

Abstract
In recent years, there has been a boom in the amount of genome-wide sequencing data that has uncovered important and unappreciated links between certain genes, families of genes and enzymatic processes and diseases such as cancer. Such studies have highlighted the impact that chromatin modifying enzymes could have in cancer and other genetic diseases. In this review, we summarize characterized mutations and single nucleotide polymorphisms (SNPs) in histone lysine methyltransferases (KMTs), histone lysine demethylases (KDMs) and histones. We primarily focus on variants with strong disease correlations and discuss how they could impact histone lysine methylation dynamics and gene regulation.
1. Introduction
Chromatin is the highly ordered structure responsible for the compaction of the eukaryotic genetic information and the regulation of processes such as DNA replication, mitosis and gene expression. The basic unit of chromatin is the nucleosome, which is composed of DNA wrapped around an octamer of histones. The histone octamer is comprised of two copies each of H2A, H2B, H3, and H4. Histone H1 is positioned in between nucleosomes and influences the higher order structure. Histones are proteins composed of a globular domain and an unstructured tail that extends away from the nucleosome core. The histone tails are extensively post-translationally modified leading to the regulation of biological processes, including transcription, cell cycle progression and DNA repair [1].
To date, there are at least 16 different classes of histones modifications that have been described [2]. One of the most studied is methylation. A main difference between this modification and others is the complexity in the number of methyl groups that can be added per residue. For example, lysine methylation can occur in a mono- (me1), di- (me2) and trimethylated (me3) state. Furthermore, the degree of methylation and the lysine being modified are associated with different consequences. For example, H3 lysine 4 monomethylation (H3K4me1) marks enhancers, while H3K4me3 peaks around the transcriptional start site of genes that are active or going to be expressed. In contrast, H3K9me3 enrichment at promoters is associated with repression, while enrichment in the body of genes associated with induction (Figure 1). These few examples highlight the importance of the lysine and degree of methylation. This modification state provides a mechanism to intricately regulate the genome and associated processes [2,3]. This topic is covered in an accompanying review in this issue (Wozniak and Strahl).
Figure 1. Putative mechanisms for how KMTs, KDMs and histone mutations could impact transcriptional regulation.

A. Mutations observed at high rate and/or associated with a functional impact in disease. These mutations are discussed in the text. B. Mutations observed in diseases at low frequency and for which no causal role or association has been reported. We apologize to all those whose work we could not depict or cite due to space limitation.
Although histone methylation has been known since the 1960’s [3], it took ~40 years for the enzymes responsible for the addition and removal to be discovered. The first histone lysine methyltransferase (KMT) was described in 2000 by Jenuwein and colleagues (KMT1A/SUV39H1) [4], and Shi and colleagues reported the first histone lysine demethylase (KDM) in 2004 (KDM1A/LSD1) [5]. Subsequent to these studies, the field of lysine methylation underwent massive expansion and numerous lysine modifying enzymes were discovered [3]. In addition to these discoveries, numerous genome-wide studies have been conducted to visualize the methylation landscape. These studies have also highlighted the importance of methylation placement in relation to gene bodies as well as regions within the genome as a whole [3]. The combined knowledge of factors modulating lysine methylation as well as the genomic distribution has now allowed the scientific community to gain more molecular insights into the regulation of nuclear processes. These data have highlighted the importance of methylation from yeast to man and established that methylation dynamics is a critical parameter to consider in disease [6].
Recent evidence from cancer genome sequencing studies and disease associated genome-wide association studies (GWAS) have begun to highlight the importance of genes involved in modulating the chromatin microenvironment (i.e., lysine methylation) in cancer and other genetic diseases. These studies are beginning to suggest that mutations in chromatin modulators may in fact be driver mutations. Driver mutations in cancer are causally implicated in the development of the disease and are the primary focus for targeted therapy. As defined by Stratton et al. [7], a driver mutation has conferred growth advantage to the cancer cell and has been positively selected by the microenvironment of the tissue in which the cancer arises. To the contrary, passenger mutations have not been selected, have not conferred growth advantage and have therefore not contributed to cancer development. These mutations will be a consequence of disease progression. However, these mutations can become drivers when the microenvironment changes (i.e., following chemotherapy) [7]. Thus, “passenger” mutations could be critical in long term treatment of diseases. This area needs more development and will require samples being collected before, during and after therapies are complete. These studies will be vital to our understanding of how chromatin associated factors are affecting cancer progression are functioning in the progression of the disease.
In addition to somatic mutations, the selection of germline variants could also result in chromatin modulators becoming important regulators in disease progression and therapeutic response. Single nucleotide polymorphisms (SNPs) are defined as a single base change in a DNA sequence that occurs in a significant proportion of a population (≥1%) [8]. Non-synonymous SNPs (i.e., changing the amino acid sequence of a protein) are less frequent in genes known to modulate diseases [9]. While SNPs can be associated with diagnosis and correlated with the risk of a disease, they can also contribute to the outcome without being a cause of the disease itself [10,11]. Therefore, there is a need to consider the impact these polymorphisms within methylation modifying enzymes have on regulation of methylation, the genomic landscape as well as on potential disorders.
In this review, we describe existing research regarding mutations and SNPs in KMTs, KDMs and histones. We focus primarily on variants with disease correlations and also enumerate diseases with observed variations (somatic mutation or SNP) in genes encoding KMTs, KDMs and histones (Table 1). These genetic variations are then discussed in relation to the histone lysine methylation state they modulate and how the variation impacts or possibly effects gene regulation (Figure 1).
Table 1.
KMTs, KDMs and histones mutations and single nucleotide polymorphism (SNP) observed in diseases.
| Protein | Mutation | SNP | |
|---|---|---|---|
| KMTs | KMT1B/SUV39H2 | Diabetes, lung cancer | |
| KMT1C/G9a/EHMT2 | Breast cancer, chronic hepatitis B, colorectal cancer | ||
| KMT1D/GLP/EHMT1 | Kleefstra syndrome, ganglioglioma | Beast cancer | |
| KMT1E/SETDB1 | Autism, plausible candidate gene for melanoma susceptibility | ||
| KMT2A / MLL | Gastric adenocarcinoma, liver cancer, bladder cancer, MM, AML, HCC | ALL | |
| KMT2B/MLL2* | HCC, OSCC, bladder urothelial carcinoma, breast adenocarcinoma, colon and rectal carcinoma, GBM, HNSCC, ccRCC, lung adenocarcinoma and SCC, ovarian serous carcinoma, uterine corpus endometrial carcinoma | ||
| KMT2C / MLL3 | Kleefstra syndrome, MB, AML, colorectal cancer, gastric adenocarcinoma, liver cancer, bladder cancer, bile duct cancer, highly agressive tumors, pancreatic cancer, breast cancer, glioblastoma, HNSCC, ccRCC, lung adenocarcinoma and SCC, ovarian serous carcinoma, uterine corpus endometrial carcinoma, adenoid cystic carcinoma, prostate cancer, MM, HCC | Breast cancer, gastric cancer | |
| KMT2D/MLL4* | Kabuki syndrome, MB, MDS, CLL, prostate cancer, CRPC, bladder cancer, DLBCL, ETP-ALL, FL, MCL, ccRCC, AML, breast adenocarcinoma, colon and rectal carcinoma, GBM, HNSCC, lung adenocarcinoma and SCC, ovarian serous carcinoma, uterine corpus endometrial carcinoma, childhood B-cell ALL, MM, HCC, OSCC | ||
| KMT2E/MLL5 | CRPC, breast cancer | ||
| KMT2F/SETD1A | Gastric adenocarcinoma | ||
| KMT2G/SETD1B | CRPC | ||
| KMT2H/ASH1L | Lung cancer (cell lines), CRPC, high grade glioma | ||
| KMT3A/SETD2 | Renal carcinoma, high-grade gliomas, ccRCC, ETP-ALL, synovial sarcoma, bladder urothelial carcinoma, breast adenocarcinoma, colon and rectal carcinoma, GBM, HNSCC, AML, lung adenocarcinoma and SCC, ovarian serous carcinoma, uterine corpus endometrial carcinoma | ||
| KMT3B / NSD1 | AML, Sotos syndrome, Weaver syndrome, Beckwith-Wiedemann syndrome, ALL, bladder urothelial carcinoma, breast adenocarcinoma, colon and rectal carcinoma, GBM, HNSCC, ccRCC, lung adenocarcinoma and SCC, ovarian serous carcinoma, uterine corpus endometrial carcinoma, OSCC | ||
| KMT3E/SMYD3 | colorectal cancer, breast cancer, HCC, NSCLC, ESCC | ||
| KMT3F/WHSC1L1 / NSD3 | MM | ||
| KMT3G / WHSC1 / NSD2 / MMSET | MCL, MM, pediatric ALL, low grade glioma | ||
| KMT4 / DOT1L | Osteoarthritis of the hip, adolescent idiopathic scoliosis and pubertal height velocity | ||
| KMT6 / EZH2 | Prostate cancer, DLBCL, FL, myelofibrosis, myeloid malignancies, T-ALL, HCC, MDS, CLL, AML, ETP-ALL, CMML, Weaver syndrome, bladder urothelial carcinoma, breast adenocarcinoma, GBM, HNSCC, ccRCC, lung adenocarcinoma and SCC, uterine corpus endometrial carcinoma | Cholangiocarcinoma, colorectal cancer, HCC, lung cancer, metastatic colorectal cancer | |
| KMT8A/PRDM2/RIZ1 # | Gastric carcinoma, endometrial carcinoma, colorectal carcinoma, pancreatic cancer, DLBCL | Breast cancer, leukemia, bone mineral density, lung cancer, Parkinson’s disease | |
| KMT8B/PRDM9 | Lung cancer (cell lines) | ||
| KDMs | KDM1A/LSD1 | Lymphoblastic leukemia, medulloblastoma | |
| KDM2B/FBXL10 | Gastric adenocarcinoma | ||
| KDM3A / JHDM2A / JMJD1A | MB | ||
| KDM3C/JMJD1C | Juvenile idiopathic arthritis | ||
| KDM4A/JMJD2A | Breast cancer | ||
| KDM4B/JMJD2B | Lung cancer (cell lines) | ||
| KDM4C/JMJD2C | MB | Autism spectrum disorders, esophageal cancer | |
| KDM5A/JARID1A# | MB, AML | Ankylosing spondylitis | |
| KDM5B/JARID1B/PLU1 | MB, breast cancer | ||
| KDM5C/JARID1C/SMCX | Renal carcinoma, ccRCC, autism spectrum disorder, X-linked mental retardation, bladder urothelial carcinoma, breast adenocarcinoma, colon and rectal carcinoma, GBM, HNSCC, lung adenocarcinoma and SCC, ovarian serous carcinoma, uterine corpus endometrial carcinoma, OSCC, high grade glioma | ||
