

Abstract
Acute pancreatitis (AP) is a disorder characterized by parenchymal injury of the pancreas controlled by immune cell-mediated inflammation. AP remains a significant challenge in the clinic due to a lack of specific and effective treatment. Knowledge of the complex mechanisms that regulate the inflammatory response in AP is needed for the development of new approaches to treatment, since immune cell-derived inflammatory cytokines have been recognized to play critical roles in the pathogenesis of the disease. Recent studies have shown that interleukin (IL)-22, a cytokine secreted by leukocytes, when applied in the severe animal models of AP, protects against the inflammation-mediated acinar injury. In contrast, in a mild AP model, endogenous IL-22 has been found to be a predominantly anti-inflammatory mediator that inhibits inflammatory cell infiltration via the induction of Reg3 proteins in acinar cells, but does not protect against acinar injury in the early stage of AP. However, constitutively over-expressed IL-22 can prevent the initial acinar injury caused by excessive autophagy through the induction of the anti-autophagic proteins Bcl-2 and Bcl-XL. Thus IL-22 plays different roles in AP depending on the severity of the AP model. This review focuses on these recently reported findings for the purpose of better understanding IL-22’s regulatory roles in AP which could help to develop a novel therapeutic strategy.
Keywords: Interleukin-22, Acute pancreatitis, Cytokine, Inflammatory response, Acinar cell
Core tip: Interleukin (IL)-22 has been recognized as a potential therapeutic agent for acute pancreatitis (AP) treatment due to its discovered beneficial effects in inflammatory diseases. However, according to recent publications and our results, IL-22 appears to have differential effects in AP depending on the severity of the disorder. In this review we discuss the different regulatory mechanisms of IL-22 in mild and severe AP models in order to promote development of an effective and efficient therapeutic approach.
INTRODUCTION
Despite recent improvements in the management of acute pancreatitis (AP), it remains a persistent challenge in clinical medicine[1]. In the United States, AP is the leading cause of hospitalizations among gastrointestinal diseases with a mortality rate which ranges from 3% to 17%. This is largely because of the unpredictable outcome early in AP, and the lack of a specific and effective treatment to block the inflammatory injury in AP[2-4]. Knowledge of the complex inflammatory regulation in AP is required to provide a basis for developing new strategies in the management of AP.
The inflammation in AP is initiated by local acinar cell injury caused by ductal obstruction, bile salts, ethanol or hyperlipidemia. The injured acinar cells release inflammatory mediators which activate immune cells in the pancreas and induce inflammatory cell infiltration to promote the tissue repair[5]. However, if the local injury cannot be resolved, escalated cytokine production by activated immune cells may bring about more severe inflammatory damage in the pancreas[6]. When the pro-inflammatory response is not sufficiently countered by an anti-inflammatory mechanism, a cascade of inflammatory reactions can lead to a systemic response and result in multiple organ dysfunction syndrome (MODS)[6,7]. Thus cytokines control the evolution of AP by mediating the progression of the pathogenesis in AP. It is known that tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, IL-8, platelet activating factor and chemokines are major pro-inflammatory cytokines in AP, whereas IL-10 is an anti-inflammatory mediator[4,6]. Recently, IL-22, a member of the IL-10 family, has been described as a key protector in rodent AP models, suggesting a potential therapeutic approach for AP patients by targeting IL-22[8-11].
IL-22 is produced by T helper 17 cells, γδ T cells, NKT cells and innate lymphoid cells (ILCs). Unlike other cytokines that act on both immune and non-immune cells, IL-22 has no effect on immune cells but primarily targets cells of epithelial origin due to the restrictive expression of its receptor[12,13]. The IL-22 receptor is a heterodimer composed of IL-22R1 and IL-10R2. While IL-10R2 is ubiquitously expressed, IL-22R1 is predominantly limited to epithelial cells, with its highest expression found in pancreatic acinar cells[8,14,15]. The main function of IL-22 is to regulate the host defense at barrier surfaces. However, in diseases of different organs and tissues, it can be either protective or pathogenic[12,13,16,17]. IL-22 has been reported to be tissue protective in pancreas[8-11], liver[18-28], intestine[29-36] and lung[9,37-40]. However, in psoriasis[41-45] and arthritis[46-49], dysregulated expression of IL-22 is pro-inflammatory and involved in the pathogenesis of the diseases. In addition, IL-22 has been found to have tumorigenic potential due to its ability to activate STAT3 signaling[23,27,35,42,50]. In AP, IL-22 appears to be involved in the pathogenesis in different animal models, but has disparate effects depending on the severity of the inflammation. As studies of the potential role of IL22 in AP prognosis are still currently lacking, in this review we focus on the different regulatory mechanisms of IL-22 in mild and severe AP models in order to help develop an effective and efficient therapeutic approach.
