

Abstract
As the life expectancy continues to increase, the cognitive decline associated with Alzheimer's disease (AD) becomes a big major issue in the world. After cellular activation upon systemic inflammation, microglia, the resident immune cells in the brain, start to release proinflammatory mediators to trigger neuroinflammation. We have found that chronic systemic inflammatory challenges induce differential age-dependent microglial responses, which are in line with the impairment of learning and memory, even in middle-aged animals. We thus raise the concept of “microglia aging.” This concept is based on the fact that microglia are the key contributor to the acceleration of cognitive decline, which is the major sign of brain aging. On the other hand, inflammation induces oxidative stress and DNA damage, which leads to the overproduction of reactive oxygen species by the numerous types of cells, including macrophages and microglia. Oxidative stress-damaged cells successively produce larger amounts of inflammatory mediators to promote microglia aging. Nutrients are necessary for maintaining general health, including the health of brain. The intake of antioxidant nutrients reduces both systemic inflammation and neuroinflammation and thus reduces cognitive decline during aging. We herein review our microglia aging concept and discuss systemic inflammation and microglia aging. We propose that a nutritional approach to controlling microglia aging will open a new window for healthy brain aging.
1. Introduction
The cognitive decline associated with aging and Alzheimer's disease (AD) will be a major issue in aging societies around the world as the life expectancy continues to increase. A better understanding of the factors that accelerate this cognitive decline will help in the development of strategies for preventing or delaying this cognitive decline. Microglia, the resident mononuclear phagocytes in the brain, are chronically or pathologically activated to influence the neuronal environment. There is increasing evidence that activated microglia produce excessive reactive oxygen species (ROS) during aging [1] and hypoxia [2–6], resulting in the nuclear factor-κB- (NF-κB-) dependent excessive production of proinflammatory mediators, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) [7–11]. Furthermore, activated microglia-mediated neuroinflammation is closely associated with the pathogenesis of AD pathogenesis [12], because activated microglia trigger neuroinflammation to promote neuronal damage and the deposition of amyloid β (Aβ) [13, 14]. Moreover, anti-inflammatory agents improve the cognitive functions of AD patients [15, 16].
It is well accepted that chronic systemic inflammation can alter the neuroinflammation in the brain [17, 18]. In addition to being associated with systemic diseases such as atherosclerosis and diabetes, rheumatoid arthritis (RA), periodontitis, and inflammatory bowel disease (IBD) also directly initiate or hasten the progression of AD [19]. A clinical study has demonstrated the impact of RA and periodontitis on AD [20], and recent experimental studies have clarified the routes of inflammatory signal transduction from chronic systemic inflammation to the brain [17, 18, 20].
We have recently found that natural products, such as propolis, inhibit the hypoxia-induced production of proinflammatory mediators by microglia through the inhibition of mitochondria-derived ROS generation and the subsequent activation of the NF-κB signaling pathway. Furthermore, we have found that RNSP, a traditional Tibetan medicine which is composed of 70 herbal components, improves the cognitive function in middle-to-moderate AD patients living at high altitude by reducing the levels of proinflammatory mediators and the deposition of Aβ [21]. In the present review, we will highlight our proposed concept of microglia aging, which refers to the fact that microglia are the potent accelerators of brain aging due to their induction of cognitive decline. We will also discuss the benefits of nutrients in preventing microglia aging and cognitive decline.
2. The Risk of Systemic Inflammatory Diseases for AD
RA is a chronic inflammatory bone disorder, which causes joint damage. A postmortem survey found that the prevalence of AD was reduced in RA patients who were long-term users of nonsteroidal anti-inflammatory agents [22–25]. More recently, patients with midlife RA were confirmed to have an increased risk of cognitive impairment, over a 21-year follow-up study, in several case-control and hospital- and register-based studies that were performed to examine the association between RA/arthritis and dementia/AD [26].
Periodontitis is a chronic inflammatory disorder in the periodontal tissues. There is growing clinical evidence to support a close link between periodontitis and the development and progression of AD [27, 28]. More recently, the three major periodontal bacteria, “Red complex,” including Treponema denticola, Tannerella forsythia, and Porphyromonas gingivalis, and their components have been detected in the brain of AD patients [29, 30]. More details have been reviewed by us recently [20].
IBD is a chronic inflammatory disorder in the gut. The gut bacteria are important for inducing systemic inflammation, and LPS is a potentially associated mediator which migrates into the intestinal capillaries [31]. Indeed, elevated LPS concentrations can be found in the plasma of AD patients [32–34], which supports a possible role of LPS in the promotion of neuroinflammation, and the triggering cognitive decline [35–37]. Furthermore, the chronically inflamed gut generates systemic proinflammatory cytokines to promote neuroinflammation, which causes cognitive decline [38, 39].
3. Oxidative Damage in Systemic Inflammatory Diseases and AD
3.1. Oxidative Damage in the Chronic Inflammatory Disorders
ROS contribute to the progression of chronic inflammatory bone disorders, including RA and periodontitis. The inflammatory cell-mediated overproduction of TNF-α is thought to be the main contributor to the increased release of ROS in RA patients [40], because TNF-α not only causes cell damage but also inhibits antioxidants, such as superoxide dismutase 1 (SOD1) and SOD3 [41, 42]. Numerous studies have indicated excess ROS levels and the depletion of antioxidant levels in the gingival crevicular fluid [43, 44]. There is further evidence of higher levels of lipid peroxidation, hydrogen peroxides, and oxidative DNA damage in animal models of periodontitis [45]. Indeed, periodontitis is associated with systemic oxidative stress and a reduced global antioxidant capacity, which suggests that oxidative stress in patients with periodontitis could be closely linked to the biomarker of inflammation, including C-reactive protein [46]. It is considered that ROS are involved in the chronic inflammatory bone disorders by regulating osteoblasts and osteoclasts [47], because the increased mitochondria-derived ROS, especially H2O2, reduces the differentiation and maturation of osteoblasts by inhibiting type 1 collagen and alkaline phosphatase, colony-forming unit-osteoprogenitor formation, and Runt-related transcription factor 2 activation [48, 49]. On the other hand, the increased ROS enhance the osteoclast numbers and resorption by stimulating receptor activator of NF-κB ligand and TNF-α expression through extracellular-signal-regulated kinase and NF-κB activation [50].
