insulin secretion remains controversial.
Insulin secretion is a tightly regulated process such that blood glucose concentrations are maintained within a narrow range at all times. Secretion of insulin is stimulated by both glucose and incretins, which include glucose-dependent insulinotropic peptide (GIP) and glucagon-like peptide-1 (GLP-1). Incretins are hormones secreted from enteroendocrine cells in response to food intake (Montrose-Rafizadeh et al., 1994). Once in circulation, GIP and GLP-1 bind to their specific Gs-protein coupled receptors (GIPR and GLP-1R, respectively) on β cells to activate adenylyl cyclase (AC) (Drucker et al., 1987; Thorens, 1992). The subsequent rise in intracellular cAMP is critical to incretin-mediated insulin secretion and accounts for approximately 50% of the total insulin secreted following an oral glucose challenge (Kim and Egan, 2008). Activation of CB1 stimulates Gi/o, which inhibits AC activity and cAMP synthesis (Turu and Hunyady, 2010). In mouse, enteroendocrine cells express CB1 receptor (Sykaras et al., 2012), and its activation inhibits GIP secretion (Moss et al., 2012; Troy-Fioramonti et al., 2014), which suggests that blockade of CB1 would at least indirectly stimulate insulin secretion through GIPR. Several recent studies have suggested a potential interaction between incretin receptors and CB1, particularly with regard to food intake (E. Bojanowska, 2011; Patel et al., 2014; Radziszewska et al., 2014). However, there is a lack of data as to how CB1 would influence the entero-insular axis if both incretin and insulin secretion are impacted by the same receptor system.
In the present study, we investigated the role of CB1 on both GLP-1 secretion and subsequent receptor-mediated insulin secretion. We demonstrate herein that mice deficient in CB1 exhibit higher postprandial incretin secretion and greater GLP-1 sensitivity than control mice. Using both genetic and pharmacological manipulations, we further show that activation of CB1 down-regulates GLP-1R signaling and, in turn, insulin secretion, in mouse insulinoma cells as well as human islets.
2. Materials and Methods
2.1. Reagents
Anandamide (AEA), 2-arachidonoylglycerol (2-AG), arachidonyl-2-chloroethylamide (ACEA), AM251 and WIN55,212-2 were obtained from Cayman Chemical (Ann Arbor, MI). JD-5037 was provided by Jenrin Discovery, Inc. (Wilmington, DE). Exendin-4 (Ex-4), GLP-1 and GIP were obtained from Bachem (Torrance, CA). Dipeptidyl peptidase-4 (DPP-4) inhibitor was purchased from Millipore (Billerica, MA). Aprotinin was obtained from Fisher-Scientific (Middletown, VA). Intralipid 20% was purchased from Fresenius-Kabi (Uppsala, Sweden). The human Cnr1 (CB1 encoding gene) cDNA was amplified by RT-PCR from a human pancreas RNA (Stratagene, Agilent Technologies, Santa Clara, CA), and cloned into the mCerulean-N1 vector (Rizzo and Piston, 2005).
2.2. Animal Models and Experimental Diets
Global CB1 receptor knockout (CB1−/−) mice and their wild-type littermates (CB1+/+) backcrossed to a C57Bl/6J background were bred as previously described (Zimmer et al., 1999). Mice (male, 2–3 months old) were provided with water and feed ad libitum on either a standard chow diet (SD; 16.7% kcal fat and 12.4% kcal sugar) or a high fat/high sugar diet (HFS; 49.2% kcal fat and 21.1% kcal sugar; Dyets Inc., Bethlehem, PA) for 15 weeks. At the end of the study, body weight was measured and animals were placed in metabolic cages in order to obtain precise measurements of food intake. All animal care and experimental procedures followed US National Institutes of Health guidelines and were approved by the National Institute on Aging Animal Care and Use Committee.
2.3. Intraperitoneal glucose tolerance tests
Mice were fasted overnight and given free access to water. Intraperitoneal glucose tolerance tests (IPGTT) were carried out as we previously described (Wang et al., 1997). After 36 h of GLP-1 (1.5 pmol/kg·min) treatment via subcutaneously-implanted Alzet microosmotic pumps (Cupertino, CA) (n=6 per genotype), a bolus of glucose (1 g/kg body weight) was administered intraperitoneally. Tail-vein blood samples were collected at 0, 15, 30, 60, and 90 min.
2.4. Mouse circulating hormone and glucose quantification
Blood glucose concentrations were determined using a glucometer (Elite, Bayer Inc.) from fresh tail-vein blood. In order to determine active levels of GLP-1, mice were orally administered a single dose of Intralipid (20%) containing D-glucose (30%) via oral gavage and blood collected 20 min post-dose (Althage et al., 2008; Lu et al., 2007) into pre-chilled tubes containing EDTA, aprotinin and DPP-4 inhibitor. Plasma insulin was measured with a mouse insulin ELISA (Crystal Chem Inc., Downers Grove, IL) and active GLP-1 determined with the GLP-1 (Active 7–36) ELISA (ALPCO, Salem, NH). Plasma GIP and leptin were analyzed in 100 μl of plasma (final bleed) using a MILLIPLEX Mouse Gut Hormone Magnetic Bead Panel (Millipore, Billerica, MA). HOMA-IR, a measure of liver insulin sensitivity, was quantified by: fasting insulin (μU/mL) x fasting glucose (mg/dL)/405 (Haffner et al., 1997).