EXPRESSION OF IL-22 AND THE IL-22 RECEPTOR IN AP
It has been shown that IL-22 is produced by the resident CD4+ T cells in normal pancreas. However, in the late stages of AP, the production of IL-22 was found to be reduced when the numbers of IL-22 producing CD4+ T cells were significantly decreased in the pancreas. On the contrary, ILCs, another type of IL-22 producing cell, were highly increased in inflamed pancreas and partially restored the IL-22 production[8]. ILCs normally reside in the gut-associated lymphoid tissue to mediate mucosal immunity in the intestine[51,52]. Increased numbers of ILCs in inflamed pancreas suggest a possible migration of ILCs from the intestine after the onset of AP[8]. In support of the activation of intestinal ILCs in AP, we found that shortly after the AP induction by cerulein overstimulation in the mouse, increased IL-22 transcription was detected in intestinal tissues but not in the pancreas or spleen (Figure 1). Cerulein-induced secreted bile in the intestine is likely associated with IL-22 expression in ILCs, as administration of biliverdin, a major pigment in bile in the intestine[53], significantly increased pancreatic IL-22 levels and ILCs via activation of the aryl hydrocarbon receptor, a ligand-dependent transcription factor for IL-22 expression in leukocytes[8,54]. Thus intestinal ILCs could provide an important source of pancreatic IL-22 in AP. Interestingly, IL-22 produced by intestinal ILCs has also been found to regulate the selective containment of lymphoid-resident bacteria to prevent systemic inflammation[55]. However, it is unknown whether the failed containment of such lymphoid-resident bacteria is associated with the development of pancreatitis.
Figure 1.
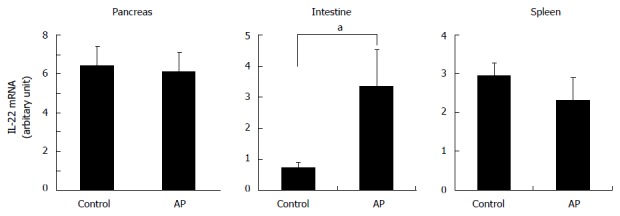
Increased interleukin-22 mRNA expression in small intestine in the early stage of acute pancreatitis. IL-22 mRNA levels in the tissues of pancreas, small intestine and spleen at 2 h after the induction of cerulein-AP were analyzed by real-time RT-PCR. In contrast to the pancreas and spleen, only the small intestine has increased IL-22 mRNA expression in the AP mice compared to the control mice. at-test, P < 0.05. IL: Interleukin; AP: Acute pancreatitis; RT-PCR: Reverse transcription polymerase chain reaction.
In contrast to IL-22, the expression of IL-22R1 on acinar cells is significantly increased in the acute phase of AP[8]. While the mechanism that regulates IL-22R1 expression in acinar cells is unknown, it has been shown that intra-peritoneal administration of LPS in mice increased IL-22R1 mRNA levels in liver, kidney and lung[56]. Thus it is possible that IL-22R1 behaves like an acute-phase protein whose expression is induced in response to the stress or injury in acinar cells, and then decreased in the late stage of AP[8]. Another study, however, showed that the IL-22R1 level was down-regulated by IL-22 in keratinocytes. This was because miR-197, a transcriptional inhibitor of IL-22R1, is actually up-regulated by IL-22[57]. Given the opposite changes in expression of IL-22 and IL-22R1 in the inflamed pancreas[8], the altered expression of IL-22R1 by IL-22 cannot be excluded.
DIFFERENT BENEFICIAL ROLES OF IL-22 IN AP
The beneficial effects of IL-22 appear to be dependent on the severity of AP as well as on the timing of IL-22’s activity in AP. In a model of mild AP induced by cerulein, administration of IL-22 or constitutively over-expressing IL-22 before AP onset prevented the AP induction. However, in the same study, IL-22 deficiency did not worsen the acinar damage[11]. Similarly, using the same AP model, we observed that IL-22 knockout mice had less acinar cell injury but significantly increased inflammatory cell infiltration in the pancreas compared to the control mice (Figure 2). In contrast to mild AP, in models of severe AP, IL-22 administration following AP onset significantly ameliorated the injury in the pancreas[8,10]. Consistently, blocking endogenous IL-22 with an anti-IL-22 antibody worsened pancreatic injury in the severe AP model[8]. These seemingly conflicting effects of IL-22 in mild and severe AP actually suggest the existence of different regulatory mechanisms by IL-22 in AP of different levels of severity.
Figure 2.
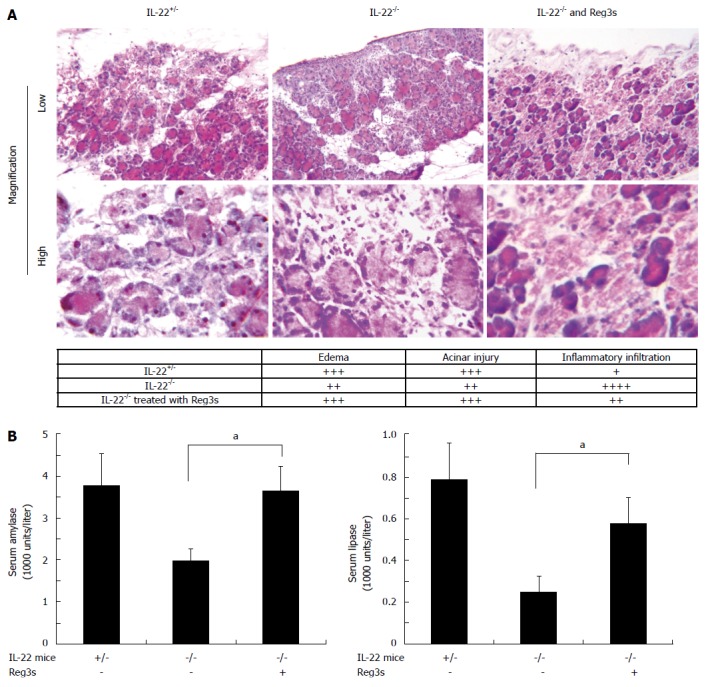
Interleukin-22 deficiency results in reduced acinar injury and increased inflammatory infiltration that are reversed by the administration of Reg3 proteins. In the upper panel of Figure 2A, H and E staining of pancreatic tissues shows that in mild AP, IL-22-/- mice have reduced tissue damage but increased inflammatory cell infiltration compared to IL-22+/- mice. These changes in IL-22-/- mice can be reversed by intravenous administration of Regα and Regβ. The lower panel in Figure 2A represents comparative severity of edema, acinar injury and inflammatory cell infiltration among the pancreata in the IL-22+/-, IL-22-/- and Reg3s treated IL-22-/- mice. Consistent with the histology study, Figure 2B shows that IL-22-/- mice have reduced serum amylase and lipase levels in AP that could be restored by the administration of Reg3s. at-test, P < 0.05. IL: Interleukin; AP: Acute pancreatitis.