| KDM6A / UTX | lung cancer (cell lines), Leukemia, Kabuki syndrome, MB, prostate cancer, bladder cancer, renal carcinoma, ccRCC, adenoid cystic carcinoma, urothelial carcinoma, CMML, AML, MM, ESCC, SCLC, T-ALL, colorectal adenocarcinoma, endometrial adenocarcinoma, glioblastoma, NSCLC, breast adenocarcinoma, HNSCC, uterine corpus endometrial carcinoma, childhood B-cell ALL, CRPC | Graves’ disease | |
| KDM6B/JMJD3 | MB, near haploid lymphoblastic leukemia | ||
| KDM7A/ JHDM1D / KIAA1718 | MB | ||
| KDM7B/PHF8 | X-linked mental retardation | ||
| Histones | H1.2 | Bladder urothelial carcinoma, breast adenocarcinoma, colon and rectal carcinoma, GBM, HNSCC, ccRCC, lung adenocarcinoma and SCC, ovarian serous carcinoma, NHLs, DLBCL | |
| H1.3 | DLBCL | ||
| H1.4 | Adenoid cystic carcinomas, DLBCL | ||
| H2A.1 | Adenoid cystic carcinomas, DLBCL | ||
| H3.1 | Paediatric diffuse intrinsic pontine gliomas, non- brainstem paediatric glioblastomas, DLBCL, OSCC, osteosarcoma | ||
| H3.3 | Childhood ALL, paediatric diffuse intrinsic pontine gliomas and non-Brainstem glioblastomas, anasplastic astrocytomas, high-grade thalamic gliomas from young adults, osteosarcoma, chondrosarcoma, giant cell tumor of bone, chondroblastoma | ||
| H3.5 | GBM, HNSCC, lung adenocarcinoma and SCC, uterine corpus endometrial carcinoma | ||
| H4 | DLBCL, HCC |
Abbreviations: AML (Acute Myeloid Leukemia), ccRCC (clear cell Renal Cell Carcinoma), CLL (Chronic Lymphocytic Leukemia), CML (Chronic Myeloid Leukemia), CMML (Chronic Myelomonocytic Leukemia), CRPC (Castrate Resistant Prostate Cancer), DLBCL (Diffuse Large B Cell Lynphoma), ESCC (Esophageal Squamous Cell Carcinoma), ETP-ALL (Early T-cell Precursor Acute Lymphoblastic Leukemia), FL (Follicular Lymphoma), GBM (glioblastoma multiforme), HCC (Hepatocellular Carcinoma), HNSCC (Head and Neck Squamous Cell Carcinoma), MB (Medulloblastoma), MCL (Mantle Cell Lymphoma), MDS (myelodysplastic syndrome), MM (Multiple Myeloma), NHLs (Non Hodgkin’s Lymphoma), NSCLC (Non Small Cell Lung Cancer), OSCC (Oral Squamous Cell Carcinoma), SCLC (Small Cell Lung Cancer), SCC (Squamous Cell Carcinoma).
KMT2B/MLL2 gene ID 9757 is located on chromosome 19, KMT2D/MLL4 gene ID 8085 is located on chromosome 12;
indicates coding SNP. This table is illustrative of variants observed in the literature and might not be fully inclusive. We apologize to all those whose work we could not depict or cite due to space limitation.
2. Mutations
Several recent genome-wide sequencing studies have revealed that chromatin modifier genes have significantly higher somatic mutation rates than other genes across different cancer types [12,13,14,15]. Interestingly, mutations have also been described for chromatin regulators in syndromes characterized by intellectual disabilities. These mutations are germline mutations (for a review see [16]). While most of the research is still at the discovery stages, some modifiers and readers have been implicated as disease events in both cancer and intellectual disabilities disorders and the mechanism of the dysfunction has begun to be elucidated [2,16]. This section focuses on these relationships in the context of histone mutations and enzymes that modify lysine methylation.
A. Histones mutations
A variety of histone mutations have been described across multiple cancer types (Table 1) [13,14,17,18,19,20,21,22,23,24,25,26,27]. In fact, recurrent mutations are found within one of the histone 3 genes: H3F3A (Figure 1; [17,22], reviewed in [28]). H3F3A encodes the H3.3 replication-independent histone H3 variant, which is enriched at actively transcribed genes and telomeres [29]. H3F3A K27M is a common mutation in pediatric and young adult gliomas and to a lesser extent in anaplastic astrocytoma [17,22,23,24,25,26]. Introduction of H3.3K27M dramatically reduces the level of lysine 27 trimethylation (H3K27me3) through inhibition of the H3K27me3 KMT- KMT6/EZH2 (Enhancer of Zeste Homolog 2) [25,30]. Furthermore, mutations of other histone lysine residues to methionine also inhibit methyltransferases containing the catalytic SET domain. The methionine competes with substrate binding and turnover so methylation is reduced in cis and trans [30]. Most recently, H3.3K36M recurrent mutations have been described in 95% of chondroblastomas, predominantly in the H3F3B gene [27]. Cells expressing the transgene H3.3K36M harbored a strong decrease in global H3K36me3 levels [30]. Interestingly, this mutation has been shown to promote site-specific copy gains, suggesting that it could be contributing to copy alterations in these tumors, which is something that needs further evaluation [31].
Pediatric gliomas also exhibit H3.3 mutations in glycine 34 to valine (G34V) or arginine (G34R); however, the frequency is less than the K27M mutation [22,25]. G34, which has not been shown to be posttranslationally modified, is adjacent to K36. This leads to the hypothesis that a mutation targeting G34 could alter the methylation of K36. Indeed, mutations of G34 lead to a decrease in the methylation of K36 on the same H3.3 molecule but do not inhibit KMTs in trans and thus do not alter global K36 methylation state [25,30]. Interestingly, G34 is part of a critical di-glycine motif that is required for KDM4A/JMJD2A to demethylate H3K36me3 (see accompanying review Del Rizzo and Trievel). Therefore, this mutation could be impacting both the addition and removal of this methylation state and the overall distribution across the genome. Consistent with this notion, a recent study demonstrated that a cell line derived from a patient carrying the G34V mutation presented altered enrichment of H3K36me3 through the genome. Gene ontology analysis of activated genes demonstrated enrichment for the processes involved in forebrain and cortex development as well as differentiation of neurons and regulation of cell proliferation. Interestingly, the most significant H3K36me3 enriched and expressed gene was MYCN, which provides an alternative mechanism for overexpressing this oncogene [32]. However, it has yet to be determined whether the alteration of K36 methylation drives the gene changes or if the genes change expression levels and thus alter the enrichment of H3K36me3.
Overall, these recurrent mutations could be affecting both H3K27 and K36 methylation by altering gene expression. Specifically, these alterations could impact gene expression through aberrant methylation at promoters modulated by H3K27me3 levels (i.e., bivalent genes; [33]) or gene bodies (see accompanying review Wozniak and Strahl). While H3K36me3 is present within the body of transcribed genes, H3K27me3 is found at inactive promoters and throughout the body of repressed genes (Figure 1), implying that mutations targeting any of these residues could alter the proper regulation of transcription (e.g., elongation and/or splicing). Since K27 and K36 methylation are diametrically opposed in the genome, there is the possibility that the mutations within these residues will allow the opposing modification to prevail and impact gene expression. However, we cannot exclude the impact that altering H3K27/36me3 patterns would have on genome organization since both modifications are associated with large domains throughout the genome [3]. They may not affect one another at all. The K27M mutation would also block the ability of K27 to be acetylated, which is a critical feature associated with enhancers [34]. This mutation would impact the enhancer elements and could in turn affect the gene expression and cellular fate. This mechanism would be independent of imbalanced H3K36 methylation. Future studies evaluating these possibilities will begin to shed light on the molecular contributions these mutations have in cancer.
B. H3K27 methylation targeted mutations
Histone mutations are not the only way to affect H3K27 methylation levels. One of the most well characterized histone methyltransferases mutated in cancer is the PRC2 (Polycomb Represive Complex 2) member KMT6/EZH2 (Figure 1 and Table 1). Loss of function mutations have been observed in 6% of myelodysplastic-myeloproliferative neoplasm patients (MDS; [35]) in the heterozygous and homozygous states. Some patients that initially present as heterozygous loss of function mutations progressed to the homozygous state [36]. Patients with loss of function mutations were associated with poor prognosis [36]. Several non-recurrent loss of function EZH2 mutations have also been described in T cell acute lymphoblastic leukemia [37,38].
EZH2 is also altered by activating somatic mutations (Figure 1 and Table 1). The recurrent Y641H mutation occurs as a heterozygous mutation and has been observed in 22% of diffuse large B cell lymphoma (DLBCL) and 7% of follicular lymphoma (FL) [39]. The heterozygosity is critical for the gain of function phenotype. While EZH2 efficiently monomethylates H3K27, EZH2-Y641H is deficient in monomethylation but has increased efficiency for di- and trimethylation reactions. The wild type and Y641H enzymes cooperate to produce a hyper-trimethylated H3K27 state [40]. Structural homology modeling lead to the hypothesis that mutation of this tyrosine would shift the product equilibrium, while reducing the steric crowding in the active site, favoring efficient trimethylation [40,41]. Indeed, this mutation increased the preference for dimethyl peptide in vitro [41].
Both gain and loss of function EZH2 mutations are early events in the disease process and maintained throughout the transformation of the disease [36,42]. These data raise the possibility that cellular states or originating cell background could be a major determinant in whether an activating or loss of function mutation would be selected for during tumorigenesis. They also raise the question of how many other chromatin regulators will have both gain and loss on function mutations. This will only be resolved as mutations within the chromatin regulators are systematically annotated and assayed in vitro and in vivo.
The gain of function mutations suggest that increasing H3K27 methylation could be important in tumorigenesis. Consistent with these possibilities, somatic mutations have been observed in the H3K27 demethylase KDM6A/UTX in multiple cancers [18,43,44,45,46,47,48,49,50,51] (Table 1). In addition to the somatic mutations, germline mutations in KDM6A have been observed in Kabuki syndrome [52,53]. Kabuki syndrome is a pediatric congenital disorder characterized by growth deficiency, a characteristic facial appearance and intellectual disabilities. Of note, germline mutations have also been observed in renal cancer, suggesting that KDM6A may contribute to susceptibility in rare instances [43,54].
