

Key Points
Many excitatory synapses in the brain release glutamate with both synchronous and asynchronous components.
Immediately following an action potential, neurons display a reduced excitability due to the post‐spike afterhyperpolarization (AHP). This gives rise to a relative refractory period.
When an action potential is evoked by glutamate synaptic input possessing asynchronous release, the delayed glutamate release events act to depolarize the neuron during the AHP and overcome the relative refractory period.
These results demonstrate a new role for asynchronous release in regulating post‐spike excitability and the relative refractory period in central neurons.
Abstract
Post‐spike afterhyperpolarizations (AHPs) functionally inhibit neuronal excitability for tens to hundreds of milliseconds following each action potential. This imposes a relative refractory period during which synaptic excitation is less effective at evoking spikes. Here we asked whether some synapses have mechanisms in place that allow them to overcome the AHP and drive spiking in target cells during this period of reduced excitability. We examined glutamate synapses onto oxytocin and vasopressin neurons in the paraventricular nucleus of the hypothalamus. These synapses can display pronounced asynchronous glutamate release following a single presynaptic spike, with the time course of release being similar to that of the post‐spike AHP. To test whether asynchronous release is more effective at overcoming the relative refractory period, we evoked a single action potential with either a brief synchronous depolarization or an asynchronous potential and then assessed excitability at multiple time points following the spike. Neurons receiving asynchronous depolarizing synaptic inputs had a shorter relative refractory period than those receiving synchronous depolarizations. Our data demonstrate that synapses releasing glutamate in an asynchronous and delayed manner are ideally adapted to counter the AHP. By effectively overcoming the relative refractory period, the kinetics of excitatory synaptic input can play an important role in controlling post‐spike excitability.
Key Points
Many excitatory synapses in the brain release glutamate with both synchronous and asynchronous components.
Immediately following an action potential, neurons display a reduced excitability due to the post‐spike afterhyperpolarization (AHP). This gives rise to a relative refractory period.
When an action potential is evoked by glutamate synaptic input possessing asynchronous release, the delayed glutamate release events act to depolarize the neuron during the AHP and overcome the relative refractory period.
These results demonstrate a new role for asynchronous release in regulating post‐spike excitability and the relative refractory period in central neurons.
Abbreviations
- AHP
afterhyperpolarization
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- ISI
inter‐stimulus interval
- MNC
magnocellular neurosecretory cell
- PVN
paraventricular nucleus
- SON
supraoptic nucleus
Introduction
The output of a neuron is governed by a complex interplay between intrinsic membrane properties, voltage‐gated conductances and synaptic inputs. Interactions between these variables not only determine the direction of changes in neuronal excitability but also alter the specific patterns of activity. These activity patterns, in particular bursts, are efficient drivers of dendritic transmitter release (Ludwig & Leng, 2006; Regehr et al. 2009) and synaptic plasticity (Lisman, 1997).
Many central neurons possess large hyperpolarizing conductances that are recruited following a single action potential (Coombs et al. 1955; Sah, 1996). These conductances inhibit excitability for tens to hundreds of milliseconds following a spike. This period of reduced excitability is known as the relative refractory period and reduces the probability that subsequent synaptic inputs will trigger a second spike or burst. While it has been previously shown that neuromodulators can regulate the magnitude of the afterhyperpolarization (AHP) (Bennett & Wilson, 1998; Savic et al. 2001; Ventura et al. 2008), it is currently unknown if other fast synaptic mechanisms exist to modify the relative refractory period.
The magnocellular neurosecretory cells (MNCs) located in the supraoptic (SON) and paraventricular nuclei (PVN) of the hypothalamus represent an ideal model system to investigate such mechanisms. These neurons exhibit synaptically driven burst firing both in vivo (Nissen et al. 1995; Brown et al. 2004) and in vitro (Jourdain et al. 1998; Israel et al. 2003; Israel et al. 2010). These burst discharges are particularly important for ensuring adequate hormone release from the nerve endings of these neurons in the posterior pituitary (Harris et al. 1969). These neurons also possess AHPs that are recruited after a single action potential (Andrew & Dudek, 1984 b; Bourque et al. 1985; Armstrong et al. 1994; Greffrath et al. 2004; Ventura et al. 2008). In addition, glutamate synaptic inputs onto these neurons also display a prolonged and desynchronized form of neurotransmitter release termed asynchronous release (Iremonger & Bains, 2007). Asynchronous neurotransmitter release has been described at many central synapses and has been proposed to regulate the duration of postsynaptic excitability changes as well as the precision of spike timing (Atluri & Regehr, 1998; Lu & Trussell, 2000; Otsu et al. 2004; Hefft & Jonas, 2005; Evstratova et al. 2014; Kaeser & Regehr, 2014). We speculated that because the temporal profile of asynchronous glutamate release onto MNCs closely matches the time course of the AHP, this form of neurotransmission may be well suited to overcoming the post‐spike refractory period and facilitating closely timed spikes.
Here we show that single action potentials evoked by depolarizing current injection are followed by a pronounced AHP which effectively reduces neuronal excitability for a period of ∼100 ms. Notably, this inhibitory period can be significantly shortened if action potentials are instead triggered by glutamate synaptic inputs with prominent asynchronous release. This effect can be replicated with injected asynchronous synaptic current waveforms but not with synchronous synaptic current waveforms. Together, these data suggest a new role for asynchronous release in controlling the duration of the relative refractory period.
Methods
Slice preparation
All protocols were approved by the University of Calgary animal care and use committee in accordance with guidelines established by the Canadian Council on Animal Care. Male Sprague–Dawley rats (postnatal day 21–30) were anaesthetized with sodium pentobarbital (0.1 ml (100 g body weight)−1) and then decapitated.