ROS are increased in the colonic mucosa of patients with the alterations in the mucosal antioxidant defenses in IBD patients [51, 52], because the body's major antioxidant, glutathione, is depleted but its oxidized form, glutathione disulfide, is increased in individuals with active IBD [53, 54]. The imbalance caused by the increase of ROS production and the decrease of antioxidant capacity-induced oxidative stress is considered to be the major pathogenic mechanism of IBD [55, 56]. Excessive levels of ROS result in damage to the cytoskeleton protein, including the temporal disruption of the barrier integrity and increasing gut permeability [57, 58]. Therefore, ROS promote oxidative damage, modulate the intra- and extracellular redox status, and interfere with the activation of proteolytic enzymes in the systemic inflammatory environment.
3.2. Oxidative Damage in AD
Oxidative stress is considered to be the main cause of AD. In microglia, mitochondrial dysfunction leads to the excess production of ROS, which promotes the redox imbalance and stimulates proinflammatory gene transcription and the release of cytokines, such as IL-1, IL-6, and TNF-α, thereby inducing neuroinflammation. The neuroinflammation-prolonged oxidative stress leads to the accumulation of Aβ and tau phosphorylation and then induces neurotoxicity in AD patients [59, 60]. Thus, microglia-mediated neuroinflammation is perceived as a cause and a consequence of chronic oxidative stress.
Extensive oxidative stress is observed in all of the cellular macromolecules of AD patients. First, lipid peroxidation is greatly enhanced in AD. The 4-hydroxynonal levels are significantly elevated in the hippocampus, entorhinal cortex, temporal cortex, amygdala, parahippocampal gyrus and ventricular fluid [61–64], and plasma [65] of AD patients. Second, the oxidative modification of proteins, which results from either a direct ROS attack or from the reactions that occur through the binding of glycation, glycoxidation, and lipid peroxidation products, has been extensively shown in AD. The most widely studied markers of protein oxidation are protein carbonyls and 3-nitrotyrosine. Significant increases of protein carbonyl are observed in the hippocampus, parietal lobe, and superior middle temporal gyrus of AD patients [66, 67]. Third, oxidative damage occurs in the DNA/RNA of AD patients. High levels of DNA breaks are found in the hippocampus and cerebral cortex of AD patients [68]. 8-Hydroxydeoxyguanosine (8-OHdG) is the most widely used DNA oxidative marker, which is increased in ventricular cerebrospinal fluid [69] and the peripheral tissues, such as sporadic fibroblasts [70], and in the lymphocytes of AD patients [71].
It has been demonstrated that the onset of AD is commonly preceded by an interim phase known as mild cognitive impairment (MCI), when there is no significant increase in senile plaques [72–74]. MCI patients exhibit significant oxidative imbalance in comparison to age-matched controls, since the elevation of overall protein peroxidation and the oxidative modification of specific proteins are detected in the brain, including hippocampus [75, 76], and reduction of the activity of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, and glutathione is observed in MCI patients [77, 78]. These facts strongly suggest that the oxidative imbalance appears at the very early stage of AD.
Chronic systemic inflammation links to neuroinflammation by the releasing of proinflammatory mediators including IL-1β to activate microglia [17, 18]. Repeated LPS-induced chronic systemic inflammation in mice induces microglial activation and prolonged IL-1β production by activated microglia [79]. Furthermore, systemic inflammatory challenge in the late gestation of mice increases the deposition of Aβ and tau phosphorylation, which resulted in the impairment of working memory during adult [36].
The four routes by which systemic immune signals can be transmitted to the brain have been intensively studied [17, 18]. In addition to these four classical routes, we have recently found that the leptomeningeal cells, which cover the surface of the brain parenchyma, release the proinflammatory cytokines to activate microglia during systemic inflammatory challenge [80, 81]. Therefore, leptomeningeal cells can transmit signals from systemic immune cells to microglia.
4. Microglia Aging Concept: Microglia and Brain Aging
As the phagocytic cells in the brain, microglia are primed during aging, even in middle age. The primed microglia can produce an exaggerated inflammatory response in the brain, because age-dependent dysfunctions of lysosomal/mitochondria system allow for the hypergeneration of ROS. The increased intracellular ROS then activates the redox-sensitive transcription factors, including NF-κB, to provoke exaggerated inflammatory responses [1]. The sensitivity to oxidative stress and activation of redox-sensitive transcription factors during aging may drive the emergence of senescent-type microglia (microglia aging). This may explain why Aβ, which cannot sufficiently activate NF-κB, is able to induce IL-1β secretion by activated microglia isolated from the aged mouse brain but not from the young adult mouse brain [82].
It is noted that chronic systemic inflammation induces age-dependent differential responses in microglia. The activated microglia produce anti-inflammatory mediators in young adult adjuvant arthritis (AA) rats, an animal model of RA [18, 80, 81]. However, the activated microglia produced proinflammatory mediators in the middle-aged AA rats [83]. Therefore, chronic systemic inflammation induces microglia aging from middle age. Furthermore, the microglia aging induces the functional outcomes during systemic inflammation. The long-term potentiation (LTP), a cellular substrate involved in learning and memory, in the hippocampus is significantly decreased in middle-aged AA rats but not in young adult rats. The systemic administration of minocycline, a known inhibitor of microglial activation, significantly restores the formation of LTP in middle-aged AA rats. These observations suggest that chronic systemic inflammation induces deficits of learning and memory through microglia aging [84].