KDM6A mutations were observed in 7–8% of chronic myelomonocytic leukemia (CML) and ~10% of derived acute myeloid leukemia (AML). Interestingly, EZH2 mutations were found in 6–8% of CML and 5–9% of AML. The mutations in EZH2 and KDM6A were not observed in the same patient samples, which suggest these mutations are mutually exclusive. However, a larger patient cohort is necessary to demonstrate this conclusively [44,45]. Because EZH2 and KDM6A modulate H3K27 methylation balance, mutations in either member may be sufficient to tip the balance and promote tumorigenesis (Figure 1 and Figure 2A). This raises the issue of how to achieve the correct balance when using KMT and KDM inhibitors for cancer therapy. It may be difficult to re-establish the balance when treating with these inhibitors and not tip the balance in the opposite direction. For example, in the case of a gain of function of EZH2 it would then be necessary to treat in such a manner to reserve some enzymatic activity. It would be beneficial to study this in cancer cell line and mouse models overtime.
Figure 2. Crosstalk between histone methylation in diseases.
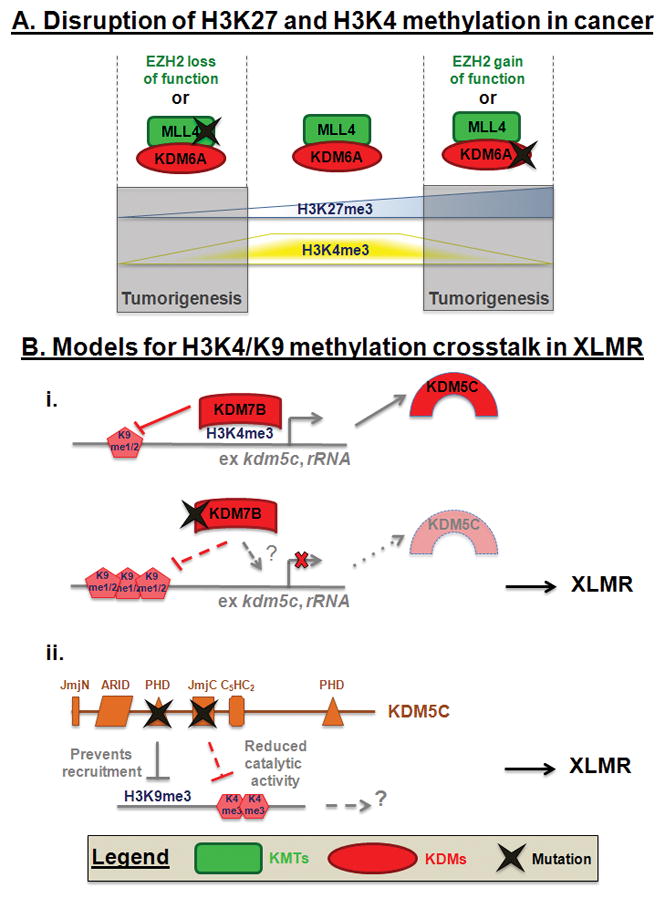
A. Mutations in EZH2 or MLL4 disrupt the H3K27 and H3K4 trimethylation crosstalk leading to tumorigenesis. B. Mutations in KDM7B or KDM5A disrupt H3K9 and H3K4 methylation leading to XLMR. (i) Schematic depicting how mutation could impact KDM7B recruitment or loss of activity. ? represents the loss of recruitment by methylation or ZNF711. (ii) Schematic depicting how mutations could impact KDM5C function.
A large study of KDM6A mutations in 1390 cancer samples implicates KDM6A as an important factor in multiple cancer types. The highest prevalence of mutations was found in multiple myeloma (MM, 10%) and esophageal squamous cell carcinoma (ESCC, 8%). Interestingly, a potential mutual exclusion was observed in MM, but with another methyltransferase KMT3G/NSD2 [43]. This observation raises an interesting question about the genetic relationship between these two enzymes, which should be further evaluated.
KDM6A is the most frequently mutated chromatin modifying gene in transitional cell carcinoma (TCC, the most common type of bladder cancer) with a frequency of 21%. Many of the mutations are predicted to truncate the catalytic domain, which would result in loss of enzymatic activity. KDM6A mutations in TCC are likely to be associated with early developmental stages of cancer as the mutation frequency was statistically associated with lower tumor grade and correlated with a lower stage [46]. Interestingly, the KDM6A mutations in adenoid cystic carcinoma (ACC, salivary gland cancers) are localized around the catalytic domain and appear to abrogate the demethylase activity [18]. These observations emphasize the selection for enzyme suppression, which raises some interesting questions about what will happen should KDM6A inhibitors be developed and clinically applied.
KDM6A is found in two of the H3K4 methylation MLL complexes (MLL3 and MLL4) [55], which are responsible for H3K4 methylation and the regulation of enhancer methylation [56]. Interestingly, MLL members are frequently mutated in human cancers as well (see H3K4 section). Since these proteins are found in complex with one another, such observations would support the notion that these protein complexes are important in tumorigenesis (Figure 2A) [55]. For example, MLL4 and KDM6A are mutated in clear cell renal cell carcinoma (CCRCC) [47], in castrate resistant prostate cancer (CRPC) [48], and so far in different tumors in 8% and 4–18% of medulloblastoma (MB), respectively [49,50,51]. These observations suggest that these tumors could have similar molecular profiles and maybe similar to one another. Taken together, these data raise the importance of knowing the biochemical characteristics of the enzymes so that the cancers can be better understood at a molecular level. This aspect will be further discussed in subsequent sections.
EZH2 and KDM6A have antagonistic functions on H3K27me3. However, both are involved in regulating gene transcription (Figure 1), especially in gene programs linked to differentiation [57]. The disruption of either of these proteins could have an impact on the same pathways and/or mechanisms. For example, one of the potential transcriptional roles of the PRC2 complex is to control engaged RNA Polymerase II during promoter escape or elongation by interacting with the RNAs transcribed by genes with paused Polymerase [58]. On the other hand, KDM6A has been directly linked to transcriptional elongation through its interaction with RNA Polymerase II via Spt6 [59]. Furthermore, PRC2 specifically resides at genes encoding developmental regulators (e.g., HOX genes) and its absence leads to embryonic stem cell differentiation defects and a delay in the loss of pluripotency [60]. On the other hand, KDM6A is needed for the re-establishment of pluripotency [61] and upon overexpression leads to demethylation of H3K27me3 at HOX gene promoters, reduced PRC2 complex association and transcriptional activation [62]. Moreover, EZH2 and KDM6A regulate adipogenesis and osteogenesis through a H3K27 methylation switch at the promoter of master regulatory genes. For example, EZH2 promotes adipogenic differentiation; while, KDM6A promotes osteogenesis [63]. Therefore, the mutations within these proteins could alter transcriptional initiation or elongation to generate variation in differentiation pathways.
C. H3K4 methylation targeted mutations
There are 5 MLL family members, MLL 1 to 5, also known as KMT2A to E. Among this family, MLL3 and MLL4 are the most frequently mutated members (Table 1). For example, MLL3 is most frequently mutated in gastric adenocarcinoma [64], cholangiocarcinoma [65], pancreatic adenocarcinoma [66], colorectal cancer [67,68], non-small cell lung cancer (NSCLC) [13,69] and bladder urothelial carcinoma (BLCA) [13]. On the other hand, MLL4 is frequently mutated in MB [70,71], DLBCL [15,19,20], FL [20,72], early T-cell precursor ALL [73], BLCA, head and neck squamous cell carcinoma and lung squamous cell carcinoma [13]. The majority of MLL4 mutations in DLBCL and FL are at the heterozygous state, but approximately half of the cases had two MLL4 mutations in trans, leading to a complete loss of MLL4 function in the tumor cells of patients [20]. Heterozygous somatic inactivation of MLL4 in DLBCL suggests a role for MLL4 as a haploinsufficient tumor suppressor, although in some patients complete inactivation of MLL4 is required [15,39]. A second, although less common mechanism, by which MLL4 function is inactivated is by loss of heterozygosity (LOH) [20]. Interestingly, truncating mutations in one allele of MLL4 have a pathogenic effect in Kabuki syndrome [74]. These data raise the possibility that MLL4 proteins dosage is a critical parameter in both cancer and Kabuki syndrome.
MLL4 is mutated in 60% of the Kabuki syndrome patients. Truncating mutations were found throughout the entire coding region, especially before the catalytic domain. Non-truncating mutations were mainly located around the LXXLL and PHD domains [52]. For example, two exons in MLL4 exhibit a higher mutation frequency, exon 39 encoding a region with LXXLL motifs and exon 48 encoding the last PHD domain and a part of the FYRN domain [74,75,76]. The localization of these mutations would suggest that the disruption of MLL4 catalytic activity and/or targeting is important to the development of the disease. Furthermore, the patients harboring MLL4 mutations may represent a clinical subgroup within the Kabuki syndrome population because they were usually more severely affected (facial appearance and growth retardation) than the patients without MLL4 mutations [77,78]. These data really highlight the selection for mutations in such genes and suggest that imbalance H3K4 methylation is a critical parameter in promoting these diseases.
In the case of MB and hepatocellular carcinoma (HCC), MLL4 is most likely a driver mutation [70,79] and the presence of a similar allelic frequency in two mantle cell lymphoma tumor samples from two distinct topographic sites or at two time points from the same patients suggests that MLL4 mutations present an early event [80]. On the contrary, the non-uniform representation of MLL4 variants in FL tumor cell subpopulations indicates that they are most likely late events during FL disease progression [72]. The early or late relationship could be the impact of the cell of origin or cell fate programs being executed. This possibility is of particular interest when considering the role of MLL4 in modulating enhancers. Indeed, the MLL4 complex methylates enhancers, which activate them to control gene expression of proteins involved in signaling cascades, embryonic development and cell differentiation [56,81]. These relationships should be further explored.