The brain was then quickly removed and placed in ice‐cold oxygenated (95% O2–5% CO2) cutting solution, which consisted of (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, 75 sucrose; saturated with 95% O2–5% CO2. The brain was then blocked and mounted on a vibrating slicer (Leica, VT1000s). Coronal sections of 300 μm were cut and subsequently incubated at 32.5°C in artificial cerebrospinal fluid (ACSF) containing (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, 10 glucose; saturated with 95% O2–5% CO2, for a minimum of 60 min.
Electrophysiology
Slices containing the PVN were placed in a recording chamber under a Zeiss Axioskop II FS Plus upright microscope and continuously superfused with 32.5°C oxygenated ACSF (95% O2–5% CO2) at a rate of 1 ml min−1. Recorded cells were confirmed to be MNCs based on their morphology and well defined electrophysiological characteristics (Luther & Tasker, 2000).
Whole‐cell recordings
Patch pipettes were pulled from borosilicate glass and had a resistance between 3–6 MΩ; they were filled with a solution containing (in mm): 116 potassium gluconate, 8 sodium gluconate, 2 MgCl2, 8 KCl, 1 potassium EGTA, 4 potassium ATP, and 0.3 sodium GTP, 10 Hepes, corrected to pH 7.2 with KOH. Series resistance was not compensated and recordings were accepted for analysis if changes in access resistance were < 20%. The liquid junction potential was calculated to be approximately −13 mV and was not compensated for.
Glutamatergic fibres were stimulated extracellularly with a monopolar glass microelectrode (3–6 MΩ) filled with ACSF and placed either within or just outside of the PVN. The perfusate always contained picrotoxin (100 μm, Sigma‐Aldrich) to block GABAA‐mediated conductances. We have previously demonstrated that under these recording conditions evoked synaptic currents are completely blocked with 10 μm 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX) and hence are AMPA receptor mediated (Iremonger & Bains, 2007).
AHP amplitude and kinetics were recorded after evoking a single action potential with a 5 ms depolarizing current step. While the fast hyperpolarizations that follow a single spike are generally termed AHPs, we note that some previous studies of magnocellular neurons have termed these events as hyperpolarizing after‐potentials (HAPs). In this study, we exclusively refer to them as AHPs. As both fast AHPs and asynchronous release are equivalent between oxytocin and vasopressin neurons (Armstrong et al. 1994; Stern & Armstrong, 1996; Iremonger & Bains, 2007), we did not attempt to distinguish between cell types in this current study.
Artificial injected synaptic currents
Synchronous or asynchronous excitatory postsynaptic currents (EPSCs) were recorded in voltage clamp (at −60 mV) in response to synaptic stimulation. The voltage clamp traces were then converted to a stimulus waveform (current) in Clampex and delivered to the neuron via the whole‐cell recording electrode.
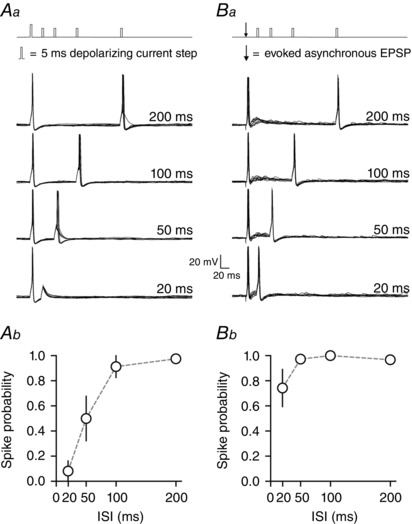
Artificial EPSCs were delivered as paired pulses with the first artificial EPSC being either synchronous or asynchronous while the second ‘test’ EPSC was always synchronous (i.e. lacking asynchronous release). Injected EPSC waveforms are shown in Fig. 4 Aa and Ba. At the start of each experiment, the amplitude of injected currents was scaled such that synchronous injected currents evoked action potentials between 70–100% of the time at the longest inter‐stimulus interval (ISI; 200 ms) but not at shorter time intervals. This procedure was similar to what was performed for square wave injected currents. Artificial synchronous injected currents had an average peak amplitude of 200 ± 19.3 pA, whereas asynchronous injected currents had an average peak amplitude of 171.6 ± 16.8 pA (n = 6).
Figure 4. Artificial injected asynchronous currents are sufficient to reduce the relative refractory period .
In order to precisely control asynchronous release without modifying other parameters of cellular excitability, we injected artificial synchronous or asynchronous current waveforms via the whole‐cell recording electrode (Aa and Ba). Panel A shows responses where both the first and second artificial EPSC were synchronous (i.e. lacking asynchronous release). Panel B shows responses where the first artificial EPSC was asynchronous, but the second was synchronous. Trials where the neuron received only synchronous artificial currents displayed a large reduction in post‐spike excitability (Ab and c) whereas trials where the first spike was evoked with an asynchronous EPSC display comparatively higher post‐spike excitability (Bb and c).
Data collection and analysis
Electrophysiological recordings were collected with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), using a low pass filter at 1 kHz and digitized with the Digidata 1322A (Molecular Devices) at 10–20 kHz. All electrophysiological data were analysed with Clampex 10 software (Molecular Devices).
For evoked currents, analysis was performed using pCLAMP 9 (Molecular Devices). The number of individual asynchronous release events was quantified by counting the occurrence of quantal EPSCs from 5 to 100 ms after the onset of the EPSC (5 ms bins) during 30 trials. Events were not counted in the first 5 ms after stimulation, because they could not be discriminated from the synchronous component of the EPSC. Baseline spontaneous release was calculated for the 100 ms before the stimulus and subtracted. The synchronous component of the EPSC charge transfer was calculated by integrating the area of the average EPSC from 0–10 ms after EPSC onset. Asynchronous charge transfer was calculated by integrating the area of the average EPSC from 10–100 ms after EPSC onset. Asynchronous release as a fraction of total charge transfer was calculated by dividing asynchronous charge by total charge transfer (total charge = 0–100 ms from EPSC onset).