Microglia is highly sensitive to excessive ROS activated NF-κB due to the increased oxidative mitochondrial DNA (mtDNA) damage [1]. The hypoxia activates the NF-κB signaling pathway to induce microglia aging [6–8, 11]. Furthermore, microglia are recognized as the major cells for NF-κB-dependent proinflammatory mediators production during stroke, the most common form of hypoxia-ischemic brain injury [85, 86]. The microglia aging mediated neuroinflammatory responses are closely associated with the pathogenesis of AD [12], because the proinflammatory mediators promote neuronal cell damage and excessive Aβ deposition [12, 87]. These observations suggest that the microglia aging is an important causative factor for AD.
5. Nutrients in Microglia Aging and Cognitive Function
5.1. Propolis
There is increasing evidence that natural nutrients can provide significant benefits in dementia patients [88]. Propolis is a resinous substance which is produced by honey as defense against intruders. It has been used therapeutically since ancient times. The chemical composition of propolis depends on the local floral at the site of collection [89–91]. In addition to the fact that propolis has antioxidative and anti-inflammatory effects [92–94], we recently found that propolis significantly inhibits the secretion of IL-1β, TNF-α, and IL-6 by microglia by inhibition of the activation of NF-κB signaling pathway [11]. Moreover, propolis was observed to significantly inhibit the increased generation of mitochondria-derived ROS, which is responsible for the activation of NF-κB signaling pathway. Moreover, propolis significantly inhibits the increased expression of 8-OHdG, a biomarker for oxidative DNA damage [95], mainly in the mitochondria of microglia after hypoxia. Since oxidative mtDNA damage impairs the respiratory chain to form a vicious cycle which promotes ROS generation [1], propolis may prevent and reverse microglia aging through its antioxidant property, both systemically and in the brain [92–96] (Figure 1).
Figure 1.
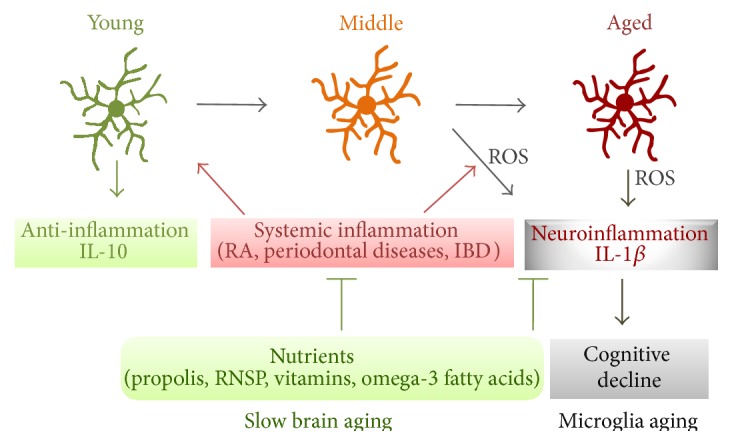
A schematic representation of the preventing and reversal of microglia aging by the antioxidant nutrients. Increased microglial mitochondria-derived ROS induce neuroinflammation which initiates cognitive decline during aging. Chronic systemic inflammation promotes microglia aging even in middle age through excessive neuroinflammation. The oral intake of antioxidant nutrients, including propolis, RNSP, vitamins, and omega-3 fatty acids, will prevent and reverse microglia aging, thereby improving cognitive function and slowing brain aging.
5.2. RNSP
RNSP is one of the Tibetan medicines composed of 70 natural components. It is used clinically for treating cerebrovascular diseases, cerebral infarction and epilepsy, and brain concussion. Our previous studies showed that RNSP improves learning and memory in a mouse model of AD (Tg2576) [21, 97] and improves the cognitive functions in mild-to-moderate AD patients living at high altitude [98]. Furthermore, RNSP reduces proinflammatory mediators, including IL-1β, TNF-α, and IL-6, in the activated macrophages and serum in humans, indicating that it also ameliorates the systemic inflammation [98, 99].
5.3. Other Nutrients
As a concept which was first introduced in 1985, oxidative stress is used to describe a condition of imbalance between oxidants and antioxidants in favor of oxidants, which potentially leads to cellular damage [100, 101]. Oxidative stress induces the oxidation of DNA, proteins, and lipids. Through detection of 8-OHdG, a marker of oxidative damage to DNA, increased mtDNA damage in the parietal cortex of AD patients was shown, indicating that mtDNA is particularly sensitive to oxidative damage [102]. Furthermore, the intake of antioxidants in patients with MCI was considered to be helpful in lowering the risk of conversion to cognitive impairment because MCI represents a prodromal stage of AD, and oxidative damage appears to occur as one of the earliest pathophysiological events in AD [103, 104].
Studies have shown the roles of the antioxidant nutrients in microglial activation. It was reported that 1,25(OH)2D3 inhibited the production of TNF-α, IL-6, and NO by the stimulated microglia in a concentration-dependent manner, because vitamin D3 receptors are expressed in microglia [105]. Another report showed that vitamin E might provide neuroprotection in vivo by attenuating microglial TNF-α and NO production by suppressing the microglial activation of p38 mitogen-activated protein kinase and NF-κB [106]. Furthermore, vitamin E reduces the LPS-induced increase in ROS and IL-6 in the primary microglia and the intraperitoneal injection of LPS has been shown to induce lipid peroxidation and IL-6 in the brain [107]. On the other hand, another study showed dramatic microglial activation, particularly in the CA1 region of the hippocampus [108]. More recent research showed that vitamin D deficiency decreases the release of TNF-α and IL-6 in cultured microglia upon stimulation with Toll-like receptor agonists [109]. The roles of n-3 fatty acids such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) in microglial activation have also been reported. It was noted that DHA and EPA decreased the inflammatory responses and increased the anti-inflammatory responses of microglia after the phagocytosis of Aβ42 and that DHA decreased TNF-α production, while EPA increased production of brain derived neurotrophic factor in cultured human CHME3 microglial cells [110]. A more recent study showed that DHA and EPA inhibited the release of TNF-α and NO from primary microglia which occurs in response to interferon-γ and myelin stimulation [111].