INCREASED IL-22 INHIBITS EXCESSIVE AUTOPHAGY IN ACINAR CELLS AND RESULTS IN RESISTANCE TO AP INDUCTION
Recent evidence has suggested that excessive autophagy leads to premature trypsinogen activation which initiates the acinar damage in AP[58,59]. Interestingly, it has been shown that genetically over-expressed IL-22 inhibited autophagosome formation in acinar cells and makes mice resistant to cerulein-induced AP[11]. This is associated with the increased levels of the anti-autophagic proteins Bcl-2 and Bcl-XL detected in the pancreas of IL-22 transgenic mice[11]. Expressions of Bcl-2 and Bcl-XL are likely regulated by IL-22 in these mice since the transcription of Bcl-2 and Bcl-XL is regulated by STAT3[60], a signaling molecule known to be activated by IL-22[23,27,35,42,50]. Similar resistance to AP induction has also been observed in mice to which IL-22 was administrated before AP onset[11]. Thus it appears that when the level of IL-22 is higher than normal it blocks cerulein-induced excessive autophagy in acinar cells, and thereby prevents the initiation of acinar injury in AP. However, this protective mechanism of IL-22 may not affect acinar survival after the initial acute phase in severe AP, when the acinar injury is mainly associated with cytokine-mediated inflammation rather than excessive autophagy[61].
PHSYOLOGICAL IL-22 BLOCKS INFLAMMATORY INFILTRATION VIA THE INDUCTION OF REG3S IN AP
The Reg3 family includes Reg3α, Reg3β and Reg3γ. These are also known as pancreatitis associated proteins (PAP2, PAP1 and PAP3 respectively). Reg3s are evolutionarily conserved C-type lectin-like proteins that are most abundantly produced by pancreatic acinar cells in AP, but are almost undetectable in the normal pancreas[62-65]. In addition to pancreatic acinar cells, Reg3 proteins are also found in α-cells of Langerhans islets[66], intestine[36,66-69], brain[70,71], hepatocellular carcinomas[66,72] and pancreatic cancers[73,74]. Our laboratory has reported that Reg3 proteins protect against pancreatic acinar cell injury, since inhibition of their expression by either antisense DNA oligos or RNAi exacerbated experimental necrotizing AP induced by sodium taurocholate[65,75].
IL-22 signaling is required for up-regulating Reg3 expression in cerulein-induced AP as we have determined that pancreatic Reg3 levels were significantly reduced in IL-22 deficient mice (Figure 3). In line with this, according to our unpublished observation, the expressions of Reg3 proteins were induced coincidentally with the peak expression of IL-22R1 in cerulein AP[8]. IL-22 likely regulates Reg3 expression via STAT3 in acinar cells. This is because that STAT3 is known to be activated by IL-22 in acinar cells[8,14,15], and that STAT3 has been found to be required for the induction of Reg3β in AP[76]. Interestingly the study of Reg3β knockout mice showed reduced acinar injury but more severe inflammatory infiltration in cerulein-induced mild AP[77]. This phenotype is almost identical to that observed in IL-22 deficient mice. Importantly, we found that the reduced acinar injury in IL-22 knockout mice could be made worse by intravenous administration of recombinant Reg3α and Reg3β (Figure 2). Additionally, similar to IL-22[8], endogenous Reg3 proteins have been found to be required to inhibit inflammatory cell infiltration in severe AP as well[65,75]. Thus we conclude that IL-22 inhibits inflammation in AP via induction of Reg3 proteins. It is noteworthy that in addition to pancreatic acinar cells, Reg3s serve as downstream effectors of IL-22 in intestinal epithelial cells. It has been shown that in the intestine, IL-22-induced expression of Reg3β and Reg3γ contributes to IL-22’s role in the innate immunity against C. rodentium[36].
Figure 3.