For current clamp experiments where action potentials were triggered with depolarizing current steps or synaptic stimulation, we only analysed trials where single spikes were evoked on the first stimuli. To avoid the confounding effects of spike jitter on the first evoked spike, only trials where the first spike was evoked within 15 ms of the stimulus onset were analysed.
All data are presented as means ± SEM. Statistical analyses were performed with either Student's paired t test or repeated measures ANOVA with a post hoc Newman–Keuls test. For two‐way repeated measures ANOVA, post hoc analysis was performed with Bonferroni's multiple comparisons test. For all statistical tests, P < 0.05 was accepted as statistically significant.
Results
The time course of asynchronous release is similar to that of the post‐spike AHP
Glutamate synapses onto MNCs in both the PVN and SON of the hypothalamus exhibit pronounced asynchronous release (Iremonger & Bains, 2007; Trudel & Bourque, 2010). We have previously shown that asynchronous glutamate release is highly calcium dependent, builds during repeated stimuli and is equivalent onto both oxytocin and vasopressin neurons (Iremonger & Bains, 2007). While asynchronous glutamate release in response to high‐frequency trains of presynaptic activity can induce prolonged spike discharges in both MNCs and other neurons (Iremonger & Bains, 2007; Peters et al. 2010), the implications of asynchronous release for synaptic integration on brief time scales (in response to 1 or 2 spikes) remains unexplored. We speculated that one such role of asynchronous glutamate release may be to overcome the post‐spike AHP which mediates the relative refractory period. To efficiently overcome the AHP, asynchronous release must have a time course similar to that of the AHP or of longer duration.
Single extracellular electrical stimulation evoked a short‐latency synchronous EPSC followed by asynchronous EPSCs in MNCs (Fig. 1 A). The frequency of asynchronous release events was highest immediately following the synchronous EPSC with event frequency decreasing until it had returned to baseline by approximately 100 ms after stimulation (Fig. 1 B). The frequency of asynchronous EPSC events was well fitted by a single exponential decay with a time constant of 16.5 ms (n = 25) similar to our previous reports (Iremonger & Bains, 2007). It is important to note, however, that due to the high membrane resistance and slow membrane time constant of MNCs, that asynchronous release exerts a depolarizing influence on the membrane potential for a period of time longer than that predicted by voltage clamp recordings (Iremonger & Bains, 2007). Indeed, our previous study reported the decay time constant of single evoked asynchronous excitatory postsynaptic potentials (EPSPs) to be 34.9 ± 6.2 ms in current clamp.
Figure 1. Time course of asynchronous release overlaps with that of the post‐spike after‐hyperpolarization .
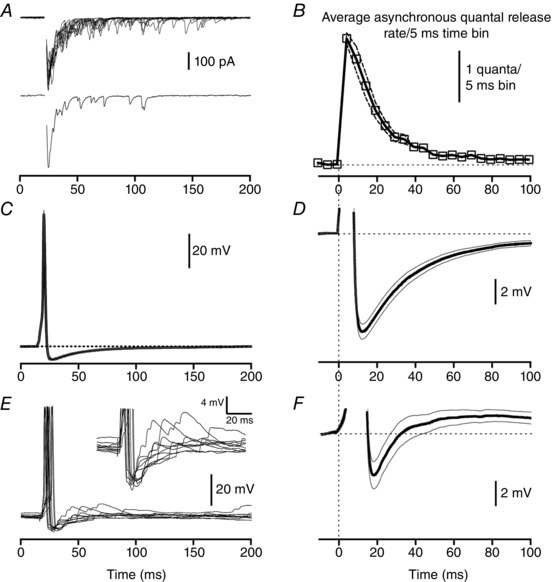
A, single extracellular stimuli evoke glutamate synaptic currents with both fast synchronous and delayed asynchronous components of the EPSC. Top panel is 10 trials overlaid, bottom panel is a single trial. Stimulus artifact has been removed for clarity. B, time course of asynchronous release plotted on same time scale as panel D. C, average ± SEM of single spike voltage averaged across 11 neurons. Spikes were evoked with a 5 ms depolarizing current step. Expanded time course of the AHP from the same data set is shown in panel D. E, voltage traces of spikes evoked with a single synaptic stimulation from a representative neuron (10 trials overlaid, stimulus artifact removed for clarity). Expanded time course of the AHP and summated asynchronous EPSPs are shown in the inset. F, expanded time course of the AHP (average ± SEM) from 11 neurons where spikes were evoked with synaptic stimulation. In panel D, E inset and F, action potentials have been truncated for clarity.
Next we measured the AHP after a single spike evoked with a 5 ms depolarizing current step. The average peak amplitude of the AHP was 7.8 ± 0.6 mV (n = 11) which occurred 7.1 ms after the peak of the action potential (Fig. 1 C). This is similar to what has been previously published (Andrew & Dudek, 1984 b; Bourque et al. 1985; Armstrong et al. 1994) but smaller in amplitude than that reported by Greffrath et al. (2004). Membrane potential returned to pre‐spike values by approximately 150–200 ms after the peak of the action potential (Fig. 1 D). The decay of the average AHP was well fitted with a two‐phase exponential with a fast and slow time constant of 22.4 ms and 783.4 ms, respectively. These two phases are likely to correspond to the fast and medium components of the AHP, respectively, similar to that described in other central neurons (Savic et al. 2001). Next we investigated whether there was any correlation between the amplitude of the AHP and the amount of asynchronous release for a given synaptic stimulation site onto a MNC. No correlation was observed between AHP amplitude and asynchronous charge transfer (as a fraction of total charge transfer; Pearson's correlation coefficient r = 0.36, P = 0.28, n = 11).