MLL proteins are part of distinct complexes. MLL1 and MLL2 are related to the Trithorax complex in Drosophila, whereas MLL3 and MLL4 are related to the Trithorax-like complex [55]. MLL3 and MLL4 have recently been demonstrated to monomethylate H3K4 at enhancers [56,81] and MLL2 is required for H3K4me3 at bivalent promoters of developmentally regulated genes [82]. Bivalent promoters are characterized by the presence of both H3K27me3 and H3K4me3 marks and exhibit low expression in ES cells. Following differentiation, bivalent promoters resolve into H3K27me3 marked promoters at genes regulating the function of unrelated lineages (non-expressed genes), and H3K4me3 at promoters of markers of the specific lineage (expressed genes) [33]. These data highlight how altered MLL complex formation or enzymatic activity could impact H3K4 methylation dynamics, cell fate and cellular responses. These data are particularly interesting when considering that mutations exist in both MLL3/4 and KDM6A, which can be found in the same multisubunit complex (i.e., COMPASS) (Figure 2A) [55]. It is interesting to see that proteins within the same complex are frequently mutated. These observations may suggest that specific genes/pathways are mis-regulated and promote/cause certain cancer types or promote/cause other genetic disorders (e.g., Kabuki syndrome). Future studies evaluating the impact that mutations within MLL3/4 and KDM6A have on gene regulation and transcription will be informative in our understanding of how they contribute to various diseases. Studying their impact in diverse cell lineages will also be of interest to determine if a unique series of gene networks are resolved and possibly associated with diseases of certain cellular lineages.
D. H3K36 methylation targeted mutations
H3K36 methylation is highly correlated with actively transcribed genes. The three states of methylation (mono-, di-, tri-) are present across transcribed regions of active genes. The level of H3K36me3 correlates with the level of gene expression [33,83]. Although this modification might also play a role in transcriptional repression, this mechanism is less well understood [83]. KMT3B/NSD1 mono and dimethylate H3K36. However, NSD1 inactivation or loss also affects H3K36me3, which is likely due to a lack of substrate availability for the trimethyltransferase KMT3A/SETD2 [83]. It is interesting to note here that the alteration of an enzyme can lead to more defects than its specific targets. For example, NSD1 effects transcriptional initiation through binding to promoters and the consequent methylation throughout the promoter proximal region of genes and facilitates recruitment of RNA Polymerase II at the promoter [83]. On the other hand, SETD2 interacts with the polymerase during elongation. In yeast, the SETD2 homolog, Set2, is important for the prevention of aberrant transcriptional initiation within the coding region [83]. Thus, the alteration of H3K36 methyltransferases in cancer and genetic disorders could lead to defects in both the transcription initiation and elongation of important genes (Figure 1).
In the case of NSD1, there are clear links between this gene and certain genetic disorders. Depending on the cohort analyzed, NSD1 has been described to be mutated or deleted in 30–100% of patients with Sotos and Weaver syndromes [84,85,86,87,88,89,90,91]. Sotos and Weaver syndromes are genetic disorders characterized by excessive physical growth during the first years of life, by a variety of intellectual disabilities as well as a host of facial characteristics [92]. NSD1 mutations are also observed in 5% of the unexplained Beckwith-Wiedemann syndrome patients (a genetic disease with a distinct overgrowth condition that is typically explained by the deregulation of imprinted growth-regulatory genes within the 11p15 region) [93]. In Sotos and Weaver syndromes, the most common NSD1 germline alterations are deletions and truncation mutations. An analysis of 530 subjects with diverse phenotypes such as facial dysmorphism, learning disabilities and childhood overgrowth showed that 99% of NSD1-positive individuals (with NSD1 mutations or 5q35 microdeletions encompassing NSD1) have Sotos syndrome and that 93% of Sotos syndromes patients present NSD1 abnormalities (83% with mutations and 10% with microdeletions). The strongest phenotype associated with NSD1 abnormalities was dysmorphism (99%) and learning disabilities (97%), while the individuals with microdeletions had more severe intellectual disabilities [94]. The fact that Sotos patients with a single mutation in the SET, PHD domains 1 through 6 or PWWP2 domains produce the same phenotype as the 5q35 microdeletion that eliminates the nsd1 gene suggests that each of these domains plays an essential role and likely represent a loss-of-function.
Eleven of the twelve mutations described in the PHD domains 4 through 6 strongly reduced the binding of NSD1 to methylated H3K4 and H3K9. Interestingly, eight out of nine mutations within PHD domains 4 and 6 reduced the binding to the transcription factor Nizp1 [95]. Nizp1 is responsible for recruitment of NSD1 to RNA polymerase II promoters, leading to transcriptional repression [96]. This repressive role for NSD1 is observed for a downstream Ras effector molecule RASIP1. NSD1 depletion increases the expression of RASIP1, which was also increased in Sotos syndrome patients with altered NSD1 [97]. These data highlight the repressive role NSD1 can have and also illustrate how mutations could increase pathways associated with disease. Taken together, these data suggest that both the positively and negatively regulated targets could be critical components in the development of NSD1-related diseases.
The methyltransferase responsible for most global H3K36me3 is SETD2 [83]. SETD2 is mutated in 15% of high grade pediatric gliomas and 8% of adult gliomas. These gliomas exhibit a substantial decrease in total H3K36me3 levels by western blot, suggesting that the mutations are loss-of-function [98]. This has also been described in CCRCC, where intron retention was dramatically increased in the H3K36me3-deficient tumors at transcripts marked by H3K36me3 in the normal kidney and by nucleosome depletion in the tumors. Many of the affected genes are part of cancer-associated pathways. Aberrant RNA processing such as intron retention, variation in exon utilization or generation of previously unannotated splice forms were detected more frequently in highly expressed genes [99]. Future studies exploring the impact H3K36 methylation-associated mutations or alterations have on gene expression, splicing, cellular growth and proliferation will be very interesting and have profound impact on our understanding of various cancers and diseases.
E. H3K9 methylation targeted mutations
Although H3K9me3 has a role in alternative splicing with a specific enrichment in the body of certain genes, H3K9 methylation is mostly correlated with the absence of transcription. Indeed, H3K9me2/3 covers the body of non-expressed genes (Figure 1). Furthermore, large domains of H3K9me2 are enriched in developmentally regulated genes, which is thought to prevent their expression in differentiated cells [3].
When compared to other chromatin regulators, the H3K9 modifying enzymes have fewer reported mutations. For example, the H3K9/27 and H4K20 demethylase KDM7B has been reported to have mutations in X-linked mental retardation (XLMR) patients [100,101], whereas two methyltransferases targeting H3K9 have been shown to be mutated in disease. The H3K9 methyltransferase KMT1E/SETDB1 presents a recurrent germline mutation in five families with autism spectrum disorder [102]; while, KMT8A/PRDM2 contains somatic mutations in 44–57% of gastric carcinomas, 33% of endometrial cancers, 26–37% of colorectal carcinomas and 10% of pancreatic cancers [103,104,105,106,107,108]
The currently identified PRDM2 mutations reside in polyA tracks within the coding sequence of the gene and were specific for tumors with microsatellite instability (MSI) [103,104,105,106,107,108]. MSI is defined by hyper mutability of microsatellites repeats, which results from impaired DNA mismatch repair (MMR). This pathway repairs errors due to DNA replication at repeated regions. Microsatellites are one to four base long repeats distributed throughout the entire genome. MSI can result in cancer through mutations in oncogenes or tumor suppressor genes such as TGFBR2 or BAX [109]. Two mechanisms have been described resulting in MSI: mutation in the MMR pathway or silencing of genes from this pathway resulting from DNA methylation [110,111]. It is not clear how methylation of H3K9 could be linked to MSI. One possibility would be a role for this mark in the MMR pathway. While a recent study linked H3K36me3 and SETD2 in proper MMR [112], H3K9 methylation has not been implicated in this pathway as of yet. A second hypothesis is that oncogenes linked to the MMR pathway are repressed by H3K9 methylation by PRDM2. The alteration of this enzyme could lead to tumorigenesis due to the misregulation of oncogene(s). A third possibility is the lack of H3K9 methylation could disrupt DNA methylation leading to the destabilization of repeats and MSI. Indeed, H3K9me2/3 is important for the maintenance of DNA methylation [113]. However, another possibility is the mutations within PRDM2 are not a cause but a consequence of MSI, and therefore, H3K9 methylation has a passenger, rather than a driver function. This possibility would not preclude H3K9 methylation imbalance from emerging as a contributor under certain selective pressures. Future studies will need to be conducted to establish if all or some of these possibilities are affiliated with the diseases.
The deregulation of H3K9 methylation has been described in several malignancies [6]. For example, a loss of H3K9 methylation has been associated with MB, together with a decreased expression of the KMT responsible for the deposition of this mark (KMT1C/G9a) and an amplification of the H3K9 demethylases KDM4B/JMJD2B and KDM4C [114]. Although very few mutations have been observed in H3K9 methylation modifying enzymes to date, further studies might uncover mutations related to disruption of H3K9 methylation in certain cancers, low frequency or less abundant cancers or in tumors subjected to therapies such as genotoxic stress. We must also consider the possibility that mutations throughout the promoters, gene bodies or 3′-untranslated regions of enzymes could contribute to an overall phenotype versus a specific amino acid being frequently mutated. Should this occur, there would be a recurrent phenotype, not a specific event. Lastly, mutations maybe less selected for in cancer, whereas, gains or losses of genes maybe more prevalent. As an example, the H3K9 methyltransferase SETDB1 is amplified in melanoma and lung cancer [115,116]. Overall, H3K9 methylation is involved in the arrangement of chromatin and global nuclear architecture, which impacts gene regulation, nuclear arrangement and cell cycle progression [3]. Therefore, future studies need to explore the potential role for mutations or altered gene function for proteins impacting H3K9 in disease.
H3K9 methylation defects are also implicated in X-linked mental retardation (XLMR) (Figure 1). For example, the H3K9/27 and H4K20 demethylase KDM7B/PHF8 [3] is truncated in some XLMR families [101,117]. Several mutations have been observed in the catalytic domain [101,117], with at least one that impairs demethylase activity [118,119,120]. Loss of KDM7B activity affects neuronal differentiation in mice [119] and regulates cell survival in Zebrafish brain and jaw development [120]. KDM7B is targeted to the chromatin via its PHD domain and binds specifically H3K4me3 positive regions [100,118,119]. Interestingly, KDM7B interacts with two MLL complexes [118] and its catalytic activity toward H3K9me1/2 at rRNA genes and consequent transcriptional activation is stimulated by the adjacent H3K4me3 [118,121], which suggest another opportunity for crosstalk between H3K4 methylation dynamics and KDM7B function. There is also another interesting link between H3K9 and H3K4 methylation in XLMR. KDM7B interacts with the XLMR protein ZNF711 that binds to some KDM7B target genes, including the H3K4 tridemethylase KDM5C (Figure 2B). The knock-down of either KDM7B or ZNF711 decreases KDM5C transcription [100]. KDM5C loss of function mutations have been described in XLMR [122,123]. Interestingly, KDM5C mutations have also been reported in CCRCC [47].