2.5. Cell culture and insulin secretion and cAMP assays from cell lines
MIN6 and βTC6 insulinoma cells were maintained in DMEM medium with 10% FBS (Life Technologies, Grand Island, NY). CHO-GLP-1R (CHO-K1 cells stably transfected with GLP-1R) (Montrose-Rafizadeh, 1997) were maintained in DMEM/F-12 medium with 10% FBS. For insulin secretion and cAMP assays, cells were plated in 12-well plates, one or three days before transfection, respectively. Cells were washed three times in PBS and were pre-incubated for 2 h in the Krebs buffer containing 4 mM glucose at 37°C. Subsequentl y, CB1 agonists or inverse agonists were pre-treated for 15 min before the subsequent addition of glucose (25 mM) or Ex-4 (10 or 25 nM) for a further 20 min. At the end of the experiment, the buffer was collected, centrifuged to remove cellular debris and saved for quantification of insulin. The cells were lysed with 0.1 M HCl and were centrifuged to remove cellular debris. The supernatant were collected for determination of cAMP and protein concentrations. cAMP was measured using a cAMP ELISA kit according to the manufacturer’s instructions. The data were normalized to protein concentration, and estimated from three independent experiments, each performed in at least triplicate. Transfections of the expression vectors and siRNA (Santa Cruz, Dallas, Texas) for Cnr1 were carried out 24 or 48 h before adding CB1 agonist using Lipofectamine 2000 and RNAiMAX (Life Technologies), respectively. Scramble siRNA (Silencer Negative Control #1; Life Technologies) or empty vector was transfected as negative control.
2.6. Insulin secretion and cAMP accumulation in isolated human islets
Human pancreatic islets were provided by the NIDDK-funded Integrated Islet Distribution Program (IIDP) at City of Hope and incubated in insulin secretion assay buffer (Montrose-Rafizadeh et al., 1994) containing 2 mM glucose for a total of 2 h at 37°C, with media being refreshed after 1 h. Islets were then pre-treated for 15 min with 7.5 mM glucose (postprandial levels), IBMX (25 μM) and increasing concentrations of ACEA before stimulation with Ex-4 (0.33 nM) for an additional 20 min at 37°C. Media were co llected for measuring insulin secretion (Mercodia, Uppsala, Sweden). Islets were processed for cAMP via an ELISA (Enzo Life Sciences, Farmingdale, NY) and total protein concentration quantified determined by a Bicinchoninic Acid Protein Assay (Pierce, Rockford, IL).
2.7. Immunoblotting
Protein samples extracted from cells using RIPA buffer (50 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA) containing protease and phosphatase inhibitor cocktails (Millipore, Billerica, Massachusetts) were subjected to Tris-glycine PAGE (Life Technologies), immunoblotted with rabbit anti-CB1 (1:500; Frontier Institute Co., Ltd, Hokkaido, Japan) and visualized by ECL (GE Healthcare, Pittsburgh, PA).
2.8. RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells using Trizol reagent (Life Technologies) according to the manufacturer’s instructions. After reverse transcription, the resulting materials were used for qRT-PCR amplification using gene-specific primer pairs (CNR1_F TCC CAC AGA AAT TCC CTC TAA CT; CNR1_R CCT TGA ACG ATG AGA GAG ACT TG) and SYBR Green PCR master mix (Life Technologies), with data normalized to 18S mRNA.
2.9. Statistical analysis
Quantitative data are presented as the mean ± SEM. Differences between mean values for variables within individual experiments were compared statistically by Student’s t-test or analysis of variance (ANOVA) as appropriate. Comparisons were performed using GraphPad Prism 6.0 software (La Jolla, CA). A p value of < 0.05 was considered statistically significant.
3. Results
3.1. CB1−/− mice are leaner and more resistant to diet-induced obesity
Mice were maintained on either a standard diet (SD) or high fat/high sugar diet (HFS). As described previously (Cota et al., 2003; Ravinet Trillou et al., 2004), CB1−/− mice were around 12% leaner than CB1+/+ mice on both diets (Figure 1A) and had lower food intake (Figure S1). CB1−/− mice had significantly lower fasting blood glucose on SD (100 ± 8 versus 118 ± 9 mg/dL for CB1+/+ mice) and on HFS (155 ± 10 versus 174 ± 9 mg/dL) (Figure S2), which was correlated with lower insulin resistance in CB1−/− mice defined by HOMA-IR index (Figure S3). After oral administration of a glucose-lipid mix bolus (30% D-glucose in 20% Intralipid), blood glucose did not rise as much in CB1−/− mice on both SD (207±15 versus 276 ± 25 mg/dL for CB1+/+ mice) or on HFS (263 ± 25 versus 373 ± 21 mg/dL) (Figure 1B). Circulating insulin levels were also decreased in CB1−/− mice (0.4 ± 0.05 versus 0.9 ± 0.1 ng/mL for CB1+/+ mice) (Figure 1C) compared to CB1+/+ mice on SD as well as on HFS (6 ± 2 versus 12 ± 2 ng/mL) 20 min after glucose-lipid challenge. Additionally, plasma leptin levels were lower in CB1−/− mice after oral administration of glucose-lipid mix bolus (Figure 1D).
Figure 1. CB1−/− mice have lower body weight and leptin levels and are more insulin sensitive than CB1+/+ mice.