Next we evoked spikes with single evoked asynchronous EPSPs (Fig. 1 E). Asynchronous EPSPs persisted beyond the evoked spike and continued to depolarize the membrane potential during the AHP (Fig. 1 E inset). When synaptically evoked spikes were peak aligned and averaged across a number of neurons (Fig. 1 F), we found that the average AHP was 4.0 ± 0.9 mV (n = 11), which was significantly smaller than the control AHP (P < 0.05). The time course of the AHP was also altered, as the recovery of the AHP was best fitted with a single phase exponential with a time constant of 10.2 ms.
Together these data demonstrate that asynchronous release and the post‐spike AHP regulate excitability over similar time periods, but in opposing directions.
Asynchronous release reduces the post‐spike relative refractory period
We next set out to determine the time window over which excitability is reduced following a single action potential due to the AHP. To assess this, we evoked a single action potential with a 5 ms depolarizing current step which was followed by a second current step 20, 50, 100 or 200 ms later (inter‐stimulus interval, ISI). The amplitude of the first current step was set to evoke spikes 100% of the time. The amplitude of the second current step was initially set such that it evoked spikes > 70% of the time at the 200 ms ISI and is referred to as the starting current pulse. Figure 2 B shows that current steps at short (20 ms) ISI that occurred close to the peak of the AHP had a low probability of evoking spikes (spike probability at 20 ms ISI with starting current pulse = 0.00 ± 0.00, n = 4). This low spike probability could, however, be significantly increased with larger current steps as would be predicated for the relative refractory period (spike probability at 20 ms ISI with +40 pA above starting current pulse = 0.78 ± 0.17, P < 0.05, n = 4, Fig. 2 B). Current steps evoked at 200 ms after the first spike, at the end of the AHP, had high probability of evoking spikes even at the starting current pulse amplitude (spike probability at 200 ms ISI with starting current pulse = 0.87 ± 0.06, n = 4). A similar reduction in post‐spike excitability during the AHP was also observed if the second test spike was evoked with synaptic stimulation (spike probability at 20 ms ISI with EPSP = 0.32 ± 0.08, spike probability at 20 ms ISI with EPSP = 0.83 ± 0.06, n = 7). Of note, spikes evoked during the period of the AHP had a higher amount of jitter (coefficient of variation in spike onset time) if evoked by synaptic stimulation as compared to spikes evoked by current injection (200 ms ISI EPSP evoked spike jitter = 40.9 ± 16.6%; current step evoked spike jitter = 20.9 ± 4.2%, P < 0.05).
Figure 2. Spiking excitability is reduced during the afterhyperpolarization .
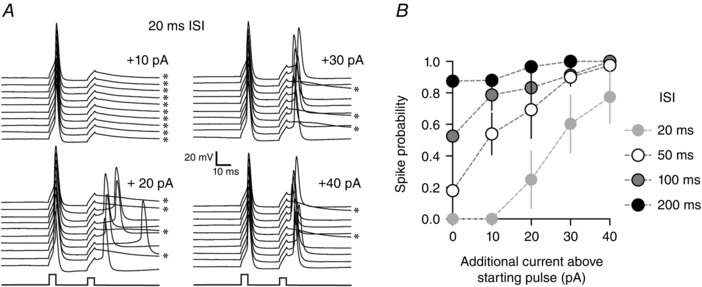
A, paired stimulation protocol where two square 5 ms depolarizing current pulses are injected into the neuron with a 20 ms inter‐stimulus interval (ISI). The amplitude of the first current pulse is set to evoke a single spike 100% of the time, while the amplitude of the second pulse is systematically varied. Panel A shows that at short ISIs, the second pulse arrives during the peak of the AHP and spike probability is reduced. By increasing the amplitude of the second pulse, spike probability can be predictably enhanced. Asterisks indicate spike failures. B, graph of spike probabilities for ISIs between 20 and 200 ms at various current amplitudes on the second pulse. This graph demonstrates that for low amplitude pulses, spike probability is only low at short ISIs and not at long ISIs.
Overall, these data show that during the period of the AHP, the probability of evoking spikes in response to small depolarizations is reduced.
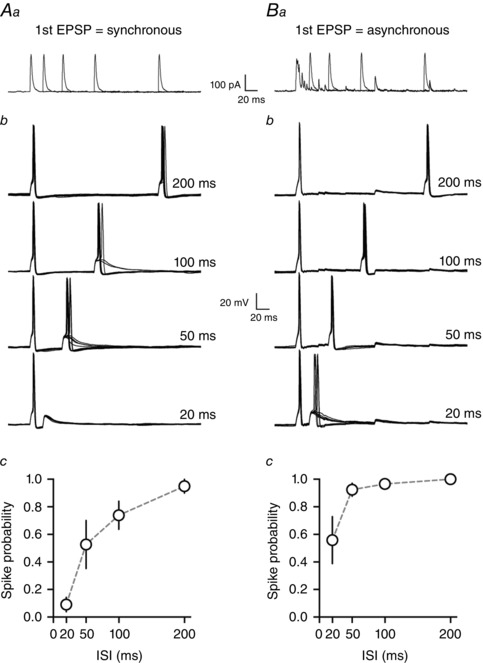
Next we investigated if the extended time course of asynchronous glutamate release could overcome the reduction in spike probability due to the AHP. We first repeated the double current step experiments (as in Fig. 2), with the current amplitude of the second step fixed such that it evoked spikes > 70% of the time only at the 200 ms post‐spike interval (average amplitude of second current step = 102.5 ± 22.1 pA, duration = 5 ms, Fig. 3 Aa). In the same cells, but separate trials, we then triggered an initial spike with an evoked asynchronous EPSP (Fig. 3 Ba) and assayed post‐spike excitability with a current step of the same duration and amplitude as used in the first part of the experiment.