A great deal of evidence exists to support the roles of antioxidant nutrients in cognitive function. Vitamin E has been reported to improve cognitive function in elderly individuals [112]. It is known that the soluble Aβ oligomers cause cognitive loss and synaptic dysfunction in AD patients. The treatment with vitamin C for 6 months attenuated Aβ oligomer formation, restored the reduced synaptophysin level, and mitigated the memory behavioral decline in an AD mouse model [113]. More recent research showed that vitamins C and E supplementation mitigated the melamine-induced impairment of hippocampal synaptic plasticity [114]. However, other studies did not find evidence to support the efficacy of vitamin E, B-6, or B-12 as a preventive therapy or treatment in individuals with AD or MCI [115, 116], and 12 months of vitamins E and C supplementation did not improve the mini-mental state examination score of elderly individuals in Iran [117]. The potential role of DHA and EPA in the prevention of cognitive decline, including the decline associated with AD, has attracted major interest over the past 20 years. Recent research showed that n-3 fatty acids supplementation ameliorated memory deficits, which increased the serum total antioxidant capacity [118]. On the other hand, EPA and DHA supplementation for 2 years was not found to affect the cognitive decline in healthy elderly individuals [119]. Further intervention studies with larger study populations should be undertaken to identify the role of antioxidants in the management of cognitive function.
Approaches with multiple antioxidant nutrients to block the oxidative stress related to the systemic and brain inflammation pathways may therefore prevent or delay the cognitive impairment associated with AD by preventing microglia aging.
6. Conclusion
We herein provided the concept of microglia aging as a brain aging accelerator, which is associated with cognitive decline during aging and in AD. Chronic systemic inflammation promotes microglia aging even at middle age. Certain nutrients may therefore be beneficial for delaying brain aging by preventing or reversing microglia aging (Figure 1).
Acknowledgment
This work was supported by a Yamada Research Grant to Zhou Wu (no. 0183).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Zhou Wu and Janchun Yu contributed equally to this work.
References
- 1.Nakanishi H., Wu Z. Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behavioural Brain Research. 2009;201(1):1–7. doi: 10.1016/j.bbr.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Kaur C., Ling E. A. Periventricular white matter damage in the hypoxic neonatal brain: role of microglial cells. Progress in Neurobiology. 2009;87(4):264–280. doi: 10.1016/j.pneurobio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Kaur C., Sivakumar V., Yip G. W., Ling E. A. Expression of syndecan-2 in the amoeboid microglial cells and its involvement in inflammation in the hypoxic developing brain. GLIA. 2009;57(3):336–349. doi: 10.1002/glia.20764. [DOI] [PubMed] [Google Scholar]
- 4.Rathnasamy G., Ling E.-A., Kaur C. Iron and iron regulatory proteins in amoeboid microglial cells are linked to oligodendrocyte death in hypoxic neonatal rat periventricular white matter through production of proinflammatory cytokines and reactive oxygen/nitrogen species. The Journal of Neuroscience. 2011;31(49):17982–17995. doi: 10.1523/jneurosci.2250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao L., Kan E. M., Lu J., et al. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: Role of TLR4 in hypoxic microglia. Journal of Neuroinflammation. 2013;10, article 23 doi: 10.1186/1742-2094-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaur C., Sivakumar V., Zou Z., Ling E.-A. Microglia-derived proinflammatory cytokines tumor necrosis factor-α and interleukin-1β induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Structure and Function. 2014;219(1):151–170. doi: 10.1007/s00429-012-0491-5. [DOI] [PubMed] [Google Scholar]
- 7.Fabbri F., Carloni S., Brigliadori G., Zoli W., Lapalombella R., Marini M. Sequential events of apoptosis involving docetaxel, a microtubule-interfering agent: a cytometric study. BMC Cell Biology. 2006;7, article 6 doi: 10.1186/1471-2121-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carloni S., Mazzoni E., Cimino M., et al. Simvastatin reduces caspase-3 activation and inflammatory markers induced by hypoxia-ischemia in the newborn rat. Neurobiology of Disease. 2006;21(1):119–126. doi: 10.1016/j.nbd.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y., Lu J., Sivakumar V., Ling E. A., Kaur C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathology. 2008;18(3):387–400. doi: 10.1111/j.1750-3639.2008.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivakumar V., Foulds W. S., Luu C. D., Ling E.-A., Kaur C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. The Journal of Pathology. 2011;224(2):245–260. doi: 10.1002/path.2858. [DOI] [PubMed] [Google Scholar]
- 11.Wu Z., Zhu A., Takayama F., et al. Brazilian green propolis suppresses the hypoxia-induced neuroinflammatory responses by inhibiting NF-κB activation in microglia. Oxidative Medicine and Cellular Longevity. 2013;2013:10. doi: 10.1155/2013/906726.906726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGeer E. G., McGeer P. L. Inflammatory processes in Alzheimer's disease. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2003;27(5):741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto M., Kiyota T., Horiba M., et al. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. The American Journal of Pathology. 2007;170(2):680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liaoi Y.-F., Wang B.-J., Cheng H.-T., Kuo L.-H., Wolfe M. S. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. The Journal of Biological Chemistry. 2004;279(47):49523–49532. doi: 10.1074/jbc.m402034200. [DOI] [PubMed] [Google Scholar]
- 15.ADAPT Research Group, Meinert C. L., McCaffrey L. D., Breitner J. C. Alzheimer's disease anti-inflammatory prevention trial: design, methods, and baseline results. Alzheimer's & Dementia. 2009;5(2):93–104. doi: 10.1016/j.jalz.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukiw W. J., Bazan N. G. Neuroinflammatory signaling upregulation in Alzheimer's disease. Neurochemical Research. 2000;25(9-10):1173–1184. doi: 10.1023/a:1007627725251. [DOI] [PubMed] [Google Scholar]