CB1+/+ (white bars/circles) and CB1−/− (black bars/circles) mice maintained on a standard diet (SD) or high fat/high sugar diet (HFS) were fasted overnight. Body weight (A) was measured. Blood glucose and plasma hormone concentrations (B–D) were determined 20 min after oral administration of a glucose and fat mix (30% D-glucose in 20% Intralipid). Plasma was assayed for glucose (B), insulin (C) and leptin (D). Data are represented as mean ± SEM (n=5–8/group). *p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001 compared to CB1+/+ mice; # p ≤ 0.01 compared to SD.
3.2. Depletion of CB1 alters incretin secretion following a glucose/lipid challenge
In order to determine if CB1 influences postprandial incretin secretion, fasted CB1+/+ and CB1−/− mice received a single bolus of glucose-lipid mix so as to stimulate incretin secretion. Blood was collected 20 min later, a time known for maximum incretin secretion. In accordance with the literature, mice deficient in CB1 had a 68% increase in plasma GIP concentrations (867 ± 71 vs. 517 ± 113 pg/mL; Figure 2A); however mice on a HFS diet already had elevated GIP levels, presumably due to the diet, and likely were at maximum GIP secretion, and so there were no differences between strains. Interestingly, postprandial active GLP-1 plasma levels were approximately 23% higher in CB1−/− mice (4.7 ± 0.2 pg/mL) than in CB1+/+ mice (3.8 ± 0.2 pg/mL) on SD, and was more pronounced (~32% increase; 4.8 ± 0.4 for CB1−/− mice versus 3.6 ± 0.2 pg/mL for CB1+/+ mice) when mice were maintained on the HFS (Figure 2B).
Figure 2. CB1−/− mice have higher re-feeding-stimulated circulating plasma incretin levels.

CB1+/+ (white bars) and CB1−/− (black bars) mice maintained on a standard diet (SD) or high fat/high sugar diet (HFS) were fasted overnight and re-fed with a mix of glucose and fat. Plasma GIP (A) and active GLP-1 (B) were determined 20 min after oral gavage. Data are represented as mean ± SEM (n=5–8/group). *p ≤ 0.05 compared to CB1+/+ mice; # p ≤ 0.05 compared to SD.
3.3. CB1−/− mice have enhanced insulin secretion over basal and are more responsive to GLP-1 treatment
Since GLP-1 concentrations were altered in CB1−/− mice independent of diet, we sought to further explore the role of CB1 in GLP-1-mediated insulin secretion. We first performed an IPGTT in CB1+/+ and CB1−/− mice in order to determine incretin-independent glucose-stimulated insulin secretion. While blood glucose clearance was comparable between strains (Figure 3A), plasma insulin levels were significantly lower in CB1−/− mice (Figure 3B). This further confirms the greater insulin sensitivity of CB1−/− mice (Figure S3). However the rise of plasma insulin 5 minutes after glucose infusion (early phase insulin secretion) over fasting levels was ~30% greater in CB1−/− mice than in CB1+/+ mice (Figure 3C). Secondly we investigated GLP-1-stimulated glucose-dependent insulin secretion. CB1+/+ and CB1−/− mice were subcutaneously infused with GLP-1 and fasted prior to receiving glucose intraperitoneally thereby avoiding endogenous incretin secretion. The fasting plasma levels of GLP-1 after 36 h of GLP-1 infusion were comparable between CB1+/+ and CB1−/− and were 2.5 fold higher than levels after fat-glucose ingestion (Figure S4). Following GLP-1 treatment, CB1−/− mice had significantly lower blood glucose levels than CB1+/+ mice (Figure 3A). Additionally, early phase insulin secretion was significantly greater in CB1−/− mice compared to CB1+/+ mice (Figure 3B). While CB1+/+ mice reached their peak insulin concentration at 30 min (5.4 ± 0.2 ng/mL), CB1−/− mice had peak insulin at 15 min, which was also greater than in CB1+/+ mice (9.4 ± 0.3 ng/mL) (Figure 3B).
Figure 3. CB1−/− mice have improved intraperitoneal glucose tolerance following GLP-1 stimulation.

Incretin-independent glucose-stimulated insulin secretion: CB1+/+ and CB1−/− mice were fasted prior to IPGTT (1 g/kg glucose). Blood glucose levels (A) and plasma insulin levels (B) were determined in CB1−/− (black boxes/bars) and in CB1+/+ mice (white boxes/bars) (n=7–8/group). Increase of plasma insulin level over basal (fold increase) is shown at 5, 15 and 30 min after glucose infusion (C). Glucose-dependent GLP-1-stimulated insulin secretion: CB1+/+ and CB1−/− mice were administered GLP-1 (1.5 pmol/kg·min) via microosmotic pumps and fasted prior to the IPGTT (1 g/kg glucose). Blood glucose levels (D) and plasma insulin levels (E) were determined in CB1−/− (black boxes/bars) and in CB1+/+ mice (white boxes/bars) (n=5–6/group). Data are represented as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
3.4. Endocannabinoids down-regulate GLP-1R-mediated cAMP accumulation and insulin secretion in mouse insulinoma cells
Incretins stimulate post-prandial insulin secretion by binding to their Gs-coupled receptors found on pancreatic β cells, which increases AC activity and in turn cAMP levels. However, elevated glucose concentrations found in the postprandial state also cause increased EC production (Kim et al., 2011), which may in turn activate the Gi/o-coupled CB1 that is present on pancreatic β cells. We therefore analyzed the effect of stimulating both the GLP-1R and CB1 on AC activity and insulin secretion in βTC6, a mouse insulinoma cell line. The potent GLP-1R agonist, Ex-4, significantly increased cAMP accumulation in βTC6 cells compared to untreated cells (Figure 4A,C). Treatment with either of the ECs, 2-AG (Figure 4A) or AEA (Figure 4C), inhibited both Ex-4-stimulated cAMP accumulation and insulin secretion by ~40% (Figure 4B,D). Importantly, such effects were prevented by pre-treating β cells with the CB1 inverse agonist, AM251 (Figure 4A,C–D). AM251 alone significantly increased intracellular cAMP (Figure S5). Additionally we confirmed that activation of CB1 blocks not only GLP-1R-mediated intracellular cAMP accumulation but also GIP action (Figure S6). These findings collectively indicate that ECs inhibit incretin-mediated insulin secretion.