Figure 3. Asynchronous release overcomes the post‐spike relative refractory period .
Aa, paired stimulation protocol where two square current pulses are injected into the neuron at different ISIs (20, 50, 100 or 200 ms). The amplitude of the second pulse is set such that it evokes spikes > 70% of the time only at the longest ISI. The mean reduction in spike probability at short ISIs is shown in panel Ab. Ba, paired stimulation protocol where the first spike is now evoked with extracellular synaptic stimulation to induce synchronous and asynchronous glutamate release. The amplitude of the second pulse (square current injection) is the same as in panel Aa. Asynchronous release is able to overcome the post‐spike refractory period to enhance excitability at short ISIs (Bb).
As expected, when the first action potential was evoked with a depolarizing current step, post‐spike excitability was significantly reduced at time intervals over which the AHP hyperpolarized the membrane potential (spike probability at 20 and 50 ms ISI was 0.08 ± 0.08 and 0.50 ± 0.18, respectively, P < 0.05 when compared to 100 or 200 ms ISI, one‐way ANOVA with Newman–Keuls test, n = 4, Fig. 3 Ab). However, if the first action potential was evoked with an asynchronous EPSP, post‐spike excitability remained high even at short ISIs (spike probability at 20 and 50 ms ISI was 0.74 ± 0.15 and 0.97 ± 0.02 respectively, n = 4, Fig. 3 Bb) and was not significantly different between different ISIs (one‐way ANOVA, P > 0.05, n = 4). Overall, the reduction in post‐spike excitability was significantly attenuated in conditions with asynchronous release for short post‐spike intervals (spike probability at 20 and 50 ms P < 0.05, comparison between conditions, 2‐way ANOVA with Bonferroni's test). These data are consistent with a role of asynchronous release for overcoming the post‐spike relative refractory period.
Artificial asynchronous injected currents overcome the post‐spike relative refractory period
While these data suggest that asynchronous release may be responsible for providing additional depolarization to negate the AHP, it is possible that synaptic stimulation mediates long‐lasting increases in excitability via other pathways, for example the release of additional neuromodulators. In order to test the specific role of asynchronous release, we needed to develop a protocol where asynchronous release could be manipulated independently of other variables. Unfortunately, pharmacological manipulations that modify asynchronous release will also modify the AHP, as both are highly calcium‐dependent processes (Bourque et al. 1985; Kirkpatrick & Bourque, 1991; Iremonger & Bains, 2007). Likewise, manipulations of potassium channels underlying the AHP will also result in changes to evoked release (Iremonger & Bains, 2009). To overcome these issues, we injected artificial EPSCs that possessed either a large amount of asynchronous release or no asynchronous release. These EPSCs were recorded in voltage clamp in response to synaptic stimulation in prior experiments. The voltage clamp traces were then converted to a stimulus waveform and delivered to the neuron via the whole‐cell recording electrode. Importantly, while the first artificial EPSC was either synchronous or asynchronous, the second ‘test’ EPSC was always synchronous (i.e. lacking asynchronous release). This means that the only variable that was different between the two conditions was the amount of asynchronous release present on the first EPSC that triggered the first action potential.
We next conducted an experiment in which the first spike was evoked with an artificial synchronous EPSP and post‐spike excitability was assayed with a second synchronous EPSP at different ISIs (Fig. 4 A). Artificial synchronous EPSPs injected during the AHP in these experiments were less effective at evoking spikes at short ISIs (spike probability at 20 ms and 50 ms ISI = 0.09 ± 0.05 and 0.53 ± 0.18, respectively, n = 6, Fig. 4 Ab and c) compared to artificial synchronous EPSPs injected at long ISIs (spike probability at 200 ms ISI = 0.95 ± 0.05, P < 0.05, one‐way ANOVA with Newman–Keuls test, n = 6, Fig. 4 Ab and c). The reduction in post‐spike excitability with artificial synchronous EPSC injected currents was not statistically different from the reduction in excitability assayed with square current steps shown in Fig. 3 (2‐way ANOVA, P > 0.05 for comparison between conditions).
Finally, we evoked the first spike with an artificial asynchronous EPSP and assayed post‐spike excitability with a synchronous EPSP at different ISIs (Fig. 4 B). Post‐spike excitability at 20 ms and 50 ms ISI was 0.56 ± 0.17 and 0.93 ± 0.05 respectively. This is significantly higher than the experiment in Fig. 4 A when no asynchronous release was present (P < 0.05 for spike probability at 20 and 50 ms between conditions, 2‐way ANOVA with Bonferroni's test, n = 6, Fig. 4 B). This illustrates that asynchronous EPSPs were much more effective than synchronous EPSPs at evoking spiking at short ISIs and shows that the depolarization from asynchronous release is sufficient to overcome the AHP and enhance excitability at short ISIs.
Discussion
It has previously been demonstrated that asynchronous glutamate release can build during trains of presynaptic activity to modify both spike jitter (Evstratova et al. 2014) and postsynaptic excitability (Kombian et al. 2000; Iremonger & Bains, 2007; Peters et al. 2010; Yang & Xu‐Friedman, 2010). We now show that asynchronous release can also have a pronounced effect on excitability over shorter time intervals. Specifically, in response to a single presynaptic spike, fast synchronous glutamate release can quickly initiate a postsynaptic spike. The subsequent slower asynchronous glutamate release, however, continues to depolarize the neurons during the period of the post‐spike AHP. This depolarization overcomes the inhibitory action of the AHP and effectively shortens the relative refractory period. These data demonstrate that synaptic release dynamics are an important factor in determining how neurons translate presynaptic activity into postsynaptic spiking.