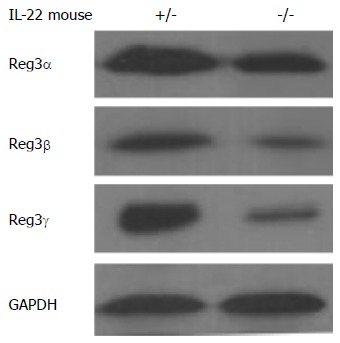
Reduced levels of Reg3 proteins in the pancreas of interleukin-22 deficient mice in acute pancreatitis. Western blot analysis of pancreatic tissues shows reduced levels of Reg3α, Reg3β and Reg3γ in IL-22-/- mice compared to IL-22+/- mice in cerulein-induced acute pancreatitis. IL: Interleukin; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
The anti-inflammatory activities of IL-22 and Reg3 in severe AP are consistent with their roles in tissue protection. On the other hand, despite their inhibition of inflammatory cell infiltration, IL-22 and Reg3 appear to make tissue injury worse in mild AP[77] (Figure 2). One possible explanation of these adverse effects in mild AP could be that their restriction of inflammatory cell infiltration leads to a reduction in the number of protective inflammatory cells such as M2 and hemin-activated macrophages in the pancreas[78,79]. If this is so, it suggests that the inflammatory infiltrates in mild AP are composed of some cells with distinctly different functions than those in severe AP, in agreement with the finding of different types of peritoneal inflammatory cells in AP of different degrees of severity[80]. An alternative explanation is that the detrimental effects of IL-22 and Reg3 in mild AP may be related to their anti-apoptotic activity which leads to a “destructive” necrosis rather than a “clean” apoptosis of injured acinar cells in mild AP[77].
ADMINISTRATION OF IL-22 PROTECTS AGAINST INFLAMMATORY INJURY IN SEVERE AP
In models of severe AP, IL-22 administration has been found to be tissue protective[8,10]. This is at least partially contributed by its anti-inflammatory activity, since reduced IL-22 serum levels were found to be associated with lung injury in a necrotizing AP model induced by sodium taurocholate[9]. In addition, IL-22 administration not only alleviated the tissue damage in the pancreas but also rescued lung injury mediated by the systemic inflammatory response in the CDE (choline-deficient diet supplemented with DL-ethionine) AP model[8]. This is also in keeping with the lack of protection against acinar injury by endogenous IL-22 in mild AP where the acinar injury is largely the result of pathogenic factors other than inflammation[58,59,61].
In addition to its anti-inflammatory activity, the potential protective role of the interplay of IL-22 with other cytokines that are highly induced in severe but not mild AP may also help to explain IL-22’s beneficial effects in severe AP. TNF-α is a pro-inflammatory cytokine that was found to be involved in AP pathogenesis and highly induced in severe AP patients[81-83]. Recent evidence, however, showed that the TNF-α expression profiles between the severe AP patients with and without MODS are different[84]. Another study claims that less TNF-α induction predicts the development of MODS and a fatal outcome in severe AP patients[85], suggesting an essential protective role of TNF-α in severe AP but with an undetermined mechanism. Interestingly, it has been shown that IL-22 and TNF-α synergistically conserve the integrity of the epidermal barrier in a skin infection model[86]. It is thus possible that the interplay between IL-22 and TNF-α could have a significant protective role in severe AP as well. In addition, IL-17, a pro-inflammatory cytokine involved in the development of necrotizing AP in rats as well as in severe AP in patients[87,88], may also coordinate IL-22 function in AP, since it has been shown that the pathological vs protective functions of IL-22 in airway inflammation were regulated by IL-17A[89]. Furthermore, IL-22 and IL-17 cooperatively enhance antimicrobial activity in keratinocytes[90]. Thus the protective function of IL-22 could be linked to other cytokines which are highly expressed in severe AP.
IL-22 IS A POTENTIAL THERAPEUTIC TARGET FOR AP TREATMENT
Immune modulation presents promising possibilities for AP treatment, since the balance between pro- and anti-inflammatory responses determines the progression and outcome of AP[2-7]. Many immune modulating strategies have been developed by either inhibiting pro-inflammatory cytokines or strengthening the effects of anti-inflammatory mediators. Although many of these strategies have been proven to be beneficial in AP animal models, their therapeutic effects in AP patients have been far from satisfactory. This could be due to the difficulties in clinical translation of animal experimental data, poor understanding of the regulation of inflammation, or the need for multimodal approaches[4]. Thus in order to achieve convincing therapeutic effects in patients, a more thorough and practical strategy that targets multiple inflammatory mediators is needed to be able to control the network of inflammation in AP.
The restrictive expression of IL-22R1 on cells of epithelial origin limits the effects of IL-22 in the body. This makes IL-22 an ideal target for drug development. Given that IL-22 has either pathogenic or beneficial effects depending on the inflammatory diseases, both anti-IL-22 antibody and recombinant IL-22 have been developed for inhibiting or strengthening IL-22 signaling respectively[17,91,92]. For example, ILV-094, an anti-IL-22 antibody that blocks IL-22 activity, is being tested in a phase II clinical trial for the treatment of atopic dermatitis; while, IL-22 IgG2-Fc, a human recombinant IL-22 developed for grade II-IV lower GI acute graft-vs-host disease, is also in a phase II clinical trial[93]. Recombinant IL-22 is a potential therapeutic agent for severe AP patients, since animal studies have proven that IL-22 administration after the onset of AP protects against inflammatory injury in different severe AP models[8,10]. Interestingly, even in a mouse model of chronic pancreatitis, IL-22 derived from an adenovirus vector ameliorates the pancreatic tissue injury[11]. It is noteworthy, however, that long-term application of IL-22 that maintains a higher than normal IL-22 level in the body is limited by its potential role in tumorigenesis[23,27,35,42,50].