Figure 4. Cannabinoids modulate GLP-1-mediated intracellular cAMP accumulation and insulin secretion in βTC6 cells.

Relative intracellular cAMP (A, C) and insulin (B, D) concentrations secreted from βTC6 cells treated with the endogenous cannabinoid 2-AG (A–B) or AEA (C–D) without or with the inverse agonist AM251 before subsequent addition of Ex-4 (25 nM). All values were normalized to protein concentration and represented as percentage to unstimulated cells. Data are mean ± SEM, n=3. *p≤0.05; **p≤0.01.
3.5. Inhibition of GLP-1-mediated adenylyl cyclase activity occurs through the CB1
As 2-AG and AEA are known to activate both CB1 and CB2, we used the highly specific, synthetic CB1 agonist ACEA in a separate mouse insulinoma cell line (MIN6) to further determine if ECs inhibit AC activity through CB1. We confirmed that the MIN6 cells were responsive to glucose-stimulated insulin secretion (Figure S7). ACEA significantly decreased Ex-4-mediated cAMP accumulation in MIN6 β cells in a dose-dependent manner (Figure 5A), with the maximum inhibition of cAMP accumulation similar to those found in non-stimulated cells. ACEA also inhibited Ex-4-stimulated insulin secretion, whereas it did not alter insulin secretion when combined with the membrane-depolarizing actions of KCl (Figure 5B), indicating that such effects are receptor-mediated. Moreover, knockdown of the CB1 encoding gene Cnr1 by siRNA (Figure 5C, D) abolished the ability of ACEA to inhibit Ex-4-stimulated cAMP accumulation (Figure 5E). To further eliminate the possibility of any non-specific effects of CB1 ligands, GLP-1R stably-transfected CHO cells, which are inherently devoid of the cannabinoid receptors, were transiently transfected with either an empty vector or Cnr1 and subsequently treated with Ex-4. The inhibitory effects of ACEA on Ex-4-stimulated cAMP accumulation were not observed in CHO-GLP-1R cells transfected with the empty vector. However, ACEA treatment in CHO-GLP-1R cells transfected with the Cnr1 vector led to reduced cAMP accumulation (Figure 5F). Moreover isolated islets from CB1−/− showed increase in glucose-mediated insulin secretion in the presence of GLP-1R agonism compared to CB1+/+ (Figure S8). Overall, these results show that ECs antagonize GLP-1-mediated effects in β cells through CB1 activation.
Figure 5. CB1 activation down-regulates GLP-1-mediated adenylyl cyclase activity.

Relative cAMP (A) and insulin (B) concentrations in MIN6 cells treated with the specific CB1 agonist ACEA before the subsequent addition of Ex-4 or KCl. (C–D) Relative Cnr1 mRNA (C) and protein (D) levels in MIN6 cells following transfection with scrambled (white bars) or Cnr1 (black bars) siRNAs for 48 h. (E) Effects of ACEA on Ex-4-stimulated cAMP accumulation in MIN6 cells following transfection with indicated siRNAs. (F) Effects of CB1 over-expression on GLP-1-mediated cAMP accumulation. CHO-K1 cells stably expressing GLP-1R were transiently transfected with empty vector (white bars) or Cnr1 (black bars) plasmids and pre-incubated with ACEA for 20 min prior to stimulation with Ex-4. All values were normalized to protein concentration and represented as percentage to unstimulated cells. Data are mean ± SEM, n=3. *p ≤ 0.05; **p ≤ 0.01, n.s.= not significant.
3.6. Blockade of CB1 prevents cannabinoid-induced inhibition of GLP-1 effect in human islets
Finally we determined if CB1 would directly influence GLP-1-mediated AC activity and therefore insulin secretion in intact human islets. Islets were pre-cultured under non-stimulatory conditions with 2 mM glucose for 2 h before being treated with increased glucose (7.5 mM .i.e. usual postprandial plasma glucose concentration), Ex-4 and/or increasing concentrations of ACEA. As expected, Ex-4 significantly increased both cAMP and insulin secretion compared to non-treated islets (Figure 6A–B). Importantly, ACEA significantly decreased Ex-4-stimulated cAMP accumulation (by ~65%; Figure 6A) and insulin secretion (by ~50%; Figure 6B) in human islets, with values similar to basal levels. This inhibitory effect was even more prominent in the human islets compared to mouse insulinoma cells since it resulted in a complete blockade of GLP-1-mediated stimulation and with much lower concentrations of ACEA (Figure 5A versus Figure 6A). Pre-treatment of islets with a CB1 inverse agonist, AM251 or JD-5037 (a peripherally-restricted CB1 blocker (Tam et al., 2012)), partially prevented (~30%) the effect of ACEA on Ex-4-stimulated AC activity (Figure 6C). Similarly, the loss in insulin secretion was partially rescued by pre-treating with AM251, and totally prevented with JD-5037 (Figure 6D).