Previous work has demonstrated that synaptic input can shorten the interspike interval, with the effect dependent on the timing of input relative to the AHP (Reyes & Fetz, 1993; Bennett & Wilson, 1998). Here we present a similar scenario in MNCs, and extend these observations to show that the mode of transmitter release that initially evokes the first spike determines the subsequent post‐spike refractory period. Other studies have demonstrated that single quantal events can effectively regulate spike probability in neurons with high membrane resistances (Carter & Regehr, 2002). Indeed, asynchronous release events are also quantal release events, whose frequency of release decays exponentially after the peak period of synchronous transmitter release. In neurons such as hypothalamic MNCs, which have high membrane resistances, these quantal events can produce large depolarizations that summate efficiently. Therefore, even though asynchronous release after a single spike lasts on average only 100 ms, the net effect on excitability lasts much longer due to the long membrane time constant and temporal summation of asynchronous postsynaptic potentials. The fact that the time course of the asynchronous EPSP is similar to that of the AHP is particularly relevant. This temporal matching means that the magnitude of synaptic drive is on average larger during the peak as compared to the tail of the AHP. While we show that MNCs both have a large AHP and are targeted by synapses that exhibit prominent asynchronous release, we found no evidence to suggest that the magnitude of the AHP and asynchronous release are correlated within this cell population. Whether there is any relationship between the amount of asynchronous release and postsynaptic cell properties, such as the AHP, in neurons in other parts of the brain is currently unknown.
AHPs have been shown to promote rhythmic spiking and enhance temporal precision during spike trains (Berry & Meister, 1998; Bennett et al. 2000; Wolfart et al. 2001; Hallworth et al. 2003; Vervaeke et al. 2006). Indeed, an alternative interpretation of our data is that in response to a single presynaptic stimulus, the post‐spike AHP prevents asynchronous release from evoking repetitive spiking, thus improving temporal precision. It is only when a second stimulus coincides with asynchronous release that spike probability is greatly enhanced. Consistent with this, it has been shown in other neurons that inhibition of potassium conductances which underlie the AHP can enhance the number of delayed spikes evoked by asynchronous release without the need for additional synaptic inputs (Yang & Xu‐Friedman, 2010).
Based on the duration of the AHP in MNCs (approximately 100–150 ms in this study), we predict that this would encourage rhythmic spiking at approximately 10 Hz in the presence of a sustained excitatory drive. Indeed, vasopressin MNCs commonly fire at approximately 6–10 Hz during the plateau phase of long duration bursts recorded in vivo (Sabatier et al. 2004). Interestingly, these phasic bursts are often associated with much higher frequency firing at burst onset, with average frequencies around 20 Hz, and instantaneous spike frequency of 30 Hz or higher (Brown & Leng, 2000; Sabatier et al. 2004). As glutamate synaptic inputs are known to be essential for both the initiation and maintenance of burst firing in vivo (Nissen et al. 1995; Brown et al. 2004), we speculate that asynchronous EPSPs may be particularly effective at overcoming AHPs and initiating these higher frequency spike discharges. These higher frequency spike discharges at burst onset efficiently recruit depolarizing after‐potentials (Andrew & Dudek, 1983, 1984 a; Ghamari‐Langroudi & Bourque, 1998) that together with ongoing excitatory synaptic drive sustain spiking during the plateau phase of the burst. Analysis of post‐spike excitability profiles of spontaneously bursting vasopressin neurons in vivo and in vitro supports this idea. Specifically, Sabatier et al. (2004) have demonstrated that vasopressin neurons in vivo have a much higher post‐spike excitability along with a shorter post‐spike refractory period when compared to vasopressin neurons recorded in acute brain slices in vitro. This shorter refractory period observed in vivo is likely to be due to the enhanced level of synaptic drive compared to the acute brain slice (Bourque & Renaud, 1991; Sabatier et al. 2004). Indeed, our current results suggest that higher levels of asynchronous synaptic drive may be responsible for this shortening of the relative refractory period in vivo and enhancing post‐spike excitability. As asynchronous glutamate release and fast AHP amplitude are equivalent between vasopressin and oxytocin neurons (Armstrong et al. 1994; Stern & Armstrong, 1996; Iremonger & Bains, 2007), this mechanism may also be important in facilitating oxytocin neuron burst firing (Wakerley & Lincoln, 1973).
While there is evidence that the molecular machinery for synchronous and asynchronous release may be distinct (Yao et al. 2011; Raingo et al. 2012; Bacaj et al. 2013; Evstratova et al. 2014), other studies suggest that these two modes of release may compete for the same pool of presynaptic vesicles (Hagler & Goda, 2001; David & Barrett, 2003; Otsu et al. 2004; Yang & Xu‐Friedman, 2010). At most central synapses, fast synchronous release predominates (Barrett & Stevens, 1972; Goda & Stevens, 1994; Atluri & Regehr, 1998); however, at the majority of glutamate synapses onto MNCS, approximately half of the charge transfer comes from asynchronous release (Iremonger & Bains, 2007) indicating that these synapses rely on both forms of neurotransmission. If a synapse were to solely use asynchronous glutamate release, its ability to trigger fast, synchronous spikes would be predicted to be lower than a synchronous synapse. One advantage of asynchronous release is that it builds during sustained presynaptic activity, allowing for longer‐lasting elevations in excitability (Lau & Bi, 2005; Iremonger & Bains, 2007). In contrast, synapses operating with purely synchronous release may be efficient at evoking precisely timed single spikes within a short temporal window. However, their ability to summate inputs over longer time windows, including during the AHP, are poor. Indeed, our experiments demonstrate that synchronous depolarizations that trigger an action potential are immediately followed by a relative refractory period that further limits integration of subsequent synaptic inputs. We speculate that by receiving glutamate inputs that possess both synchronous and asynchronous forms of release, MNCs are able to integrate synaptic inputs over variable temporal windows. This is likely to allow MNCs to efficiently integrate afferent information that is temporally dispersed, allowing them to efficiently transform synaptic inputs into a hormonal output signal. Releasing in both a synchronous and asynchronous manner does, however, represent an increased energy cost, since the overall amount of neurotransmitter that is released is increased.