KDM5C binds H3K9me3 through its PHD domain. XLMR-related point mutations have been shown to reduce catalytic activity and disrupt binding to H3K9me3 peptides [124]. Furthermore, KDM5C activity is important in neuronal survival and dendritic development [124]. These data highlight an interesting crosstalk between the H3K4 and H3K9 methylation pathways within the same genetic disease. On the one hand, the recruitment of KDM5C to H3K9 methylation and its catalytic activity towards H3K4; and on the other hand, by the recruitment of KDM7B to H3K4 methylation and its catalytic activity toward H3K9 (Figure 2B). Thus, mutations in either KDM5C or KDM7B may yield similar phenotypes and could therefore be considered a phenotypic group for mutational frequency analysis. These relationships emphasize the need to consider the target and molecular relationships between the genes that are mutated in cancer and diseases. These relationships will ultimately give us the molecular fingerprint that could allow patient stratification.
3. Single Nucleotide Polymorphism (SNP)
The first systematic studies of Single Nucleotide Polymorphisms (SNPs) in the coding region of human genes reveal that among all SNPs, half are predicted to be non coding SNPs. The synonymous (silent, without modification of the amino acid in the protein) and the non synonymous SNPs within the coding region (coding SNP or cSNPs) are equally represented. While the average diversity is one difference per 1,200 bp when comparing two human genomes, the average gene contains approximately four cSNPs [125,126]. There is a high fraction of non-synonymous SNPs that are predicted to affect the structure and thus probably function of the protein [127]. For example, these variants may affect protein stability or folding, ligand binding or post-translational modifications [127]. This suggests that some cSNPs might have a phenotype. While, at least in cancer, personalized therapy against individual mutations is starting to be considered as necessary if not essential, few studies have been done implicating SNPs of KMTs and KDMs in diseases or response to treatment. Even as it has become clear that KDMs and KMTs are involved in diseases, and that SNPs in these enzymes can correlate with disease (Table 1), it remains unclear how these SNPs affect the function or regulation of the enzymes.
SNPs located outside of the coding region of genes could impact gene function through a multitude of mechanisms. For example, SNPs in introns could impact splice site recognition, binding of proteins which regulate alternative splicing or affect mRNA stability. SNPs could also be located in promoter or enhancer elements and effect binding of transcription factors. In fact, many SNPs correlated with disease are found in regions with histone modifications consistent with enhancer function [128]. In agreement with this observation, a SNP in the regulatory region of KMT3E/SMYD3, creates a third E2F binding site resulting in increased SMYD3 expression. The frequency of the allele with three binding sites was higher in individuals with colorectal, HCC and breast cancers. Furthermore, elevated expression of SMYD3 is important in carcinogenesis of the colon, liver and breast [129]. The ability of SNPs to impact transcription suggests they could be important discriminators in response to chemotherapies or disease progression. Indeed, a SNP in EZH2 associated with progression free survival and overall survival in metastatic colorectal cancer patients treated with first-line irinotecan-based chemotherapy [130,131].
One of the issues of studying SNPs and their potential implication in disease mechanism and/or response to therapy is deciding which SNP to look at. Based on the fact that non synonymous cSNPs are more likely to affect gene function, models have been developed for the action of missense cSNPs providing a basis for understanding the impact of a cSNP at the molecular level, and thus an approach of predicting which cSNP are potentially involved in disease [127,132]. A computational method evaluating candidate SNPs that are likely to affect the protein and their function identified a SNP in MLL1 (Q1198P), which is predicted to alter the solvent accessibility of seven residues within MLL1 [133]. Although further studies are necessary to definitively link this SNP and any potential effect on MLL function/stability to any disease, such methods could be helpful to identify important cSNPs for study.
So far, very few cSNPs in KMTs and KDMs have been associated with diseases (Table 1). A KDM5A/JARID1A SNP is associated with ankylosing spondylitis [134]. PRDM2 N283E, located between the conserved E1A-related motifs CR1 and CR2, presents a biased distribution between leukemic cells and normal subjects. The T allele encoding an Asparagine (N283) is present in the homozygous state in 81% of leukemic samples and in 10% of the normal samples [135]. However, it is not clear if these SNPs exert any functional consequences on KDM5A or PRDM2 catalytic activity or targeting to genomic locations. Understanding how cSNPs impact the function of KMTs and KDMs and if these functionally important cSNPs associate with disease or allow stratification of patient outcome is an important area of future research.
4. Conclusion
Recent advances in sequencing technologies have allowed chromatin-modifying enzymes to emerge as important genes mutated in diseases such as cancer. This review emphasizes that chromatin modifier genes, especially enzymes involved in lysine methylation, are mutated at the same intermediate frequency as most cancer genes (i.e., 2–20%) [136]. An emerging theme from the genome-wide sequencing studies in various diseases is the presence of common mutations in related protein complex components or in enzymes targeting a certain amino acid residue such as H3K4 and H3K27. However, a few major questions remain: 1) if larger patient cohorts are evaluated, will mutations within chromatin modifying enzymes co-occur in patient samples or will they be mutually exclusive? 2) Are mutations within genes associated with specific complexes or certain lysines able to connect cancer subtypes or predict tumor or patient responses? As genome-wide sequencing data expands and information about treatment responses and patient outcomes are annotated these questions can be addressed.
The catalog of cancer genes is still under construction and is far from complete as many more sequenced patient samples will be necessary in order to have the power to detect genes mutated at lower frequency (<2%) [136]. Genes mutated at a lower frequency may still be important in cancer when considering phenotypic recurrence across enzymes involved in methylation balance (e.g., KDM6A and MLL4 loss of function mutations or the KDM7B and KDM5C mutations in XLMR). These alterations could alter the same pathways, and in turn, result in the same molecular and cellular phenotypes. In that regard, the trithorax-related histone modifications could be considered to have a more pronounced role because the combined mutations within interactors or enzymes modulating the methylation state would increase the tumor frequency. Future studies that explore these types of relationships will be essential in our understanding of how to molecularly classify tumors and to completely appreciate the impact of mutations affecting lysine methylation on disease.
The recent exponential increase in the amount of data generated by new sequencing technologies has increased the power to detect variants that increase risk factor for a specific disease. For decades, SNPs have been linked to and involved in genetic diseases. Another important path that research needs to take is to study the implication of variants in response to treatment and/or with disease outcome. This analysis would facilitate the development of personalized therapy. In that regard, GWAS methodology might prove useful, allowing large scale comparison of patients with or without resistance to therapy. These data should be applied to KMTs and KDMs, which is a clearly under developed area.
In conclusion, it is becoming clear that KMTs and KDMs are altered in many diseases, at both germline and somatic levels, and that these enzymes are associated with disease mechanisms. However, while a number of recent studies uncovered the alteration of the genes encoding KMTs and KDMs, a lot of work is left to do in order to understand the mechanisms by which KMTs and KDMs are involved in the onset of diseases and response to treatments. Additionally, years of research have proven the importance of studying SNPs in relationship to genetic diseases; however, the understanding of the importance and functional impact of SNPs in KMTs and KDMs is still in the nascent stages. Improving our understanding of SNPs and mutations in KMTs and KDMs will help to assess risk and develop personalized medicine.
Highlights.
KMTs, KDMs and histones are significantly mutated in cancer and other diseases.
SNPs in KMTs and KDMs would be important to study in relation to diseases.
Mutations are observed in KMTs and KDMs associated with the same complexes.
Mutation in KMTs and KDMs can tip the methylation balance and promote disease.
Acknowledgments
We are grateful to Dr. Joshua C. Black for his comments and suggestions. The studies conducted in this manuscript were funded by the following agencies: American Cancer Society Basic Scholar Grant, MGH Proton Beam Federal Share Grant (CA059267) and NIH R01GM097360 to J.R.W. A post-doctoral fellowship was provided by the Fund for Medical Discovery (C.V.R). This research was supported in part by a grant from the Marsha Rivkin Center for Ovarian Cancer Research (C.V.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allis CD, Jenuwein T, Reinberg D. Epigenetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2007. [Google Scholar]