Figure 6. CB1 blockade reduces GLP-1-stimulated AC activity and insulin secretion in human islets.

(A–B) Relative concentrations of intracellular cAMP (A) and insulin secretion (B) from isolated human islets pre-treated for 15 min with increasing concentrations of ACEA before stimulation with Ex-4 for an additional 20 min. (C–D) Effects of blocking CB1 on GLP-1 response in human islets. Relative levels of cAMP (C) and insulin secretion (D) in islets pre-treated with a CB1 inverse agonist (AM251 or JD-5037) prior to ACEA and Ex-4 as described above. All values were normalized to protein concentration and represented as percentage to unstimulated cells. Data are represented as mean ± SEM, n=3–5. *p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001.
4. Discussion
In the present report, we provide evidence for a regulatory mechanism by which the ECS indirectly regulates insulin secretion through influencing both GLP-1 release and GLP-1R signaling in β cells. We report that CB1−/− mice not only have enhanced early phase glucose-mediated insulin secretion but also are more sensitive to GLP-1-stimulated glucose-dependent insulin secretion. Additionally CB1−/− mice have higher circulating incretin levels upon fat-glucose ingestion, and GLP-1R agonists are known to improve early phase glucose-mediated insulin secretion by elevating intracellular cAMP levels in β cells (Wang et al., 1997). Furthermore, CB1 activation negatively impacts GLP-1R-mediated insulin secretion. This was prevented by blockade of the CB1 receptor in both insulinoma cells and in human islets.
Global CB1−/− mice are known to exhibit reduced fasting plasma insulin levels, low leptin levels and food intake and are resistant to diet-induced obesity (Cota et al., 2003; Ravinet Trillou et al., 2004). A very recent study reported a comparable phenotype in mice lacking DAGLα, a lipase responsible for 2-AG biosynthesis, suggesting 2-AG as the EC responsible for regulating body weight and not AEA (Powell et al., 2015). Similarly, the blockade of peripheral CB1 in both genetic and diet-induced obese mice results in decreased plasma insulin concentrations (Nam et al., 2012; Tam et al., 2012, 2010). Accordingly CB1−/− mice eat less and weigh less with both SD and HFS, which in turn would result in lower mean circulating glucose levels. Insulin and leptin levels were also significantly lower than wild-type and did not rise to the same extent with HFS. Interestingly, although circulating leptin, which is known to stimulate GLP-1 secretion (Anini and Brubaker, 2003), was lower in CB1−/− mice in SD and this strain is significantly smaller, a bolus of glucose/lipid stimulated incretin secretion ~20–70% more than in wild-type mice. This effect was constant in HFS only for GLP-1 but not for GIP, probably because secretion of GIP was at maximum capacity in wild-type when fed HFS. Previous studies have already documented a relationship between ECS and enteroendocrine cells. Duodenal AEA and 2-AG levels are increased with fasting and decreased by re-feeding in both lean and obese rats (Izzo et al., 2009) in a manner opposite of what would be expected for incretin secretion. Indeed CB1 is expressed in enteroendocrine cells and ECs have been shown to inhibit GIP release (Moss et al., 2012; Sykaras et al., 2012). Not only was GLP-1 secretion greater in CB1−/− mice, which must mean that L cells are normally under tonic inhibition by CB1, but the mice were more sensitive to GLP-1-mediated insulin secretion even under regular chow conditions.
The role of CB1 in insulin secretion and islet function has long been a point of contention. While several in vitro studies have found increased insulin secretion following CB1 activation (Bermúdez-Silva et al., 2008; Duvivier et al., 2009; Getty-Kaushik et al., 2009; Juan-Pico et al., 2006; Li et al., 2010; Malenczyk et al., 2013; Matias et al., 2006), others found decreases in insulin release (Anderson et al., 2013; Li et al., 2011; Nakata and Yada, 2008). One potential explanation for these discrepancies is that ECs, which are elevated under obese conditions, act on CB1 and it in turn regulates factors, such as incretins, found only in the whole organism that are absent in isolated islet and β cell cultures. Pharmacologic blockade of CB1 with rimonabant along with co-administration of Ex-4 reduced hyperglycemia in diet-induced obese mice (Patel et al., 2014). It is also plausible that human islets have not only different morphology from mouse islets, but have different physiological responses to CB1 modulation. In our study we show that cannabinoids reduced GLP-1-mediated intracellular cAMP accumulation and subsequent insulin secretion in a concentration-dependent manner in insulinoma cells and human islets. Indeed, both synthetic (ACEA) and endogenous (2-AG and AEA) ligands of CB1 dose-dependently reduced Ex-4-stimulated cAMP accumulation and insulin secretion in mouse insulinoma cells. These effects undoubtedly occurred through CB1 as demonstrated by the use of CB1 siRNA as well as a CHO cell line that exclusively expresses GLP-1R and CB1. ACEA blockade of Ex-4 action was prevented when islets were pre-treated with either a global (AM251) or peripherally-restricted (JD-5037) CB1 inverse agonist, suggesting that peripherally-restricted compounds might have translational relevance.