- 17.Perry V. H., Newman T. A., Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nature Reviews Neuroscience. 2003;4(2):103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 18.Perry V. H. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain, Behavior, and Immunity. 2004;18(5):407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Pischon N., Heng N., Bernimoulin J.-P., Kleber B.-M., Willich S. N., Pischon T. Obesity, inflammation, and periodontal disease. Journal of Dental Research. 2007;86(5):400–409. doi: 10.1177/154405910708600503. [DOI] [PubMed] [Google Scholar]
- 20.Wu Z., Nakanishi H. Connection between periodontitis and Alzheimer's disease: possible roles of microglia and leptomeningeal cells. Journal of Pharmacological Sciences. 2014;126(1):8–13. doi: 10.1254/jphs.14r11cp. [DOI] [PubMed] [Google Scholar]
- 21.Zhu A.-Q., Chu Y.-D., Li Q.-X., Masters C. L. Tibet-medicine effects on β-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Chinese Pharmacological Bulletin. 2009;25(6):720–724. [Google Scholar]
- 22.Mcgeer P. L., Mcgeer E., Rogers J., Sibley J. Anti-inflammatory drugs and Alzheimer disease. The Lancet. 1990;335(8696):p. 1037. doi: 10.1016/0140-6736(90)91101-f. [DOI] [PubMed] [Google Scholar]
- 23.McGeer P. L., Schulzer M., McGeer E. G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 24.In't Veld B. A., Ruitenberg A., Hofman A., et al. Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer's disease. The New England Journal of Medicine. 2001;345(21):1515–1521. doi: 10.1056/nejmoa010178. [DOI] [PubMed] [Google Scholar]
- 25.Szekely C. A., Thorne J. E., Zandi P. P., et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23(4):159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- 26.Wallin K., Solomon A., Kåreholt I., Tuomilehto J., Soininen H., Walin K. Midlife rheumatoid arthritis increases the risk of cognitive impairment two decades later: a population-based study. Journal of Alzheimer's Disease. 2012;31(3):669–676. doi: 10.3233/jad-2012-111736. [DOI] [PubMed] [Google Scholar]
- 27.Noble J. M., Borrell L. N., Papapanou P. N., Elkind M. S. V., Scarmeas N., Wright C. B. Periodontitis is associated with cognitive impairment among older adults: analysis of NHANES-III. Journal of Neurology, Neurosurgery and Psychiatry. 2009;80(11):1206–1211. doi: 10.1136/jnnp.2009.174029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein P. S., Steffen M. J., Smith C., et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer's disease. Alzheimer's and Dementia. 2012;8(3):196–203. doi: 10.1016/j.jalz.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poole S., Singhrao S. K., Kesavalu L., Curtis M. A., Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. Journal of Alzheimer's Disease. 2013;36(4):665–677. doi: 10.3233/jad-121918. [DOI] [PubMed] [Google Scholar]
- 30.Riviere G., Riviere K. H., Smith K. S. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease. Oral Microbiology and Immunology. 2002;17(2):113–118. doi: 10.1046/j.0902-0055.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- 31.Neal M. D., Leaphart C., Levy R., et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. The Journal of Immunology. 2006;176(5):3070–3079. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- 32.Zhang R., Miller R. G., Gascon R., et al. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS) Journal of Neuroimmunology. 2009;206(1-2):121–124. doi: 10.1016/j.jneuroim.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang R., Miller R. G., Madison C., et al. Systemic immune system alterations in early stages of Alzheimer's disease. Journal of Neuroimmunology. 2013;256(1-2):38–42. doi: 10.1016/j.jneuroim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall J. R., Wiechmann A. R., Johnson L. A., et al. Biomarkers of vascular risk, systemic inflammation, and microvascular pathology and neuropsychiatric symptoms in Alzheimer's disease. Journal of Alzheimer's Disease. 2013;35(2):363–371. doi: 10.3233/JAD-122359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelhart M. J., Geerlings M. I., Meijer J., et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Archives of Neurology. 2004;61(5):668–672. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- 36.Krstic D., Madhusudan A., Doehner J., et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. Journal of Neuroinflammation. 2012;9, article 151 doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoozemans J. J. M., Rozemuller A. J. M., van Haastert E. S., Eikelenboom P., van Gool W. A. Neuroinflammation in Alzheimer's disease wanes with age. Journal of Neuroinflammation. 2011;8, article 171 doi: 10.1186/1742-2094-8-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eikelenboom P., Van Exel E., Hoozemans J. J. M., Veerhuis R., Rozemuller A. J. M., Van Gool W. A. Neuroinflammation—an early event in both the history and pathogenesis of Alzheimer's disease. Neurodegenerative Diseases. 2010;7(1–3):38–41. doi: 10.1159/000283480. [DOI] [PubMed] [Google Scholar]
- 39.Eikelenboom P., Hoozemans J. J. M., Veerhuis R., Van Exel E., Rozemuller A. J. M., Van Gool W. A. Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer's disease. Alzheimer's Research & Therapy. 2012;4(3, article no. 15) doi: 10.1186/alzrt118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afonso V., Champy R., Mitrovic D., Collin P., Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine. 2007;74(4):324–329. doi: 10.1016/j.jbspin.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Afonso V., Santos G., Collin P., et al. Tumor necrosis factor-alpha down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radical Biology and Medicine. 2006;41(5):709–721. doi: 10.1016/j.freeradbiomed.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Strålin P., Marklund S. L. Multiple cytokines regulate the expression of extracellular superoxide dismutase in human vascular smooth muscle cells. Atherosclerosis. 2000;151(2):433–441. doi: 10.1016/S0021-9150(99)00427-X. [DOI] [PubMed] [Google Scholar]