- 2.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Molecular cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Black JC, Whetstine JR. Tipping the lysine methylation balance in disease. Biopolymers. 2013;99:127–135. doi: 10.1002/bip.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brookes AJ. The essence of SNPs. Gene. 1999;234:177–186. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- 9.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nature genetics. 2008;40:340–345. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 10.Lee JC, Espeli M, Anderson CA, Linterman MA, Pocock JM, Williams NJ, Roberts R, Viatte S, Fu B, Peshu N, Hien TT, Phu NH, Wesley E, Edwards C, Ahmad T, Mansfield JC, Gearry R, Dunstan S, Williams TN, Barton A, Vinuesa CG, Parkes M, Lyons PA, Smith KG. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell. 2013;155:57–69. doi: 10.1016/j.cell.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, Davies B, Peterse EF, Sacilotto N, Walker GJ, Terzian T, Tomlinson IP, Box NF, Meinshausen N, De Val S, Bell DA, Bond GL. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell. 2013;155:410–422. doi: 10.1016/j.cell.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Lee W, Jiang Z, Chen Z, Jhunjhunwala S, Haverty PM, Gnad F, Guan Y, Gilbert HN, Stinson J, Klijn C, Guillory J, Bhatt D, Vartanian S, Walter K, Chan J, Holcomb T, Dijkgraaf P, Johnson S, Koeman J, Minna JD, Gazdar AF, Stern HM, Hoeflich KP, Wu TD, Settleman J, de Sauvage FJ, Gentleman RC, Neve RM, Stokoe D, Modrusan Z, Seshagiri S, Shames DS, Zhang Z. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome research. 2012;22:2315–2327. doi: 10.1101/gr.140988.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi S, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature genetics. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 15.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, Dalla-Favera R. Analysis of the coding genome of diffuse large B-cell lymphoma. Nature genetics. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kramer JM, van Bokhoven H. Genetic and epigenetic defects in mental retardation. The international journal of biochemistry & cell biology. 2009;41:96–107. doi: 10.1016/j.biocel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Baker SJ. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, Zhang J, Dolgalev I, Huberman K, Heguy A, Viale A, Drobnjak M, Leversha MA, Rice CE, Singh B, Iyer NG, Leemans CR, Bloemena E, Ferris RL, Seethala RR, Gross BE, Liang Y, Sinha R, Peng L, Raphael BJ, Turcan S, Gong Y, Schultz N, Kim S, Chiosea S, Shah JP, Sander C, Lee W, Chan TA. The mutational landscape of adenoid cystic carcinoma. Nature genetics. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, Saksena G, Stransky N, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Hernandez-Lemus E, Schwarz-Cruz y Celis A, Imaz-Rosshandler I, Ojesina AI, Jung J, Pedamallu CS, Lander ES, Habermann TM, Cerhan JR, Shipp MA, Getz G, Golub TR. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maitra A, Biswas NK, Amin K, Kowtal P, Kumar S, Das S, Sarin R, Majumder PP, Bagchi I, Bairagya BB, Basu A, Bhan MK, Chaturvedi P, Das D, D’Cruz A, Dhar R, Dutta D, Ganguli D, Gera P, Gupta T, Mahapatra S, Mujawar MH, Mukherjee S, Nair S, Nikam S, Nobre M, Patil A, Patra S, Rama-Gowtham M, Rao TS, Roy B, Roychowdhury B, Sarkar D, Sarkar S, Sarkar-Roy N, Sutradhar D. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nature communications. 2013;4:2873. doi: 10.1038/ncomms3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 23.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta neuropathologica. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gielen GH, Gessi M, Hammes J, Kramm CM, Waha A, Pietsch T. H3F3A K27M mutation in pediatric CNS tumors: a marker for diffuse high-grade astrocytomas. American journal of clinical pathology. 2013;139:345–349. doi: 10.1309/AJCPABOHBC33FVMO. [DOI] [PubMed] [Google Scholar]
- 25.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, Sarkaria J, Zhang Z. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & development. 2013;27:985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aihara K, Mukasa A, Gotoh K, Saito K, Nagae G, Tsuji S, Tatuno K, Yamamoto S, Takayanagi S, Narita Y, Shibui S, Aburatani H, Saito N. H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro-oncology. 2013;16:140–146. doi: 10.1093/neuonc/not144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, Goodie V, Robinson B, Butler A, Teague JW, Halai D, Khatri B, Myklebost O, Baumhoer D, Jundt G, Hamoudi R, Tirabosco R, Amary MF, Futreal PA, Stratton MR, Campbell PJ, Flanagan AM. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature genetics. 2013;45:1479–1482. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuen BT, Knoepfler PS. Histone h3.3 mutations: a variant path to cancer. Cancer cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, Lewis PW, Glass K, Allis CD, Dyson NJ, Getz G, Whetstine JR. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell. 2013;154:541–555. doi: 10.1016/j.cell.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M, Bajrami I, McGonnell IM, Lord CJ, Reis RM, Hargrave D, Ashworth A, Workman P, Jones C. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer discovery. 2013 doi: 10.1158/2159-8290.CD-12-0426. Ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetics. 2010;42:665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 36.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nature genetics. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 37.Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, De Keersmaecker K, Patel J, Huynh T, Utro F, Poglio S, Samon JB, Paietta E, Racevskis J, Rowe JM, Rabadan R, Levine RL, Brown S, Pflumio F, Dominguez M, Ferrando A, Aifantis I. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nature medicine. 2012;18:298–301. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S, Bobba KC, Song G, Pei D, Cheng C, Roberts S, Barbato MI, Campana D, Coustan-Smith E, Shurtleff SA, Raimondi SC, Kleppe M, Cools J, Shimano KA, Hermiston ML, Doulatov S, Eppert K, Laurenti E, Notta F, Dick JE, Basso G, Hunger SP, Loh ML, Devidas M, Wood B, Winter S, Dunsmore KP, Fulton RS, Fulton LL, Hong X, Harris CC, Dooling DJ, Ochoa K, Johnson KJ, Obenauer JC, Evans WE, Pui CH, Naeve CW, Ley TJ, Mardis ER, Wilson RK, Downing JR, Mullighan CG. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature genetics. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, Aparicio SA. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodor C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, Marzec J, Araf S, Wang J, Lee AM, Clear A, Montoto S, Matthews J, Iqbal S, Rajnai H, Rosenwald A, Ott G, Campo E, Rimsza LM, Smeland EB, Chan WC, Braziel RM, Staudt LM, Wright G, Lister TA, Elemento O, Hills R, Gribben JG, Chelala C, Matolcsy A, Kohlmann A, Haferlach T, Gascoyne RD, Fitzgibbon J. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122:3165–3168. doi: 10.1182/blood-2013-04-496893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O’Meara S, Teague J, Butler A, Hinton J, Latimer C, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Cole J, Forbes S, Jia M, Jones D, Kok CY, Leroy C, Lin ML, McBride DJ, Maddison M, Maquire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, Pleasance E, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Collins VP, Ichimura K, Law S, Wong J, Yuen ST, Leung SY, Tonon G, DePinho RA, Tai YT, Anderson KC, Kahnoski RJ, Massie A, Khoo SK, Teh BT, Stratton MR, Futreal PA. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nature genetics. 2009;41:521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, Visconte V, Sugimoto Y, Prince C, O’Keefe C, Hsi ED, List A, Sekeres MA, Rao A, McDevitt MA, Maciejewski JP. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118:3932–3941. doi: 10.1182/blood-2010-10-311019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kar SA, Jankowska A, Makishima H, Visconte V, Jerez A, Sugimoto Y, Muramatsu H, Traina F, Afable M, Guinta K, Tiu RV, Przychodzen B, Sakaguchi H, Kojima S, Sekeres MA, List AF, McDevitt MA, Maciejewski JP. Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica. 2013;98:107–113. doi: 10.3324/haematol.2012.064048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, Jia W, Chen J, Yang S, Zhou F, Zhao X, Wan S, Ye R, Liang C, Liu Z, Huang P, Liu C, Jiang H, Wang Y, Zheng H, Sun L, Liu X, Jiang Z, Feng D, Wu S, Zou J, Zhang Z, Yang R, Zhao J, Xu C, Yin W, Guan Z, Ye J, Zhang H, Li J, Kristiansen K, Nickerson ML, Theodorescu D, Li Y, Zhang X, Li S, Wang J, Yang H, Cai Z. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature genetics. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O’Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DT, Unterberger A, Morrissy AS, Shih D, Peacock J, Ramaswamy V, Rolider A, Wang X, Witt H, Hielscher T, Hawkins C, Vibhakar R, Croul S, Rutka JT, Weiss WA, Jones SJ, Eberhart CG, Marra MA, Pfister SM, Taylor MD. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta neuropathologica. 2013;125:373–384. doi: 10.1007/s00401-012-1070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, Kriwacki R, Curley N, Thiruvenkatam R, Wang J, Wu G, Rusch M, Hong X, Becksfort J, Gupta P, Ma J, Easton J, Vadodaria B, Onar-Thomas A, Lin T, Li S, Pounds S, Paugh S, Zhao D, Kawauchi D, Roussel MF, Finkelstein D, Ellison DW, Lau CC, Bouffet E, Hassall T, Gururangan S, Cohn R, Fulton RS, Fulton LL, Dooling DJ, Ochoa K, Gajjar A, Mardis ER, Wilson RK, Downing JR, Zhang J, Gilbertson RJ. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, Rausch T, Warnatz HJ, Ryzhova M, Bender S, Sturm D, Pleier S, Cin H, Pfaff E, Sieber L, Wittmann A, Remke M, Witt H, Hutter S, Tzaridis T, Weischenfeldt J, Raeder B, Avci M, Amstislavskiy V, Zapatka M, Weber UD, Wang Q, Lasitschka B, Bartholomae CC, Schmidt M, von Kalle C, Ast V, Lawerenz C, Eils J, Kabbe R, Benes V, van Sluis P, Koster J, Volckmann R, Shih D, Betts MJ, Russell RB, Coco S, Tonini GP, Schuller U, Hans V, Graf N, Kim YJ, Monoranu C, Roggendorf W, Unterberg A, Herold-Mende C, Milde T, Kulozik AE, von Deimling A, Witt O, Maass E, Rossler J, Ebinger M, Schuhmann MU, Fruhwald MC, Hasselblatt M, Jabado N, Rutkowski S, von Bueren AO, Williamson D, Clifford SC, McCabe MG, Collins VP, Wolf S, Wiemann S, Lehrach H, Brors B, Scheurlen W, Felsberg J, Reifenberger G, Northcott PA, Taylor MD, Meyerson M, Pomeroy SL, Yaspo ML, Korbel JO, Korshunov A, Eils R, Pfister SM, Lichter P. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyake N, Koshimizu E, Okamoto N, Mizuno S, Ogata T, Nagai T, Kosho T, Ohashi H, Kato M, Sasaki G, Mabe H, Watanabe Y, Yoshino M, Matsuishi T, Takanashi J, Shotelersuk V, Tekin M, Ochi N, Kubota M, Ito N, Ihara K, Hara T, Tonoki H, Ohta T, Saito K, Matsuo M, Urano M, Enokizono T, Sato A, Tanaka H, Ogawa A, Fujita T, Hiraki Y, Kitanaka S, Matsubara Y, Makita T, Taguri M, Nakashima M, Tsurusaki Y, Saitsu H, Yoshiura K, Matsumoto N, Niikawa N. MLL2 and KDM6A mutations in patients with Kabuki syndrome. American journal of medical genetics Part A. 2013;161:2234–2243. doi: 10.1002/ajmg.a.36072. [DOI] [PubMed] [Google Scholar]
- 53.Miyake N, Mizuno S, Okamoto N, Ohashi H, Shiina M, Ogata K, Tsurusaki Y, Nakashima M, Saitsu H, Niikawa N, Matsumoto N. KDM6A point mutations cause Kabuki syndrome. Human mutation. 2013;34:108–110. doi: 10.1002/humu.22229. [DOI] [PubMed] [Google Scholar]