A new intriguing mechanism for prolonging GLP-1 action has recently been described. Lipids, such as oleoylethanolamide (OEA) and 2-oleoylglycerol appear to bind GLP-1 itself and this modification enhances GLP-1 action at its receptor (Cheng et al., 2015). Therefore it is possible to hypothesize that diets with differing lipid compositions will alter EC synthesis and other lipid mediators, which might also then impact GLP-1 action in either a negative or a positive manner.
Peripheral CB1 blockade has previously been shown to protect against CB1-induced macrophage infiltration and its associated β cell loss in a rodent model of type 2 diabetes, thereby improving β cell health (Jourdan et al., 2013). In addition, CB1 inverse agonists enhance insulin receptor signaling and increased survival in β cells (Kim et al., 2012, 2011). Together with the results presented herein, blockade of CB1 may prove to be a beneficial therapeutic for type 2 diabetes in a multi-fold fashion by increasing insulin responsiveness, preventing macrophage infiltration and improving incretin-mediated insulin secretion.
5. Conclusion
Mice lacking CB1 receptor secrete greater amounts of both GIP and GLP-1 and diet-induced obesity does not further increase the already higher GIP levels present in CB1−/− mice. Mice lacking CB1 receptor are also more sensitive to the insulin secretory effects of GLP-1. CB1 receptor indeed negatively regulates GLP-1R in β cells, which in turns regulates insulin secretion in mouse and also in human islets. In summary, we contribute to further evidence that CB1 tonically inhibits incretin secretion in the fasting state and acts as a brake on the entero-insular axis to limit the secretion of incretins as well as their ability to stimulate insulin secretion in the fed state (illustrated schematically in the graphical abstract). Further studies examining the relationship between the CB1 and incretin receptors on β cells under hyperglycemic and/or obese conditions are warranted and may provide insight as to the potential benefit for using peripherally-restricted CB1 inverse agonists in combination with exenatide, liraglutide and lixisenatide (as well as dipeptidyl peptidase-4 inhibitors) for the treatment of type 2 diabetes.
Supplementary Material
Highlights.
CB1−/− mice have improved glucose homeostasis in regular diet and high fat diet.
Knocking out CB1 increases incretin secretion in mice.
Cannabinoids inhibit GLP-1R-dependent insulin secretion in insulinoma cells via CB1.
ACEA inhibition of insulin secretion is rescued by CB1 blockade in human islets.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute on Aging, NIH. WK is supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2012R1A1A1041352). Dr. J. Pickel, NIMH Transgenic Core/NIH, provided the CB1−/− mice and the animal facilities of NIA/NIH carried out the genotyping and husbandry. The authors wish to thank Jenrin Discovery (Drs. McElroy and Chorvat) for generously providing the JD-5037 compound.
8. Funding
This work was supported by the Intramural Research Program of National Institute on Aging within the National Institutes of Health. WK was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future (NRF-2012R1A1A1041352) and the Ministry of Education (No. 2009-0093826).
Abbreviations
- AC
Adenylyl cyclase
- ACEA
arachidonoyl 2′-chloroethylamide
- AEA
anandamide
- 2-AG
2-arachidonoylglycerol
- EC
endocannabinoid
- GIP
glucose-dependent insulinotropic peptide
- GLP-1
glucagon-like peptide-1
- DPP-4
Dipeptidyl peptidase-4
- Ex-4
exendin-4
- IPGTT
Intraperitoneal glucose tolerance test
- SD
standard diet
- HFS
high fat/high sugar diet
- Glut2
glucose transporter 2
- PKA
protein kinase A
- NAPE-PLD
N-Acyl phosphatidylethanolamine-specific phospholipase D
- DAGL
Diacylglycerol lipase
Footnotes
9. Competing interest
The authors have nothing to disclose.
10. Author contribution
IGM and SMKW designed/performed experiments, analyzed data and wrote the manuscript. WK and MR performed experiments and analyzed data. JME contributed to design of experiments, interpretation of data and writing of manuscript. JME is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Althage MC, Ford EL, Wang S, Tso P, Polonsky KS, Wice BM. Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J Biol Chem. 2008;283:18365–76. doi: 10.1074/jbc.M710466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RL, Randall MD, Chan SLF. The complex effects of cannabinoids on insulin secretion from rat isolated islets of Langerhans. Eur J Pharmacol. 2013;706:56–62. doi: 10.1016/j.ejphar.2013.02.034. [DOI] [PubMed] [Google Scholar]
- Anini Y, Brubaker PL. Role of leptin in the regulation of glucagon-like peptide-1 secretion. Diabetes. 2003;52:252–9. doi: 10.2337/diabetes.52.2.252. [DOI] [PubMed] [Google Scholar]
- Bermúdez-Silva FJ, Suárez J, Baixeras E, Cobo N, Bautista D, Cuesta-Muñoz AL, Fuentes E, Juan-Pico P, Castro MJ, Milman G, Mechoulam R, Nadal A, Rodríguez de Fonseca F. Presence of functional cannabinoid receptors in human endocrine pancreas. Diabetologia. 2008;51:476–87. doi: 10.1007/s00125-007-0890-y. [DOI] [PubMed] [Google Scholar]