- 43.Tsai C. C., Chen H. S., Chen S. L., et al. Lipid peroxidation: a possible role in the induction and progression of chronic periodontitis. Journal of Periodontal Research. 2005;40(5):378–384. doi: 10.1111/j.1600-0765.2005.00818.x. [DOI] [PubMed] [Google Scholar]
- 44.Konopka T., Król K., Kopeć W., Gerber H. Total antioxidant status and 8-hydroxy-2′-deoxyguanosine levels in gingival and peripheral blood of periodontitis patients. Archivum Immunologiae et Therapiae Experimentalis. 2007;55(6):417–422. doi: 10.1007/s00005-007-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto T., Tomofuji T., Tamaki N., Ekuni D., Azuma T., Sanbe T. Effects of topical application of lipopolysaccharide and proteases on hepatic injury induced by high-cholesterol diet in rats. Journal of Periodontal Research. 2010;45(1):129–135. doi: 10.1111/j.1600-0765.2009.01212.x. [DOI] [PubMed] [Google Scholar]
- 46.D'Aiuto F., Nibali L., Parkar M., Patel K., Suvan J., Donos N. Oxidative stress, systemic inflammation, and severe periodontitis. Journal of Dental Research. 2010;89(11):1241–1246. doi: 10.1177/0022034510375830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Almeida M., Han L., Martin-Millan M., O'Brien C. A., Manolagas S. C. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting β-catenin from T cell factor- to forkhead box O-mediated transcription. The Journal of Biological Chemistry. 2007;282(37):27298–27305. doi: 10.1074/jbc.m702811200. [DOI] [PubMed] [Google Scholar]
- 48.Bai X.-C., Lu D., Bai J., et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-κB. Biochemical and Biophysical Research Communications. 2004;314(1):197–207. doi: 10.1016/j.bbrc.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 49.Mody N., Parhami F., Sarafian T. A., Demer L. L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radical Biology and Medicine. 2001;31(4):509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 50.Manolagas S. C. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocrine Reviews. 2010;31(3):266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simmonds N. J., Allen R. E., Stevens T. R. J., Van Someren R. N. M., Blake D. R., Rampton D. S. Chemiluminescence assay of mucosal reactive oxygen metabolites in inflammatory bowel disease. Gastroenterology. 1992;103(1):186–196. doi: 10.1016/0016-5085(92)91112-h. [DOI] [PubMed] [Google Scholar]
- 52.Grisham M. B., MacDermott R. P., Deitch E. A. Oxidant defense mechanisms in the human colon. Inflammation. 1990;14(6):669–680. doi: 10.1007/BF00916370. [DOI] [PubMed] [Google Scholar]
- 53.Sido B., Hack V., Hochlehnert A., Lipps H., Herfarth C., Dröge W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut. 1998;42(4):485–492. doi: 10.1136/gut.42.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lih-Brody L., Powell S. R., Collier K. P., et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Digestive Diseases and Sciences. 1996;41(10):2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 55.Grisham M. B. Oxidants and free radicals in inflammatory bowel disease. The Lancet. 1994;344(8926):859–861. doi: 10.1016/S0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 56.Gross V., Arndt H., Andus T., Palitzsch K. D., Scholmerich J. Free radicals in inflammatory bowel diseases pathophysiology and therapeutic implications. Hepatogastroenterology. 1994;41(4):320–327. [PubMed] [Google Scholar]
- 57.Banan A., Choudhary S., Zhang Y., Fields J. Z., Keshavarzian A. Oxidant-induced intestinal barrier disruption and its prevention by growth factors in a human colonic cell line: role of the microtubule cytoskeleton. Free Radical Biology and Medicine. 2000;28(5):727–738. doi: 10.1016/s0891-5849(00)00160-x. [DOI] [PubMed] [Google Scholar]
- 58.Bellomo G., Mirabelli F., Vairetti M., Iosi F., Malorni W. Cytoskeleton as a target in menadione-induced oxidative stress in cultured mammalian cells. I. Biochemical and immunocytochemical features. Journal of Cellular Physiology. 1990;143(1):118–128. doi: 10.1002/jcp.1041430116. [DOI] [PubMed] [Google Scholar]
- 59.Zhao Y., Zhao B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxidative Medicine and Cellular Longevity. 2013;2013:10. doi: 10.1155/2013/316523.316523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sochocka M., Koutsouraki E. S., Gasiorowski K., Leszek J. Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer's disease: a new approach to therapy. CNS and Neurological Disorders—Drug Targets. 2013;12(6):870–881. doi: 10.2174/18715273113129990072. [DOI] [PubMed] [Google Scholar]
- 61.Lovell M. A., Ehmann W. D., Butler S. M., Markesbery W. R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology. 1995;45(8):1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 62.Lovell M. A., Ehmann W. D., Mattson M. P., Markesbery W. R. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiology of Aging. 1997;18(5):457–461. doi: 10.1016/S0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 63.Markesbery W. R., Lovell M. A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiology of Aging. 1998;19(1):33–36. doi: 10.1016/S0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 64.Montine K. S., Reich E., Neely M. D., et al. Distribution of reducible 4-hydroxynonenal adduct immunoreactivity in Alzheimer disease is associated with APOE genotype. Journal of Neuropathology & Experimental Neurology. 1998;57(5):415–425. doi: 10.1097/00005072-199805000-00005. [DOI] [PubMed] [Google Scholar]