- 54.Gossage L, Murtaza M, Slatter AF, Lichtenstein CP, Warren A, Haynes B, Marass F, Roberts I, Shanahan SJ, Claas A, Dunham A, May AP, Rosenfeld N, Forshew T, Eisen T. Clinical and pathological impact of VHL, PBRM1, BAP1, SETD2, KDM6A, and JARID1c in clear cell renal cell carcinoma. Genes, chromosomes & cancer. 2014;53:38–51. doi: 10.1002/gcc.22116. [DOI] [PubMed] [Google Scholar]
- 55.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annual review of biochemistry. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Molecular and cellular biology. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen YH, Hung MC, Li LY. EZH2: a pivotal regulator in controlling cell differentiation. American journal of translational research. 2012;4:364–375. [PMC free article] [PubMed] [Google Scholar]
- 58.Kanhere A, Viiri K, Araujo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira CF, Brookes E, Walker K, Bell GW, Pombo A, Fisher AG, Young RA, Jenner RG. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Molecular cell. 2010;38:675–688. doi: 10.1016/j.molcel.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang AH, Zare H, Mousavi K, Wang C, Moravec CE, Sirotkin HI, Ge K, Gutierrez-Cruz G, Sartorelli V. The histone chaperone Spt6 coordinates histone H3K27 demethylation and myogenesis. The EMBO journal. 2013;32:1075–1086. doi: 10.1038/emboj.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mansour AA, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, Krupalnik V, Zerbib M, Amann-Zalcenstein D, Maza I, Geula S, Viukov S, Holtzman L, Pribluda A, Canaani E, Horn-Saban S, Amit I, Novershtern N, Hanna JH. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488:409–413. doi: 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- 62.Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 63.Hemming S, Cakouros D, Isenmann S, Cooper L, Menicanin D, Zannettino A, Gronthos S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem cells. 2013 doi: 10.1002/stem.1573. [DOI] [PubMed] [Google Scholar]
- 64.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, Chin SY, Tan IB, Ng CC, Yu W, Wu Y, Lee M, Wu J, Poh D, Wan WK, Rha SY, So J, Salto-Tellez M, Yeoh KG, Wong WK, Zhu YJ, Futreal PA, Pang B, Ruan Y, Hillmer AM, Bertrand D, Nagarajan N, Rozen S, Teh BT, Tan P. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nature genetics. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 65.Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, Myint SS, Rajasegaran V, Heng HL, Gan A, Zang ZJ, Wu Y, Wu J, Lee MH, Huang D, Ong P, Chan-on W, Cao Y, Qian CN, Lim KH, Ooi A, Dykema K, Furge K, Kukongviriyapan V, Sripa B, Wongkham C, Yongvanit P, Futreal PA, Bhudhisawasdi V, Rozen S, Tan P, Teh BT. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nature genetics. 2012;44:690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 66.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watanabe Y, Castoro RJ, Kim HS, North B, Oikawa R, Hiraishi T, Ahmed SS, Chung W, Cho MY, Toyota M, Itoh F, Estecio MR, Shen L, Jelinek J, Issa JP. Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PloS one. 2011;6:e23320. doi: 10.1371/journal.pone.0023320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shin N, You KT, Lee H, Kim WK, Song M, Choi HJ, Rhee H, Nam SW, Kim H. Identification of frequently mutated genes with relevance to nonsense mediated mRNA decay in the high microsatellite instability cancers. International journal of cancer Journal international du cancer. 2011;128:2872–2880. doi: 10.1002/ijc.25641. [DOI] [PubMed] [Google Scholar]
- 69.Liu P, Morrison C, Wang L, Xiong D, Vedell P, Cui P, Hua X, Ding F, Lu Y, James M, Ebben JD, Xu H, Adjei AA, Head K, Andrae JW, Tschannen MR, Jacob H, Pan J, Zhang Q, Van den Bergh F, Xiao H, Lo KC, Patel J, Richmond T, Watt MA, Albert T, Selzer R, Anderson M, Wang J, Wang Y, Starnes S, Yang P, You M. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012;33:1270–1276. doi: 10.1093/carcin/bgs148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, Kela I, Hopmans ES, Myklebust JH, Ji H, Plevritis SK, Levy R, Alizadeh AA. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604–1611. doi: 10.1182/blood-2012-09-457283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A, Krebs S, Blum H, Raff T, Bruggemann M, Hofmann WK, Hecht J, Bohlander SK, Greif PA, Baldus CD. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121:4749–4752. doi: 10.1182/blood-2012-11-465138. [DOI] [PubMed] [Google Scholar]
- 74.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature genetics. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hannibal MC, Buckingham KJ, Ng SB, Ming JE, Beck AE, McMillin MJ, Gildersleeve HI, Bigham AW, Tabor HK, Mefford HC, Cook J, Yoshiura K, Matsumoto T, Matsumoto N, Miyake N, Tonoki H, Naritomi K, Kaname T, Nagai T, Ohashi H, Kurosawa K, Hou JW, Ohta T, Liang D, Sudo A, Morris CA, Banka S, Black GC, Clayton-Smith J, Nickerson DA, Zackai EH, Shaikh TH, Donnai D, Niikawa N, Shendure J, Bamshad MJ. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. American journal of medical genetics Part A. 2011;155A:1511–1516. doi: 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Banka S, Veeramachaneni R, Reardon W, Howard E, Bunstone S, Ragge N, Parker MJ, Crow YJ, Kerr B, Kingston H, Metcalfe K, Chandler K, Magee A, Stewart F, McConnell VP, Donnelly DE, Berland S, Houge G, Morton JE, Oley C, Revencu N, Park SM, Davies SJ, Fry AE, Lynch SA, Gill H, Schweiger S, Lam WW, Tolmie J, Mohammed SN, Hobson E, Smith A, Blyth M, Bennett C, Vasudevan PC, Garcia-Minaur S, Henderson A, Goodship J, Wright MJ, Fisher R, Gibbons R, Price SM, CdS D, Temple IK, Collins AL, Lachlan K, Elmslie F, McEntagart M, Castle B, Clayton-Smith J, Black GC, Donnai D. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. European journal of human genetics : EJHG. 2012;20:381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Makrythanasis P, van Bon B, Steehouwer M, Rodriguez-Santiago B, Simpson M, Dias P, Anderlid B, Arts P, Bhat M, Augello B, Biamino E, Bongers E, Del Campo M, Cordeiro I, Cueto-Gonzalez A, Cusco I, Deshpande C, Frysira E, Izatt L, Flores R, Galan E, Gener B, Gilissen C, Granneman S, Hoyer J, Yntema H, Kets C, Koolen D, Marcelis C, Medeira A, Micale L, Mohammed S, de Munnik S, Nordgren A, Psoni S, Reardon W, Revencu N, Roscioli T, Ruiterkamp-Versteeg M, Santos H, Schoumans J, Schuurs-Hoeijmakers J, Silengo M, Toledo L, Vendrell T, van der Burgt I, van Lier B, Zweier C, Reymond A, Trembath R, Perez-Jurado L, Dupont J, de Vries B, Brunner H, Veltman J, Merla G, Antonarakis S, Hoischen A. MLL2 mutation detection in 86 patients with Kabuki syndrome: a genotype-phenotype study. Clinical genetics. 2013;6:539–545. doi: 10.1111/cge.12081. [DOI] [PubMed] [Google Scholar]
- 78.Paulussen AD, Stegmann AP, Blok MJ, Tserpelis D, Posma-Velter C, Detisch Y, Smeets EE, Wagemans A, Schrander JJ, van den Boogaard MJ, van der Smagt J, van Haeringen A, Stolte-Dijkstra I, Kerstjens-Frederikse WS, Mancini GM, Wessels MW, Hennekam RC, Vreeburg M, Geraedts J, de Ravel T, Fryns JP, Smeets HJ, Devriendt K, Schrander-Stumpel CT. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Human mutation. 2011;32:E2018–2025. doi: 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- 79.Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, Parker JS, Li J, Powers S, Kim H, Fischer S, Guindi M, Ghanekar A, Chiang DY. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013 doi: 10.1002/hep.26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, Vizan P, Di Croce L, Puente DA, Lopez-Guerra M, Moros A, Roue G, Aymerich M, Villamor N, Colomo L, Martinez A, Valera A, Martin-Subero JI, Amador V, Hernandez L, Rozman M, Enjuanes A, Forcada P, Muntanola A, Hartmann EM, Calasanz MJ, Rosenwald A, Ott G, Hernandez-Rivas JM, Klapper W, Siebert R, Wiestner A, Wilson WH, Colomer D, Lopez-Guillermo A, Lopez-Otin C, Puente XS, Campo E. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, Ge K. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu D, Garruss AS, Gao X, Morgan MA, Cook M, Smith ER, Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nature structural & molecular biology. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nature reviews Molecular cell biology. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, Sugimoto T, Sonoda T, Hasegawa T, Chinen Y, Tomita Ha HA, Kinoshita A, Mizuguchi T, Yoshiura Ki K, Ohta T, Kishino T, Fukushima Y, Niikawa N, Matsumoto N. Haploinsufficiency of NSD1 causes Sotos syndrome. Nature genetics. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 85.Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TR, Rahman N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. American journal of human genetics. 2003;72:132–143. doi: 10.1086/345647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turkmen S, Gillessen-Kaesbach G, Meinecke P, Albrecht B, Neumann LM, Hesse V, Palanduz S, Balg S, Majewski F, Fuchs S, Zschieschang P, Greiwe M, Mennicke K, Kreuz FR, Dehmel HJ, Rodeck B, Kunze J, Tinschert S, Mundlos S, Horn D. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. European journal of human genetics : EJHG. 2003;11:858–865. doi: 10.1038/sj.ejhg.5201050. [DOI] [PubMed] [Google Scholar]
- 87.Kurotaki N, Harada N, Shimokawa O, Miyake N, Kawame H, Uetake K, Makita Y, Kondoh T, Ogata T, Hasegawa T, Nagai T, Ozaki T, Touyama M, Shenhav R, Ohashi H, Medne L, Shiihara T, Ohtsu S, Kato Z, Okamoto N, Nishimoto J, Lev D, Miyoshi Y, Ishikiriyama S, Sonoda T, Sakazume S, Fukushima Y, Kurosawa K, Cheng JF, Yoshiura K, Ohta T, Kishino T, Niikawa N, Matsumoto N. Fifty microdeletions among 112 cases of Sotos syndrome: low copy repeats possibly mediate the common deletion. Human mutation. 2003;22:378–387. doi: 10.1002/humu.10270. [DOI] [PubMed] [Google Scholar]
- 88.Rio M, Clech L, Amiel J, Faivre L, Lyonnet S, Le Merrer M, Odent S, Lacombe D, Edery P, Brauner R, Raoul O, Gosset P, Prieur M, Vekemans M, Munnich A, Colleaux L, Cormier-Daire V. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. Journal of medical genetics. 2003;40:436–440. doi: 10.1136/jmg.40.6.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kamimura J, Endo Y, Kurotaki N, Kinoshita A, Miyake N, Shimokawa O, Harada N, Visser R, Ohashi H, Miyakawa K, Gerritsen J, Innes AM, Lagace L, Frydman M, Okamoto N, Puttinger R, Raskin S, Resic B, Culic V, Yoshiura K, Ohta T, Kishino T, Ishikawa M, Niikawa N, Matsumoto N. Identification of eight novel NSD1 mutations in Sotos syndrome. Journal of medical genetics. 2003;40:e126. doi: 10.1136/jmg.40.11.e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Melchior L, Schwartz M, Duno M. dHPLC screening of the NSD1 gene identifies nine novel mutations--summary of the first 100 Sotos syndrome mutations. Annals of human genetics. 2005;69:222–226. doi: 10.1046/j.1529-8817.2004.00150.x. [DOI] [PubMed] [Google Scholar]