CONCLUSION
Treatment of severe AP by temporally strengthening IL-22 signaling in acinar cells is an attractive concept supported by several independent studies of animal AP. The highest expression level of the IL-22 receptor on acinar cells strengthens IL-22 signaling in AP. This enables the acinar cells to abundantly produce Reg3s to combat inflammatory cell infiltration, and other pro-survival mediators to protect against tissue injury. However, in order to prepare IL-22 as a therapeutic agent for AP treatment, more thorough research is needed to comprehend the detailed mechanisms of IL-22-mediated anti-inflammatory and tissue protective mechanisms in severe AP, particularly the interplay of IL-22 with other pro-inflammatory cytokines. This will enable the development of a complex immune-modulating strategy to effectively control the cytokine-orchestrated inflammation network in severe AP.
ACKNOWLEDGMENTS
We thank Dr. Wenjun Ouyang at Genentech for providing us with the IL-22 knockout mice and other experimental reagents. We appreciate his comments to our manuscript as well. We also thank Cathy Mueller at SUNY Downstate for the technical support.
Footnotes
Conflict-of-interest statement: The authors declare no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 11, 2015
First decision: September 28, 2015
Article in press: November 25, 2015
P- Reviewer: Carrasco C, Poma EM, Vujasinovic M S- Editor: Gong XM L- Editor: A E- Editor: Jiao XK
References
- 1.Working Group IAP/APA Acute Pancreatitis Guidelines. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology. 2013;13:e1–15. doi: 10.1016/j.pan.2013.07.063. [DOI] [PubMed] [Google Scholar]
- 2.Singh VK, Bollen TL, Wu BU, Repas K, Maurer R, Yu S, Mortele KJ, Conwell DL, Banks PA. An assessment of the severity of interstitial pancreatitis. Clin Gastroenterol Hepatol. 2011;9:1098–1103. doi: 10.1016/j.cgh.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 3.Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet. 2015;386:85–96. doi: 10.1016/S0140-6736(14)60649-8. [DOI] [PubMed] [Google Scholar]
- 4.Akinosoglou K, Gogos C. Immune-modulating therapy in acute pancreatitis: fact or fiction. World J Gastroenterol. 2014;20:15200–15215. doi: 10.3748/wjg.v20.i41.15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raraty MG, Murphy JA, Mcloughlin E, Smith D, Criddle D, Sutton R. Mechanisms of acinar cell injury in acute pancreatitis. Scand J Surg. 2005;94:89–96. doi: 10.1177/145749690509400202. [DOI] [PubMed] [Google Scholar]
- 6.Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2002;9:401–410. doi: 10.1007/s005340200049. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia M. Inflammatory response on the pancreatic acinar cell injury. Scand J Surg. 2005;94:97–102. doi: 10.1177/145749690509400203. [DOI] [PubMed] [Google Scholar]
- 8.Xue J, Nguyen DT, Habtezion A. Aryl hydrocarbon receptor regulates pancreatic IL-22 production and protects mice from acute pancreatitis. Gastroenterology. 2012;143:1670–1680. doi: 10.1053/j.gastro.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huai JP, Sun XC, Chen MJ, Jin Y, Ye XH, Wu JS, Huang ZM. Melatonin attenuates acute pancreatitis-associated lung injury in rats by modulating interleukin 22. World J Gastroenterol. 2012;18:5122–5128. doi: 10.3748/wjg.v18.i36.5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Shemi AG, Basalamah MA, Kensara OA, Ashshi AM. Interleukin-22 therapy attenuates the development of acute pancreatitis in rats. J Clin Med Res. 2011;3:82–88. [Google Scholar]
- 11.Feng D, Park O, Radaeva S, Wang H, Yin S, Kong X, Zheng M, Zakhari S, Kolls JK, Gao B. Interleukin-22 ameliorates cerulein-induced pancreatitis in mice by inhibiting the autophagic pathway. Int J Biol Sci. 2012;8:249–257. doi: 10.7150/ijbs.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17–31. doi: 10.1007/s00281-009-0188-x. [DOI] [PubMed] [Google Scholar]
- 13.Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–785. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggarwal S, Xie MH, Maruoka M, Foster J, Gurney AL. Acinar cells of the pancreas are a target of interleukin-22. J Interferon Cytokine Res. 2001;21:1047–1053. doi: 10.1089/107999001317205178. [DOI] [PubMed] [Google Scholar]
- 15.Gurney AL. IL-22, a Th1 cytokine that targets the pancreas and select other peripheral tissues. Int Immunopharmacol. 2004;4:669–677. doi: 10.1016/j.intimp.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Mühl H, Scheiermann P, Bachmann M, Härdle L, Heinrichs A, Pfeilschifter J. IL-22 in tissue-protective therapy. Br J Pharmacol. 2013;169:761–771. doi: 10.1111/bph.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. 2014;13:21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 18.Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Mühl H. Application of interleukin-22 mediates protection in experimental acetaminophen-induced acute liver injury. Am J Pathol. 2013;182:1107–1113. doi: 10.1016/j.ajpath.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, Gao B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–1159. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chestovich PJ, Uchida Y, Chang W, Ajalat M, Lassman C, Sabat R, Busuttil RW, Kupiec-Weglinski JW. Interleukin-22: implications for liver ischemia-reperfusion injury. Transplantation. 2012;93:485–492. doi: 10.1097/TP.0b013e3182449136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–766.e1-3. doi: 10.1053/j.gastro.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xing WW, Zou MJ, Liu S, Xu T, Gao J, Wang JX, Xu DG. Hepatoprotective effects of IL-22 on fulminant hepatic failure induced by d-galactosamine and lipopolysaccharide in mice. Cytokine. 2011;56:174–179. doi: 10.1016/j.cyto.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 23.Xing WW, Zou MJ, Liu S, Xu T, Wang JX, Xu DG. Interleukin-22 protects against acute alcohol-induced hepatotoxicity in mice. Biosci Biotechnol Biochem. 2011;75:1290–1294. doi: 10.1271/bbb.110061. [DOI] [PubMed] [Google Scholar]
- 24.Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, Ki SH, Yoo SH, Dooley S, Wang FS, et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology. 2011;54:252–261. doi: 10.1002/hep.24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L, Zhang Y, Wang L, Fan F, Zhu L, Li Z, Ruan X, Huang H, Wang Z, Huang Z, et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. 2010;53:339–347. doi: 10.1016/j.jhep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Pan H, Hong F, Radaeva S, Gao B. Hydrodynamic gene delivery of interleukin-22 protects the mouse liver from concanavalin A-, carbon tetrachloride-, and Fas ligand-induced injury via activation of STAT3. Cell Mol Immunol. 2004;1:43–49. [PubMed] [Google Scholar]
- 28.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 29.Sovran B, Loonen LM, Lu P, Hugenholtz F, Belzer C, Stolte EH, Boekschoten MV, van Baarlen P, Kleerebezem M, de Vos P, et al. IL-22-STAT3 pathway plays a key role in the maintenance of ileal homeostasis in mice lacking secreted mucus barrier. Inflamm Bowel Dis. 2015;21:531–542. doi: 10.1097/MIB.0000000000000319. [DOI] [PubMed] [Google Scholar]
- 30.Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tumanov AV, Koroleva EP, Guo X, Wang Y, Kruglov A, Nedospasov S, Fu YX. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. 2011;10:44–53. doi: 10.1016/j.chom.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leung JM, Davenport M, Wolff MJ, Wiens KE, Abidi WM, Poles MA, Cho I, Ullman T, Mayer L, Loke P. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 2014;7:124–133. doi: 10.1038/mi.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rendon JL, Li X, Akhtar S, Choudhry MA. Interleukin-22 modulates gut epithelial and immune barrier functions following acute alcohol exposure and burn injury. Shock. 2013;39:11–18. doi: 10.1097/SHK.0b013e3182749f96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 37.Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, Alcorn JF. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol. 2013;182:1286–1296. doi: 10.1016/j.ajpath.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi K, Hirose K, Kawashima S, Niwa Y, Wakashin H, Iwata A, Tokoyoda K, Renauld JC, Iwamoto I, Nakayama T, et al. IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. J Allergy Clin Immunol. 2011;128:1067–1076.e1-6. doi: 10.1016/j.jaci.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 39.Taube C, Tertilt C, Gyülveszi G, Dehzad N, Kreymborg K, Schneeweiss K, Michel E, Reuter S, Renauld JC, Arnold-Schild D, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS One. 2011;6:e21799. doi: 10.1371/journal.pone.0021799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP. γδ T cells protect against lung fibrosis via IL-22. J Exp Med. 2010;207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, Senices M, Gill D, Dunussi-Joannopoulos K, Collins M, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 43.Boniface K, Guignouard E, Pedretti N, Garcia M, Delwail A, Bernard FX, Nau F, Guillet G, Dagregorio G, Yssel H, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407–415. doi: 10.1111/j.1365-2249.2007.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakajima H, Nakajima K, Tarutani M, Morishige R, Sano S. Kinetics of circulating Th17 cytokines and adipokines in psoriasis patients. Arch Dermatol Res. 2011;303:451–455. doi: 10.1007/s00403-011-1159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, Eidsmo L. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. 2014;192:3111–3120. doi: 10.4049/jimmunol.1302313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ikeuchi H, Kuroiwa T, Hiramatsu N, Kaneko Y, Hiromura K, Ueki K, Nojima Y. Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum. 2005;52:1037–1046. doi: 10.1002/art.20965. [DOI] [PubMed] [Google Scholar]
- 47.Leipe J, Schramm MA, Grunke M, Baeuerle M, Dechant C, Nigg AP, Witt MN, Vielhauer V, Reindl CS, Schulze-Koops H, et al. Interleukin 22 serum levels are associated with radiographic progression in rheumatoid arthritis. Ann Rheum Dis. 2011;70:1453–1457. doi: 10.1136/ard.2011.152074. [DOI] [PubMed] [Google Scholar]