- 65.McGrath L. T., McGleenon B. M., Brennan S., McColl D., McIlroy S., Passmore A. P. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM—Monthly Journal of the Association of Physicians. 2001;94(9):485–490. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 66.Hensley K., Hall N., Subramaniam R., et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. Journal of Neurochemistry. 1995;65(5):2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 67.Aksenov M. Y., Aksenova M. V., Butterfield D. A., Geddes J. W., Markesbery W. R. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103(2):373–383. doi: 10.1016/S0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 68.Violet M., Delattre L., Tardivel M., et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Frontiers in Cellular Neuroscience. 2014;8, article 84 doi: 10.3389/fncel.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lovell M. A., Gabbita S. P., Markesbery W. R. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. Journal of Neurochemistry. 1999;72(2):771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 70.Ramamoorthy M., Sykora P., Scheibye-Knudsen M., et al. Sporadic Alzheimer disease fibroblasts display an oxidative stress phenotype. Free Radical Biology and Medicine. 2012;53(6):1371–1380. doi: 10.1016/j.freeradbiomed.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mórocz M., Kálmán J., Juhász A., et al. Elevated levels of oxidative DNA damage in lymphocytes from patients with Alzheimer's disease. Neurobiology of Aging. 2002;23(1):47–53. doi: 10.1016/S0197-4580(01)00257-3. [DOI] [PubMed] [Google Scholar]
- 72.Mufson E. J., Chen E.-Y., Cochran E. J., Beckett L. A., Bennett D. A., Kordower J. H. Entorhinal cortex β-amyloid load in individuals with mild cognitive impairment. Experimental Neurology. 1999;158(2):469–490. doi: 10.1006/exnr.1999.7086. [DOI] [PubMed] [Google Scholar]
- 73.Markesbery W. R., Schmitt F. A., Kryscio R. J., Davis D. G., Smith C. D., Wekstein D. R. Neuropathologic substrate of mild cognitive impairment. Archives of Neurology. 2006;63(1):38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 74.Price J. L., McKeel D. W., Jr., Buckles V. D., et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiology of Aging. 2009;30(7):1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Butterfield D. A., Poon H. F., St Clair D., et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiology of Disease. 2006;22(2):223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 76.Keller J. N., Schmitt F. A., Scheff S. W., et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64(7):1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 77.Rinaldi P., Polidori M. C., Metastasio A., et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiology of Aging. 2003;24(7):915–919. doi: 10.1016/s0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 78.Torres L. L., Quaglio N. B., De Souza G. T., et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer's disease. Journal of Alzheimer's Disease. 2011;26(1):59–68. doi: 10.3233/jad-2011-110284. [DOI] [PubMed] [Google Scholar]
- 79.Püntener U., Booth S. G., Perry V. H., Teeling J. L. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. Journal of Neuroinflammation. 2012;9, article 146 doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu Z., Zhang J., Nakanishi H. Leptomeningeal cells activate microglia and astrocytes to induce IL-10 production by releasing pro-inflammatory cytokines during systemic inflammation. Journal of Neuroimmunology. 2005;167(1-2):90–98. doi: 10.1016/j.jneuroim.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 81.Wu Z., Hayashi Y., Zhang J., Nakanishi H. Involvement of prostaglandin E2 released from leptomeningeal cells in increased expression of transforming growth factor-β in glial cells and cortical neurons during systemic inflammation. Journal of Neuroscience Research. 2007;85(1):184–192. doi: 10.1002/jnr.21100. [DOI] [PubMed] [Google Scholar]
- 82.Wu Z., Sun L., Hashioka S., et al. Differential pathways for interleukin-1β production activated by chromogranin A and amyloid β in microglia. Neurobiology of Aging. 2013;34(12):2715–2725. doi: 10.1016/j.neurobiolaging.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 83.Wu Z., Tokuda Y., Zhang X.-W., Nakanishi H. Age-dependent responses of glial cells and leptomeninges during systemic inflammation. Neurobiology of Disease. 2008;32(3):543–551. doi: 10.1016/j.nbd.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 84.Liu X., Wu Z., Hayashi Y., Nakanishi H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience. 2012;216:133–142. doi: 10.1016/j.neuroscience.2012.04.050. [DOI] [PubMed] [Google Scholar]
- 85.Gerhard A., Neumaier B., Elitok E., et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. NeuroReport. 2000;11(13):2957–2960. doi: 10.1097/00001756-200009110-00025. [DOI] [PubMed] [Google Scholar]
- 86.Price C. J. S., Wang D., Menon D. K., et al. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37(7):1749–1753. doi: 10.1161/01.STR.0000226980.95389.0b. [DOI] [PubMed] [Google Scholar]
- 87.Ruan L., Kang Z., Pei G., Le Y. Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer's disease. Current Alzheimer Research. 2009;6(6):531–540. doi: 10.2174/156720509790147070. [DOI] [PubMed] [Google Scholar]
- 88.Perry E., Howes M.-J. R. Medicinal plants and dementia therapy: herbal hopes for brain aging? CNS Neuroscience & Therapeutics. 2011;17(6):683–698. doi: 10.1111/j.1755-5949.2010.00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bankova V. S., de Castro S. L., Marcucci M. C. Propolis: recent advances in chemistry and plant origin. Apidologie. 2000;31(1):3–15. doi: 10.1051/apido:2000102. [DOI] [Google Scholar]
- 90.Marcucci M. C. Propolis: chemical composition, biological properties and therapeutic activity. Apidologie. 1995;26(2):83–99. doi: 10.1051/apido:19950202. [DOI] [Google Scholar]
- 91.Burdock G. A. Review of the biological properties and toxicity of bee propolis (propolis) Food and Chemical Toxicology. 1998;36(4):347–363. doi: 10.1016/S0278-6915(97)00145-2. [DOI] [PubMed] [Google Scholar]
- 92.Banskota A. H., Tezuka Y., Kadota S. Recent progress in pharmacological research of propolis. Phytotherapy Research. 2001;15(7):561–571. doi: 10.1002/ptr.1029. [DOI] [PubMed] [Google Scholar]
- 93.Sforcin J. M. Propolis and the immune system: a review. Journal of Ethnopharmacology. 2007;113(1):1–14. doi: 10.1016/j.jep.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 94.Ramos A. F. N., Miranda J. L. Propolis: a review of its anti-inflammatory and healing actions. Journal of Venomous Animals and Toxins including Tropical Diseases. 2007;13(4):697–710. doi: 10.1590/s1678-91992007000400002. [DOI] [Google Scholar]
- 95.Englander E. W., Hu Z., Sharma A., Lee H.-M., Wu Z.-H., Greeley G. H. Rat MYH, a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. Journal of Neurochemistry. 2002;83(6):1471–1480. doi: 10.1046/j.1471-4159.2002.01259.x. [DOI] [PubMed] [Google Scholar]