- Cheng YH, Ho MS, Huang WT, Chou YT, King K. Modulation of Glucagon-like Peptide-1 (GLP-1) Potency by Endocannabinoid-like Lipids Represents a Novel Mode of Regulating GLP-1 Receptor Signaling. J Biol Chem. 2015;290:14302–13. doi: 10.1074/jbc.M115.655662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thone-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112:423–431. doi: 10.1172/JCI17725. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–5. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A. 1987;84:3434–8. doi: 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvivier VF, Delafoy-Plasse L, Delion V, Lechevalier P, Le Bail JC, Guillot E, Pruniaux MP, Galzin AM. Beneficial effect of a chronic treatment with rimonabant on pancreatic function and beta-cell morphology in Zucker Fatty rats. Eur J Pharmacol. 2009;616:314–20. doi: 10.1016/j.ejphar.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Bojanowska EER. Combined stimulation of glucagon-like peptide-1 receptor and inhibition of cannabinoid CB1 receptor act synergistically to reduce food intake and body weight in the rat. J Physiol Pharmacol. 2011;62:395–402. [PubMed] [Google Scholar]
- Engeli S. Dysregulation of the endocannabinoid system in obesity. J Neuroendocrinol. 2008;20(Suppl 1):110–5. doi: 10.1111/j.1365-2826.2008.01683.x. [DOI] [PubMed] [Google Scholar]
- Esposito I, Proto MC, Gazzerro P, Laezza C, Miele C, Alberobello AT, D’Esposito V, Beguinot F, Formisano P, Bifulco M. The cannabinoid CB1 receptor antagonist rimonabant stimulates 2-deoxyglucose uptake in skeletal muscle cells by regulating the expression of phosphatidylinositol-3-kinase. Mol Pharmacol. 2008;74:1678–86. doi: 10.1124/mol.108.049205. [DOI] [PubMed] [Google Scholar]
- Getty-Kaushik L, Richard AMT, Deeney JT, Krawczyk S, Shirihai O, Corkey BE. The CB1 antagonist rimonabant decreases insulin hypersecretion in rat pancreatic islets. Obesity (Silver Spring) 2009;17:1856–60. doi: 10.1038/oby.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner SM, Miettinen H, Stern MP. The homeostasis model in the San Antonio Heart Study. Diabetes Care. 1997;20:1087–92. doi: 10.2337/diacare.20.7.1087. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Piscitelli F, Capasso R, Aviello G, Romano B, Borrelli F, Petrosino S, Di Marzo V. Peripheral endocannabinoid dysregulation in obesity: relation to intestinal motility and energy processing induced by food deprivation and re-feeding. Br J Pharmacol. 2009;158:451–61. doi: 10.1111/j.1476-5381.2009.00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, Ju C, Aouadi M, Czech MP, Kunos G. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19:1132–40. doi: 10.1038/nm.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan-Pico P, Fuentes E, Bermudez-Silva FJ, Javier Diaz-Molina F, Ripoll C, Rodriguez de Fonseca F, Nadal A, Juan-Picó P, Bermúdez-Silva FJ, Javier Díaz-Molina F, Rodríguez de Fonseca F. Cannabinoid receptors regulate Ca(2+) signals and insulin secretion in pancreatic beta-cell. Cell Calcium. 2006;39:155–62. doi: 10.1016/j.ceca.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Kim W, Doyle ME, Liu Z, Lao Q, Shin YK, Carlson OD, Kim HS, Thomas S, Napora JK, Lee EK, Moaddel R, Wang Y, Maudsley S, Martin B, Kulkarni RN, Egan JM. Cannabinoids inhibit insulin receptor signaling in pancreatic β-cells. Diabetes. 2011;60:1198–209. doi: 10.2337/db10-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60:470–512. doi: 10.1124/pr.108.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Lao Q, Shin YK, Carlson OD, Lee EK, Gorospe M, Kulkarni RN, Egan JM. Cannabinoids induce pancreatic beta-cell death by directly inhibiting insulin receptor activation. Sci Signal. 2012;5:ra23. doi: 10.1126/scisignal.20025195/216/ra23. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Bowe JE, Huang GC, Amiel SA, Jones PM, Persaud SJ. Cannabinoid receptor agonists and antagonists stimulate insulin secretion from isolated human islets of Langerhans. Diabetes Obes Metab. 2011;13:903–10. doi: 10.1111/j.1463-1326.2011.01422.x. [DOI] [PubMed] [Google Scholar]
- Li C, Jones PM, Persaud SJ. Cannabinoid receptors are coupled to stimulation of insulin secretion from mouse MIN6 beta-cells. Cell Physiol Biochem. 2010;26:187–96. doi: 10.1159/000320527. [DOI] [PubMed] [Google Scholar]
- Lu WJ, Yang Q, Sun W, Woods SC, D’Alessio D, Tso P. The regulation of the lymphatic secretion of glucagon-like peptide-1 (GLP-1) by intestinal absorption of fat and carbohydrate. Am J Physiol Gastrointest Liver Physiol. 2007;293:G963–71. doi: 10.1152/ajpgi.00146.2007. [DOI] [PubMed] [Google Scholar]
- Malenczyk K, Jazurek M, Keimpema E, Silvestri C, Janikiewicz J, Mackie K, Di Marzo V, Redowicz MJ, Harkany T, Dobrzyn A. CB1 cannabinoid receptors couple to focal adhesion kinase to control insulin release. J Biol Chem. 2013;288:32685–99. doi: 10.1074/jbc.M113.478354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias I, Di Marzo V. Endocannabinoids and the control of energy balance. Trends Endocrinol Metab. 2007;18:27–37. doi: 10.1016/j.tem.2006.11.006. S1043-2760(06)00243-8 [pii] [DOI] [PubMed] [Google Scholar]