- 48.Carrión M, Juarranz Y, Martínez C, González-Álvaro I, Pablos JL, Gutiérrez-Cañas I, Gomariz RP. IL-22/IL-22R1 axis and S100A8/A9 alarmins in human osteoarthritic and rheumatoid arthritis synovial fibroblasts. Rheumatology (Oxford) 2013;52:2177–2186. doi: 10.1093/rheumatology/ket315. [DOI] [PubMed] [Google Scholar]
- 49.Carrión M, Juarranz Y, Seoane IV, Martínez C, González-Álvaro I, Pablos JL, Gutiérrez-Cañas I, Gomariz RP. VIP modulates IL-22R1 expression and prevents the contribution of rheumatoid synovial fibroblasts to IL-22-mediated joint destruction. J Mol Neurosci. 2014;52:10–17. doi: 10.1007/s12031-013-0177-3. [DOI] [PubMed] [Google Scholar]
- 50.Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. 2002;277:33676–33682. doi: 10.1074/jbc.M204204200. [DOI] [PubMed] [Google Scholar]
- 51.Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol. 2012;5:658–669. doi: 10.1038/mi.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 53.Bulmer AC, Coombes JS, Blanchfield JT, Toth I, Fassett RG, Taylor SM. Bile pigment pharmacokinetics and absorption in the rat: therapeutic potential for enteral administration. Br J Pharmacol. 2011;164:1857–1870. doi: 10.1111/j.1476-5381.2011.01413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tachiiri A, Imamura R, Wang Y, Fukui M, Umemura M, Suda T. Genomic structure and inducible expression of the IL-22 receptor alpha chain in mice. Genes Immun. 2003;4:153–159. doi: 10.1038/sj.gene.6363934. [DOI] [PubMed] [Google Scholar]
- 57.Lerman G, Sharon M, Leibowitz-Amit R, Sidi Y, Avni D. The crosstalk between IL-22 signaling and miR-197 in human keratinocytes. PLoS One. 2014;9:e107467. doi: 10.1371/journal.pone.0107467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mareninova OA, Hermann K, French SW, O’Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–3355. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fortunato F, Bürgers H, Bergmann F, Rieger P, Büchler MW, Kroemer G, Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350–360, 360.e1-5. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 60.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 61.Abdulla A, Awla D, Thorlacius H, Regnér S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J Leukoc Biol. 2011;90:975–982. doi: 10.1189/jlb.0411195. [DOI] [PubMed] [Google Scholar]
- 62.Narushima Y, Unno M, Nakagawara K, Mori M, Miyashita H, Suzuki Y, Noguchi N, Takasawa S, Kumagai T, Yonekura H, et al. Structure, chromosomal localization and expression of mouse genes encoding type III Reg, RegIII alpha, RegIII beta, RegIII gamma. Gene. 1997;185:159–168. doi: 10.1016/s0378-1119(96)00589-6. [DOI] [PubMed] [Google Scholar]
- 63.Keim V, Iovanna JL, Rohr G, Usadel KH, Dagorn JC. Characterization of a rat pancreatic secretory protein associated with pancreatitis. Gastroenterology. 1991;100:775–782. doi: 10.1016/0016-5085(91)80025-5. [DOI] [PubMed] [Google Scholar]
- 64.Iovanna J, Orelle B, Keim V, Dagorn JC. Messenger RNA sequence and expression of rat pancreatitis-associated protein, a lectin-related protein overexpressed during acute experimental pancreatitis. J Biol Chem. 1991;266:24664–24669. [PubMed] [Google Scholar]
- 65.Zhang H, Kandil E, Lin YY, Levi G, Zenilman ME. Targeted inhibition of gene expression of pancreatitis-associated proteins exacerbates the severity of acute pancreatitis in rats. Scand J Gastroenterol. 2004;39:870–881. doi: 10.1080/00365520410006477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Christa L, Carnot F, Simon MT, Levavasseur F, Stinnakre MG, Lasserre C, Thepot D, Clement B, Devinoy E, Brechot C. HIP/PAP is an adhesive protein expressed in hepatocarcinoma, normal Paneth, and pancreatic cells. Am J Physiol. 1996;271:G993–1002. doi: 10.1152/ajpgi.1996.271.6.G993. [DOI] [PubMed] [Google Scholar]
- 67.McKie AT, Simpson RJ, Ghosh S, Peters TJ, Farzaneh F. Regulation of pancreatitis-associated protein (HIP/PAP) mRNA levels in mouse pancreas and small intestine. Clin Sci (Lond) 1996;91:213–218. doi: 10.1042/cs0910213. [DOI] [PubMed] [Google Scholar]
- 68.Masciotra L, Lechêne de la Porte P, Frigerio JM, Dusetti NJ, Dagorn JC, Iovanna JL. Immunocytochemical localization of pancreatitis-associated protein in human small intestine. Dig Dis Sci. 1995;40:519–524. doi: 10.1007/BF02064359. [DOI] [PubMed] [Google Scholar]
- 69.Dieckgraefe BK, Stenson WF, Korzenik JR, Swanson PE, Harrington CA. Analysis of mucosal gene expression in inflammatory bowel disease by parallel oligonucleotide arrays. Physiol Genomics. 2000;4:1–11. doi: 10.1152/physiolgenomics.2000.4.1.1. [DOI] [PubMed] [Google Scholar]
- 70.Ozturk M, de la Monte SM, Gross J, Wands JR. Elevated levels of an exocrine pancreatic secretory protein in Alzheimer disease brain. Proc Natl Acad Sci USA. 1989;86:419–423. doi: 10.1073/pnas.86.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duplan L, Michel B, Boucraut J, Barthellémy S, Desplat-Jego S, Marin V, Gambarelli D, Bernard D, Berthézène P, Alescio-Lautier B, et al. Lithostathine and pancreatitis-associated protein are involved in the very early stages of Alzheimer’s disease. Neurobiol Aging. 2001;22:79–88. doi: 10.1016/s0197-4580(00)00182-2. [DOI] [PubMed] [Google Scholar]