- 96.Sforcin J. M., Bankova V. Propolis: is there a potential for the development of new drugs? The Journal of Ethnopharmacology. 2011;133(2):253–260. doi: 10.1016/j.jep.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 97.Zhu A. Q., Masters C. L., Li Q. X. Tibet-medicine ratanasampil modulates amyloid precursor protein cleavage and C-terminal fragments (CTFS) in Tg2576 transgenic mice brain of Alzheimer's disease. Chinese Journal of Geriatric Dentistry. 2009;11:950–954. [Google Scholar]
- 98.Zhu A. Q., Xia Q., Li G. F., et al. Ratanasampil (tibetan medicine, RNSP) reduces A-amyloid protein and pro-inflammatory factor levels and improves cognitive functions in mild-to-moderate Alzheimer's disease (AD) patients living at high altitude. Journal of Behavioral and Brain Science. 2012;2:82–91. [Google Scholar]
- 99.Liu Y., Wu Z., Zhang X., et al. Leptomeningeal cells transduce peripheral macrophages inflammatory signal to microglia in reponse to Porphyromonas gingivalis LPS. Mediators of Inflammation. 2013;2013:11. doi: 10.1155/2013/407562.407562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sies H., Stahl W., Sevanian A. Nutritional, dietary and postprandial oxidative stress. Journal of Nutrition. 2005;135(5):96–972. doi: 10.1093/jn/135.5.969. [DOI] [PubMed] [Google Scholar]
- 101.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biology. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mecocci P., MacGarvey U., Beal M. F. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Annals of Neurology. 1994;36(5):747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 103.Rinaldi P., Polidori M. C., Metastasio A., et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiology of Aging. 2003;24(7):915–919. doi: 10.1016/S0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 104.Mecocci P., Mariani E., Cornacchiola V., Polidori M. C. Antioxidants for the treatment of mild cognitive impairment. Neurological Research. 2004;26(5):598–602. doi: 10.1179/016164104225017659. [DOI] [PubMed] [Google Scholar]
- 105.Lefebvre d'Hellencourt C., Montero-Menei C. N., Bernard R., Couez D. Vitamin D3 inhibits proinflammatory cytokines and nitric oxide production by the EOC13 microglial cell line. Journal of Neuroscience Research. 2003;71(4):575–582. doi: 10.1002/jnr.10491. [DOI] [PubMed] [Google Scholar]
- 106.Li Y., Liu L., Barger S. W., Mrak R. E., Griffin W. S. T. Vitamin E suppression of microglial activation is neuroprotective. Journal of Neuroscience Research. 2001;66(2):163–170. doi: 10.1002/jnr.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Godbout J. P., Berg B. M., Kelley K. W., Johnson R. W. α-tocopherol reduces lipopolysaccharide-induced peroxide radical formation and interleukin-6 secretion in primary murine microglia and in brain. Journal of Neuroimmunology. 2004;149(1-2):101–109. doi: 10.1016/j.jneuroim.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 108.Bialowas-McGoey L. A., Lesicka A., Whitaker-Azmitia P. M. Vitamin E increases S100b-mediated microglial activation in an S100b-overexpressing mouse model of pathological aging. Glia. 2008;56(16):1780–1790. doi: 10.1002/glia.20727. [DOI] [PubMed] [Google Scholar]
- 109.Djukic M., Onken M. L., Schütze S., et al. Vitamin D deficiency reduces the immune response, phagocytosis rate, and intracellular killing rate of microglial cells. Infection and Immunity. 2014;82(6):2585–2594. doi: 10.1128/IAI.01814-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hjorth E., Zhu M., Toro V. C., et al. Omega-3 fatty acids enhance phagocytosis of alzheimer's disease-related amyloid-β42 by human microglia and decrease inflammatory markers. Journal of Alzheimer's Disease. 2013;35(4):697–713. doi: 10.3233/jad-130131. [DOI] [PubMed] [Google Scholar]
- 111.Chen S., Zhang H., Pu H., et al. n-3 PUFA supplementation benefits microglial responses to myelin pathology. Scientific Reports. 2014;4, article 7458 doi: 10.1038/srep07458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ortega R. M., Requejo A. M., López-Sobaler A. M., et al. Cognitive function in elderly people is influenced by vitamin E status. The Journal of Nutrition. 2002;132(7):2065–2068. doi: 10.1093/jn/132.7.2065. [DOI] [PubMed] [Google Scholar]
- 113.Murakami K., Murata N., Ozawa Y., et al. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of alzheimer's disease. Journal of Alzheimer's Disease. 2011;26(1):7–18. doi: 10.3233/jad-2011-101971. [DOI] [PubMed] [Google Scholar]
- 114.An L., Zhang T. Vitamins C and E reverse melamine-induced deficits in spatial cognition and hippocampal synaptic plasticity in rats. Neurotoxicology. 2014;44:132–139. doi: 10.1016/j.neuro.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 115.Isaac M. G., Quinn R., Tabet N. Vitamin E for Alzheimer's disease and mild cognitive impairment. Cochrane Database Systematic Reviews. 2008;16(3) doi: 10.1002/14651858.CD002854.pub2.CD002854 [DOI] [PubMed] [Google Scholar]
- 116.Agnew-Blais J. C., Wassertheil-Smoller S., Kang J. H., et al. Folate, vitamin B-6, and vitamin B-12 intake and mild cognitive impairment and probable dementia in the Women's Health Initiative Memory study. Journal of the Academy of Nutrition and Dietetics. 2015;115(2):231–241. doi: 10.1016/j.jand.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alavi Naeini A. M., Elmadfa I., Djazayery A., et al. The effect of antioxidant vitamins E and C on cognitive performance of the elderly with mild cognitive impairment in Isfahan, Iran: a double-blind, randomized, placebo-controlled trial. European Journal of Nutrition. 2014;53(5):1255–1262. doi: 10.1007/s00394-013-0628-1. [DOI] [PubMed] [Google Scholar]
- 118.Abd Allah E. S., Gomaa A. M., Sayed M. M. The effect of omega-3 on cognition in hypothyroid adult male rats. Acta Physiologica Hungarica. 2014;101(3):362–376. doi: 10.1556/aphysiol.101.2014.3.11. [DOI] [PubMed] [Google Scholar]
- 119.Cederholm T., Salem N., Jr., Palmblad J. ω-3 Fatty acids in the prevention of cognitive decline in humans. Advances in Nutrition. 2013;4(6):672–676. doi: 10.3945/an.113.004556. [DOI] [PMC free article] [PubMed] [Google Scholar]