It is also important to consider that MNCs integrate paracrine and humoral signals simultaneously with synaptic inputs. These non‐synaptic signals include local neuromodulators and neuropeptides (Price et al. 2008; Price et al. 2009) as well as osmosensitive signals acting through TRPV1 ion channels (Sharif Naeini et al. 2006). These slow graded potentials would not be expected to modify post‐spike excitability on short time scales as shown for asynchronous release. However, these signals may instead function to increase spike frequency in a slow graded manner as opposed to triggering bursts of spiking.
Previous work has suggested that asynchronous release contributes to MNCs functioning as integrators of synaptic inputs (Iremonger & Bains, 2007). This current work extends this idea by demonstrating that asynchronous release also allows MNCs to integrate synaptic inputs that occur during the AHP. If not for asynchronous release, small excitatory synaptic inputs that coincided with the AHP would be insufficient to drive the neuron to fire a subsequent spike. Together, these data suggest a new mechanism by which asynchronous glutamate release can modify post‐spike excitability and the relative refractory period.
Additional information
Competing interests
None declared.
Author contributions
KJI and JSB designed the project, KJI performed the experiments and data analysis, KJI and JSB wrote the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by an operating grant to J.S.B. from The Canadian Institute for Health Research. K.I. is currently a Sir Charles Hercus Health Research Fellow.
Acknowledgements
The authors would like to thank Allan Herbison and Colin Brown for comments on the manuscript.
References
- Andrew RD & Dudek FE (1983). Burst discharge in mammalian neuroendocrine cells involves an intrinsic regenerative mechanism. Science 221, 1050–1052. [DOI] [PubMed] [Google Scholar]
- Andrew RD & Dudek FE (1984. a). Analysis of intracellularly recorded phasic bursting by mammalian neuroendocrine cells. J Neurophysiol 51, 552–566. [DOI] [PubMed] [Google Scholar]
- Andrew RD & Dudek FE (1984. b). Intrinsic inhibition in magnocellular neuroendocrine cells of rat hypothalamus. J Physiol 353, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong WE, Smith BN & Tian M (1994). Electrophysiological characteristics of immunochemically identified rat oxytocin and vasopressin neurones in vitro . J Physiol 475, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP & Regehr WG (1998). Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci 18, 8214–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC & Sudhof TC (2013). Synaptotagmin‐1 and synaptotagmin‐7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF & Stevens CF (1972). The kinetics of transmitter release at the frog neuromuscular junction. J Physiol 227, 691–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC & Wilson CJ (2000). Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci 20, 8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD & Wilson CJ (1998). Synaptic regulation of action potential timing in neostriatal cholinergic interneurons. J Neurosci 18, 8539–8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MJ 2nd & Meister M (1998). Refractoriness and neural precision. J Neurosci 18, 2200–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW, Randle JC & Renaud LP (1985). Calcium‐dependent potassium conductance in rat supraoptic nucleus neurosecretory neurons. J Neurophysiol 54, 1375–1382. [DOI] [PubMed] [Google Scholar]
- Bourque CW & Renaud LP (1991). Membrane properties of rat magnocellular neuroendocrine cells in vivo. Brain Res 540, 349–352. [DOI] [PubMed] [Google Scholar]
- Brown CH, Bull PM & Bourque CW (2004). Phasic bursts in rat magnocellular neurosecretory cells are not intrinsically regenerative in vivo. Eur J Neurosci 19, 2977–2983. [DOI] [PubMed] [Google Scholar]
- Brown CH & Leng G (2000). In vivo modulation of post‐spike excitability in vasopressin cells by kappa‐opioid receptor activation. J Neuroendocrinol 12, 711–714. [DOI] [PubMed] [Google Scholar]
- Carter AG & Regehr WG (2002). Quantal events shape cerebellar interneuron firing. Nat Neurosci 5, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Coombs JS, Eccles JC & Fatt P (1955). The electrical properties of the motoneurone membrane. J Physiol 130, 291–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G & Barrett EF (2003). Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol 548, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evstratova A, Chamberland S, Faundez V & Toth K (2014). Vesicles derived via AP‐3‐dependent recycling contribute to asynchronous release and influence information transfer. Nat Commun 5, 5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari‐Langroudi M & Bourque CW (1998). Caesium blocks depolarizing after‐potentials and phasic firing in rat supraoptic neurones. J Physiol 510, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y & Stevens CF (1994). Two components of transmitter release at a central synapse. Proc Natl Acad Sci USA 91, 12942–12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greffrath W, Magerl W, Disque‐Kaiser U, Martin E, Reuss S & Boehmer G (2004). Contribution of Ca2+‐activated K+ channels to hyperpolarizing after‐potentials and discharge pattern in rat supraoptic neurones. J Neuroendocrinol 16, 577–588. [DOI] [PubMed] [Google Scholar]
- Hagler DJ Jr & Goda Y (2001). Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol 85, 2324–2334. [DOI] [PubMed] [Google Scholar]
- Hallworth NE, Wilson CJ & Bevan MD (2003). Apamin‐sensitive small conductance calcium‐activated potassium channels, through their selective coupling to voltage‐gated calcium channels, are critical determinants of the precision, pace, and pattern of action potential generation in rat subthalamic nucleus neurons in vitro. J Neurosci 23, 7525–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GW, Manabe Y & Ruf KB (1969). A study of the parameters of electrical stimulation of unmyelinated fibres in the pituitary stalk. J Physiol 203, 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S & Jonas P (2005). Asynchronous GABA release generates long‐lasting inhibition at a hippocampal interneuron‐principal neuron synapse. Nat Neurosci 8, 1319–1328. [DOI] [PubMed] [Google Scholar]