- Matias I, Gonthier MP, Orlando P, Martiadis V, De Petrocellis L, Cervino C, Petrosino S, Hoareau L, Festy F, Pasquali R, Roche R, Maj M, Pagotto U, Monteleone P, Di Marzo V. Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J Clin Endocrinol Metab. 2006;91:3171–80. doi: 10.1210/jc.2005-2679. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C. High Potency Antagonists of the Pancreatic Glucagon-like Peptide-1 Receptor. J Biol Chem. 1997;272:21201–21206. doi: 10.1074/jbc.272.34.21201. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Egan JM, Roth J. Incretin hormones regulate glucose-dependent insulin secretion in RIN 1046-38 cells: mechanisms of action. Endocrinology. 1994;135:589–94. doi: 10.1210/endo.135.2.8033807. [DOI] [PubMed] [Google Scholar]
- Moss CE, Marsh WJ, Parker HE, Ogunnowo-Bada E, Riches CH, Habib AM, Evans ML, Gribble FM, Reimann F. Somatostatin receptor 5 and cannabinoid receptor 1 activation inhibit secretion of glucose-dependent insulinotropic polypeptide from intestinal K cells in rodents. Diabetologia. 2012;55:3094–103. doi: 10.1007/s00125-012-2663-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata M, Yada T. Cannabinoids inhibit insulin secretion and cytosolic Ca2+ oscillation in islet beta-cells via CB1 receptors. Regul Pept. 2008;145:49–53. doi: 10.1016/j.regpep.2007.08.009. S0167-0115(07)00157-7 [pii] [DOI] [PubMed] [Google Scholar]
- Nam DH, Lee MH, Kim JE, Song HK, Kang YS, Lee JE, Kim HW, Cha JJ, Hyun YY, Kim SH, Han SY, Han KH, Han JY, Cha DR. Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology. 2012;153:1387–1396. doi: 10.1210/en.2011-1423. pii. [DOI] [PubMed] [Google Scholar]
- Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Bátkai S, Harvey-White J, Mackie K, Offertáler L, Wang L, Kunos G. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KN, Joharapurkar AA, Patel V, Kshirsagar SG, Bahekar R, Srivastava BK, Jain MR. Cannabinoid receptor 1 antagonist treatment induces glucagon release and shows an additive therapeutic effect with GLP-1 agonist in diet-induced obese mice. Can J Physiol Pharmacol. 2014;92:975–83. doi: 10.1139/cjpp-2014-0310. [DOI] [PubMed] [Google Scholar]
- Powell DR, Gay JP, Wilganowski N, Doree D, Savelieva KV, Lanthorn TH, Read R, Vogel P, Hansen GM, Brommage R, Ding ZM, Desai U, Zambrowicz B. Diacylglycerol Lipase α Knockout Mice Demonstrate Metabolic and Behavioral Phenotypes Similar to Those of Cannabinoid Receptor 1 Knockout Mice. Front Endocrinol (Lausanne) 2015;6:86. doi: 10.3389/fendo.2015.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziszewska E, Wolak M, Bojanowska E. Concurrent pharmacological modification of cannabinoid-1 and glucagon-like peptide-1 receptor activity affects feeding behavior and body weight in rats fed a free-choice, high-carbohydrate diet. Behav Pharmacol. 2014;25:53–60. doi: 10.1097/FBP.0000000000000018. [DOI] [PubMed] [Google Scholar]
- Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrié P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord. 2004;28:640–8. doi: 10.1038/sj.ijo.0802583. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Piston DW. High-Contrast Imaging of Fluorescent Protein FRET by Fluorescence Polarization Microscopy. Biophys J. 2005;88:L14–L16. doi: 10.1529/biophysj.104.055442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri C, Di Marzo V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013;17:475–490. doi: 10.1016/j.cmet.2013.03.001. pii. [DOI] [PubMed] [Google Scholar]
- Starowicz KM, Cristino L, Matias I, Capasso R, Racioppi A, Izzo AA, Di Marzo V. Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with a high-fat diet. Obesity (Silver Spring) 2008;16:553–65. doi: 10.1038/oby.2007.106. [DOI] [PubMed] [Google Scholar]
- Sykaras AG, Demenis C, Case RM, McLaughlin JT, Smith CP. Duodenal enteroendocrine I-cells contain mRNA transcripts encoding key endocannabinoid and fatty acid receptors. PLoS One. 2012;7:e42373. doi: 10.1371/journal.pone.0042373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, Szanda G, Mukhopadhyay B, Chedester L, Liow JS, Innis RB, Cheng K, Rice KC, Deschamps JR, Chorvat RJ, McElroy JF, Kunos G. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–79. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, Makriyannis A, Kunos G. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–2966. doi: 10.1172/JCI42551. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U S A. 1992;89:8641–5. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy-Fioramonti S, Demizieux L, Gresti J, Muller T, Vergès B, Degrace P. Acute Activation of Cannabinoid Receptors by Anandamide Reduces Gastro-Intestinal Motility and Improves Postprandial Glycemia in Mice. Diabetes. 2014 doi: 10.2337/db14-0721. [DOI] [PubMed] [Google Scholar]
- Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rössner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365:1389–97. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- Wang Y, Perfetti R, Greig NH, Holloway HW, DeOre KA, Montrose-Rafizadeh C, Elahi D, Egan JM. Glucagon-like peptide-1 can reverse the age-related decline in glucose tolerance in rats. J Clin Invest. 1997;99:2883–9. doi: 10.1172/JCI119482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.