- 72.Christa L, Simon MT, Brezault-Bonnet C, Bonte E, Carnot F, Zylberberg H, Franco D, Capron F, Roskams T, Bréchot C. Hepatocarcinoma-intestine-pancreas/pancreatic associated protein (HIP/PAP) is expressed and secreted by proliferating ductules as well as by hepatocarcinoma and cholangiocarcinoma cells. Am J Pathol. 1999;155:1525–1533. doi: 10.1016/S0002-9440(10)65468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie MJ, Motoo Y, Iovanna JL, Su SB, Ohtsubo K, Matsubara F, Sawabu N. Overexpression of pancreatitis-associated protein (PAP) in human pancreatic ductal adenocarcinoma. Dig Dis Sci. 2003;48:459–464. doi: 10.1023/a:1022520212447. [DOI] [PubMed] [Google Scholar]
- 74.Rosty C, Christa L, Kuzdzal S, Baldwin WM, Zahurak ML, Carnot F, Chan DW, Canto M, Lillemoe KD, Cameron JL, et al. Identification of hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein I as a biomarker for pancreatic ductal adenocarcinoma by protein biochip technology. Cancer Res. 2002;62:1868–1875. [PubMed] [Google Scholar]
- 75.Lin YY, Viterbo D, Mueller CM, Stanek AE, Smith-Norowitz T, Drew H, Wadgaonkar R, Zenilman ME, Bluth MH. Small-interference RNA gene knockdown of pancreatitis-associated proteins in rat acute pancreatitis. Pancreas. 2008;36:402–410. doi: 10.1097/MPA.0b013e31815f3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shigekawa M, Hikita H, Kodama T, Shimizu S, Li W, Uemura A, Miyagi T, Hosui A, Kanto T, Hiramatsu N, et al. Pancreatic STAT3 protects mice against caerulein-induced pancreatitis via PAP1 induction. Am J Pathol. 2012;181:2105–2113. doi: 10.1016/j.ajpath.2012.08.038. [DOI] [PubMed] [Google Scholar]
- 77.Gironella M, Folch-Puy E, LeGoffic A, Garcia S, Christa L, Smith A, Tebar L, Hunt SP, Bayne R, Smith AJ, et al. Experimental acute pancreatitis in PAP/HIP knock-out mice. Gut. 2007;56:1091–1097. doi: 10.1136/gut.2006.116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakamichi I, Habtezion A, Zhong B, Contag CH, Butcher EC, Omary MB. Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J Clin Invest. 2005;115:3007–3014. doi: 10.1172/JCI24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mikami Y, Takeda K, Shibuya K, Qiu-Feng H, Egawa S, Sunamura M, Matsuno S. Peritoneal inflammatory cells in acute pancreatitis: Relationship of infiltration dynamics and cytokine production with severity of illness. Surgery. 2002;132:86–92. doi: 10.1067/msy.2002.125171. [DOI] [PubMed] [Google Scholar]
- 81.Malleo G, Mazzon E, Siriwardena AK, Cuzzocrea S. Role of tumor necrosis factor-alpha in acute pancreatitis: from biological basis to clinical evidence. Shock. 2007;28:130–140. doi: 10.1097/shk.0b013e3180487ba1. [DOI] [PubMed] [Google Scholar]
- 82.Kaufmann P, Tilz GP, Lueger A, Demel U. Elevated plasma levels of soluble tumor necrosis factor receptor (sTNFRp60) reflect severity of acute pancreatitis. Intensive Care Med. 1997;23:841–848. doi: 10.1007/s001340050420. [DOI] [PubMed] [Google Scholar]
- 83.Granell S, Pereda J, Gómez-Cambronero L, Cassinello N, Sabater L, Closa D, Sastre J. Circulating TNF-alpha and its soluble receptors during experimental acute pancreatitis. Cytokine. 2004;25:187–191. doi: 10.1016/j.cyto.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 84.Shen Y, Cui N, Miao B, Zhao E. Immune dysregulation in patients with severe acute pancreatitis. Inflammation. 2011;34:36–42. doi: 10.1007/s10753-010-9205-4. [DOI] [PubMed] [Google Scholar]
- 85.Surbatovic M, Radakovic S. Tumor necrosis factor-α levels early in severe acute pancreatitis: is there predictive value regarding severity and outcome? J Clin Gastroenterol. 2013;47:637–643. doi: 10.1097/MCG.0b013e31828a6cfc. [DOI] [PubMed] [Google Scholar]
- 86.Eyerich S, Wagener J, Wenzel V, Scarponi C, Pennino D, Albanesi C, Schaller M, Behrendt H, Ring J, Schmidt-Weber CB, et al. IL-22 and TNF-α represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur J Immunol. 2011;41:1894–1901. doi: 10.1002/eji.201041197. [DOI] [PubMed] [Google Scholar]
- 87.Ni J, Hu G, Xiong J, Shen J, Shen J, Yang L, Tang M, Zhao Y, Ying G, Yu G, et al. Involvement of interleukin-17A in pancreatic damage in rat experimental acute necrotizing pancreatitis. Inflammation. 2013;36:53–65. doi: 10.1007/s10753-012-9519-5. [DOI] [PubMed] [Google Scholar]
- 88.Dai SR, Li Z, Zhang JB. Serum interleukin 17 as an early prognostic biomarker of severe acute pancreatitis receiving continuous blood purification. Int J Artif Organs. 2015;38:192–198. doi: 10.5301/ijao.5000406. [DOI] [PubMed] [Google Scholar]
- 89.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207:1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Warren RB, Griffiths CE. The future of biological therapies. Semin Cutan Med Surg. 2010;29:63–66. doi: 10.1016/j.sder.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 92.Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–241. doi: 10.1038/nature13564. [DOI] [PubMed] [Google Scholar]
- 93.Wang X ClinicalTrials. gov. A service of the U.S. National Institutes of Health. Available from: https://clinicaltrials.gov.