- 91.Sohn YB, Lee CG, Ko JM, Yang JA, Yun JN, Jung EJ, Jin HS, Park SJ, Jeong SY. Clinical and genetic spectrum of 18 unrelated Korean patients with Sotos syndrome: frequent 5q35 microdeletion and identification of four novel NSD1 mutations. Journal of human genetics. 2013;58:73–77. doi: 10.1038/jhg.2012.135. [DOI] [PubMed] [Google Scholar]
- 92.Opitz JM, Weaver DW, Reynolds JF., Jr The syndromes of Sotos and Weaver: reports and review. American journal of medical genetics. 1998;79:294–304. doi: 10.1002/(sici)1096-8628(19981002)79:4<294::aid-ajmg12>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 93.Baujat G, Rio M, Rossignol S, Sanlaville D, Lyonnet S, Le Merrer M, Munnich A, Gicquel C, Cormier-Daire V, Colleaux L. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. American journal of human genetics. 2004;74:715–720. doi: 10.1086/383093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. American journal of human genetics. 2005;77:193–204. doi: 10.1086/432082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pasillas MP, Shah M, Kamps MP. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Human mutation. 2011;32:292–298. doi: 10.1002/humu.21424. [DOI] [PubMed] [Google Scholar]
- 96.Nielsen AL, Jorgensen P, Lerouge T, Cervino M, Chambon P, Losson R. Nizp1, a novel multitype zinc finger protein that interacts with the NSD1 histone lysine methyltransferase through a unique C2HR motif. Molecular and cellular biology. 2004;24:5184–5196. doi: 10.1128/MCB.24.12.5184-5196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Visser R, Landman EB, Goeman J, Wit JM, Karperien M. Sotos syndrome is associated with deregulation of the MAPK/ERK-signaling pathway. PloS one. 2012;7:e49229. doi: 10.1371/journal.pone.0049229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, Jones DT, Witt H, Kool M, Albrecht S, Fleming A, Hadjadj D, Busche S, Lepage P, Montpetit A, Staffa A, Gerges N, Zakrzewska M, Zakrzewski K, Liberski PP, Hauser P, Garami M, Klekner A, Bognar L, Zadeh G, Faury D, Pfister SM, Jabado N, Majewski J. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta neuropathologica. 2013;125:659–669. doi: 10.1007/s00401-013-1095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, Prins JF, Lieb JD, Rathmell WK, Davis IJ. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome research. 2013 doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kleine-Kohlbrecher D, Christensen J, Vandamme J, Abarrategui I, Bak M, Tommerup N, Shi X, Gozani O, Rappsilber J, Salcini AE, Helin K. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Molecular cell. 2010;38:165–178. doi: 10.1016/j.molcel.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, Lespinasse J, Van Esch H, Lacombe D, Goizet C, Phan-Dinh Tuy F, van Bokhoven H, Fryns JP, Chelly J, Ropers HH, Moraine C, Hamel BC, Briault S. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. Journal of medical genetics. 2005;42:780–786. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cukier HN, Lee JM, Ma D, Young JI, Mayo V, Butler BL, Ramsook SS, Rantus JA, Abrams AJ, Whitehead PL, Wright HH, Abramson RK, Haines JL, Cuccaro ML, Pericak-Vance MA, Gilbert JR. The expanding role of MBD genes in autism: identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism research : official journal of the International Society for Autism Research. 2012;5:385–397. doi: 10.1002/aur.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Piao Z, Fang W, Malkhosyan S, Kim H, Horii A, Perucho M, Huang S. Frequent frameshift mutations of RIZ in sporadic gastrointestinal and endometrial carcinomas with microsatellite instability. Cancer research. 2000;60:4701–4704. [PubMed] [Google Scholar]
- 104.Chadwick RB, Jiang GL, Bennington GA, Yuan B, Johnson CK, Stevens MW, Niemann TH, Peltomaki P, Huang S, de la Chapelle A. Candidate tumor suppressor RIZ is frequently involved in colorectal carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2662–2667. doi: 10.1073/pnas.040579497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tokumaru Y, Nomoto S, Jeronimo C, Henrique R, Harden S, Trink B, Sidransky D. Biallelic inactivation of the RIZ1 gene in human gastric cancer. Oncogene. 2003;22:6954–6958. doi: 10.1038/sj.onc.1206403. [DOI] [PubMed] [Google Scholar]
- 106.Sakurada K, Furukawa T, Kato Y, Kayama T, Huang S, Horii A. RIZ, the retinoblastoma protein interacting zinc finger gene, is mutated in genetically unstable cancers of the pancreas, stomach, and colorectum. Genes chromosomes & cancer. 2001;30:207–211. [PubMed] [Google Scholar]
- 107.Emterling A, Wallin A, Arbman G, Sun XF. Clinicopathological significance of microsatellite instability and mutated RIZ in colorectal cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2004;15:242–246. doi: 10.1093/annonc/mdh045. [DOI] [PubMed] [Google Scholar]
- 108.Pan KF, Lu YY, Liu WG, Zhang L, You WC. Detection of frameshift mutations of RIZ in gastric cancers with microsatellite instability. World journal of gastroenterology : WJG. 2004;10:2719–2722. doi: 10.3748/wjg.v10.i18.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grady WM. Genomic instability and colon cancer. Cancer metastasis reviews. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 110.Putiri EL, Robertson KD. Epigenetic mechanisms and genome stability. Clinical epigenetics. 2011;2:299–314. doi: 10.1007/s13148-010-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nature reviews Clinical oncology. 2010;7:153–162. doi: 10.1038/nrclinonc.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, Koseki H, Wong J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nature communications. 2013;4:1563. doi: 10.1038/ncomms2562. [DOI] [PubMed] [Google Scholar]
- 114.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nature genetics. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rodriguez-Paredes M, Martinez de Paz A, Simo-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A, Vazquez-Cedeira M, Lazo PA, Carneiro F, Moura CS, Vieira J, Teixeira MR, Esteller M. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2013 doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Koivisto AM, Ala-Mello S, Lemmela S, Komu HA, Rautio J, Jarvela I. Screening of mutations in the PHF8 gene and identification of a novel mutation in a Finnish family with XLMR and cleft lip/cleft palate. Clinical genetics. 2007;72:145–149. doi: 10.1111/j.1399-0004.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- 118.Feng W, Yonezawa M, Ye J, Jenuwein T, Grummt I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nature structural & molecular biology. 2010;17:445–450. doi: 10.1038/nsmb.1778. [DOI] [PubMed] [Google Scholar]
- 119.Qiu J, Shi G, Jia Y, Li J, Wu M, Dong S, Wong J. The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell research. 2010;20:908–918. doi: 10.1038/cr.2010.81. [DOI] [PubMed] [Google Scholar]
- 120.Qi HH, Sarkissian M, Hu GQ, Wang Z, Bhattacharjee A, Gordon DB, Gonzales M, Lan F, Ongusaha PP, Huarte M, Yaghi NK, Lim H, Garcia BA, Brizuela L, Zhao K, Roberts TM, Shi Y. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu Z, Wang Y, Li X, Xu L, Wang X, Sun T, Dong X, Chen L, Mao H, Yu Y, Li J, Chen PA, Chen CD. PHF8 is a histone H3K9me2 demethylase regulating rRNA synthesis. Cell research. 2010;20:794–801. doi: 10.1038/cr.2010.75. [DOI] [PubMed] [Google Scholar]
- 122.Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, Van Esch H, Kleefstra T, Hamel B, Moraine C, Gecz J, Turner G, Reinhardt R, Kalscheuer VM, Ropers HH, Lenzner S. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. American journal of human genetics. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Santos C, Rodriguez-Revenga L, Madrigal I, Badenas C, Pineda M, Mila M. A novel mutation in JARID1C gene associated with mental retardation. European journal of human genetics : EJHG. 2006;14:583–586. doi: 10.1038/sj.ejhg.5201608. [DOI] [PubMed] [Google Scholar]
- 124.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 125.Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nature genetics. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- 126.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nature genetics. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 127.Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends in genetics : TIG. 2000;16:198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- 128.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tsuge M, Hamamoto R, Silva FP, Ohnishi Y, Chayama K, Kamatani N, Furukawa Y, Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nature genetics. 2005;37:1104–1107. doi: 10.1038/ng1638. [DOI] [PubMed] [Google Scholar]
- 130.Crea F, Fornaro L, Paolicchi E, Masi G, Frumento P, Loupakis F, Salvatore L, Cremolini C, Schirripa M, Graziano F, Ronzoni M, Ricci V, Farrar WL, Falcone A, Danesi R. An EZH2 polymorphism is associated with clinical outcome in metastatic colorectal cancer patients. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23:1207–1213. doi: 10.1093/annonc/mdr387. [DOI] [PubMed] [Google Scholar]
- 131.Fornaro L, Crea F, Masi G, Paolicchi E, Loupakis F, Graziano F, Salvatore L, Ronzoni M, Ricci V, Cremolini C, Schirripa M, Danesi R, Falcone A. EZH2 polymorphism and benefit from bevacizumab in colorectal cancer: another piece to the puzzle. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23:1370–1371. doi: 10.1093/annonc/mds031. [DOI] [PubMed] [Google Scholar]
- 132.Wang Z, Moult J. SNPs, protein structure, and disease. Human mutation. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- 133.George Priya Doss C, Rajasekaran R, Sethumadhavan R. Computational identification and structural analysis of deleterious functional SNPs in MLL gene causing acute leukemia. Interdisciplinary sciences computational life sciences. 2010;2:247–255. doi: 10.1007/s12539-010-0007-z. [DOI] [PubMed] [Google Scholar]
- 134.Pointon JJ, Harvey D, Karaderi T, Appleton LH, Farrar C, Wordsworth BP. The histone demethylase JARID1A is associated with susceptibility to ankylosing spondylitis. Genes and immunity. 2011;12:395–398. doi: 10.1038/gene.2011.23. [DOI] [PubMed] [Google Scholar]
- 135.Sasaki O, Meguro K, Tohmiya Y, Funato T, Shibahara S, Sasaki T. Nucleotide alteration of retinoblastoma protein-interacting zinc finger gene, RIZ, in human leukemia. The Tohoku journal of experimental medicine. 2002;196:193–201. doi: 10.1620/tjem.196.193. [DOI] [PubMed] [Google Scholar]
- 136.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014 doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]