- Iremonger KJ & Bains JS (2007). Integration of asynchronously released quanta prolongs the postsynaptic spike window. J Neurosci 27, 6684–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iremonger KJ & Bains JS (2009). Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. J Neurosci 29, 7349–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel JM, Le MG, Theodosis DT & Poulain DA (2003). Glutamatergic input governs periodicity and synchronization of bursting activity in oxytocin neurons in hypothalamic organotypic cultures. Eur J Neurosci 17, 2619–2629. [DOI] [PubMed] [Google Scholar]
- Israel JM, Poulain DA & Oliet SH (2010). Glutamatergic inputs contribute to phasic activity in vasopressin neurons. J Neurosci 30, 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Israel JM, Dupouy B, Oliet SH, Allard M, Vitiello S, Theodosis DT & Poulain DA (1998). Evidence for a hypothalamic oxytocin‐sensitive pattern‐generating network governing oxytocin neurons in vitro. J Neurosci 18, 6641–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS & Regehr WG (2014). Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76, 333–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick K & Bourque CW (1991). Dual role for calcium in the control of spike duration in rat supraoptic neuroendocrine cells. Neurosci Lett 133, 271–274. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Hirasawa M, Mouginot D, Chen X & Pittman QJ (2000). Short‐term potentiation of miniature excitatory synaptic currents causes excitation of supraoptic neurons. J Neurophysiol 83, 2542–2553. [DOI] [PubMed] [Google Scholar]
- Lau PM & Bi GQ (2005). Synaptic mechanisms of persistent reverberatory activity in neuronal networks. Proc Natl Acad Sci USA 102, 10333–10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (1997). Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20, 38–43. [DOI] [PubMed] [Google Scholar]
- Lu T & Trussell LO (2000). Inhibitory transmission mediated by asynchronous transmitter release. Neuron 26, 683–694. [DOI] [PubMed] [Google Scholar]
- Ludwig M & Leng G (2006). Dendritic peptide release and peptide‐dependent behaviours. Nat Rev Neurosci 7, 126–136. [DOI] [PubMed] [Google Scholar]
- Luther JA & Tasker JG (2000). Voltage‐gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol 523, 193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen R, Hu B & Renaud LP (1995). Regulation of spontaneous phasic firing of rat supraoptic vasopressin neurones in vivo by glutamate receptors. J Physiol 484, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Shahrezaei V, Li B, Raymond LA, Delaney KR & Murphy TH (2004). Competition between phasic and asynchronous release for recovered synaptic vesicles at developing hippocampal autaptic synapses. J Neurosci 24, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM & Andresen MC (2010). Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron 65, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Hoyda TD, Samson WK & Ferguson AV (2008). Nesfatin‐1 influences the excitability of paraventricular nucleus neurones. J Neuroendocrinol 20, 245–250. [DOI] [PubMed] [Google Scholar]
- Price CJ, Samson WK & Ferguson AV (2009). Neuropeptide W has cell phenotype‐specific effects on the excitability of different subpopulations of paraventricular nucleus neurones. J Neuroendocrinol 21, 850–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J, Khvotchev M, Liu P, Darios F, Li YC, Ramirez DM, Adachi M, Lemieux P, Toth K, Davletov B & Kavalali ET (2012). VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci 15, 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Carey MR & Best AR (2009). Activity‐dependent regulation of synapses by retrograde messengers. Neuron 63, 154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes AD & Fetz EE (1993). Two modes of interspike interval shortening by brief transient depolarizations in cat neocortical neurons. J Neurophysiol 69, 1661–1672. [DOI] [PubMed] [Google Scholar]
- Sabatier N, Brown CH, Ludwig M & Leng G (2004). Phasic spike patterning in rat supraoptic neurones in vivo and in vitro . J Physiol 558, 161–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P (1996). Ca2+‐activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci 19, 150–154. [DOI] [PubMed] [Google Scholar]
- Savic N, Pedarzani P & Sciancalepore M (2001). Medium afterhyperpolarization and firing pattern modulation in interneurons of stratum radiatum in the CA3 hippocampal region. J Neurophysiol 85, 1986–1997. [DOI] [PubMed] [Google Scholar]
- Sharif Naeini R, Witty MF, Seguela P & Bourque CW (2006). An N‐terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci 9, 93–98. [DOI] [PubMed] [Google Scholar]
- Stern JE & Armstrong WE (1996). Changes in the electrical properties of supraoptic nucleus oxytocin and vasopressin neurons during lactation. J Neurosci 16, 4861–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel E & Bourque CW (2010). Central clock excites vasopressin neurons by waking osmosensory afferents during late sleep. Nat Neurosci 13, 467–474. [DOI] [PubMed] [Google Scholar]
- Ventura RR, Aguiar JF, Antunes‐Rodrigues J & Varanda WA (2008). Nitric oxide modulates the firing rate of the rat supraoptic magnocellular neurons. Neuroscience 155, 359–365. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Hu H, Graham LJ & Storm JF (2006). Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron 49, 257–270. [DOI] [PubMed] [Google Scholar]
- Wakerley JB & Lincoln DW (1973). The milk‐ejection reflex of the rat: a 20‐ to 40‐fold acceleration in the firing of paraventricular neurones during oxytocin release. J Endocrinol 57, 477–493. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Neuhoff H, Franz O & Roeper J (2001). Differential expression of the small‐conductance, calcium‐activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci 21, 3443–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H & Xu‐Friedman MA (2010). Developmental mechanisms for suppressing the effects of delayed release at the endbulb of Held. J Neurosci 30, 11466–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE & Chapman ER (2011). Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]