

Key points
In the dorsal raphe nucleus, it is known that serotonin release activates metabotropic 5‐HT1A autoreceptors located on serotonin neurons that leads to an inhibition of firing through the activation of G‐protein‐coupled inwardly rectifying potassium channels.
We found that in mouse brain slices evoked serotonin release produced a 5‐HT1A receptor‐mediated inhibitory postsynaptic current (IPSC) that resulted in only a transient pause in firing.
While spillover activation of receptors contributed to evoked IPSCs, serotonin reuptake transporters prevented pooling of serotonin in the extrasynaptic space from activating 5‐HT1A‐IPSCs.
As a result, the decay of 5‐HT1A‐IPSCs was independent of the intensity of stimulation or the probability of transmitter release.
These results indicate that evoked serotonin transmission in the dorsal raphe nucleus mediated by metabotropic 5‐HT1A autoreceptors may occur via point‐to‐point synapses rather than by paracrine mechanisms.
Abstract
In the dorsal raphe nucleus (DRN), feedback activation by Gαi/o‐coupled 5‐HT1A autoreceptors reduces the excitability of serotoninergic neurons, which decreases serotonin release both locally within the DRN and in projection regions. Serotonin transmission within the DRN is thought to occur via transmitter spillover and paracrine activation of extrasynaptic receptors. Here, we tested the volume transmission hypothesis in mouse DRN brain slices by recording 5‐HT1A receptor‐mediated inhibitory postsynaptic currents (5‐HT1A‐IPSCs) generated by the activation of G‐protein‐coupled inwardly rectifying potassium channels (GIRKs). We found that in the DRN of ePET1‐EYFP mice, which selectively express enhanced yellow fluorescent protein in serontonergic neurons, the local release of serotonin generated 5‐HT1A‐IPSCs in serotonin neurons that rose and fell within a second. The transient activation of 5‐HT1A autoreceptors resulted in brief pauses in neuron firing that did not alter the overall firing rate. The duration of 5‐HT1A‐IPSCs was primarily shaped by receptor deactivation due to clearance via serotonin reuptake transporters. Slowing diffusion with dextran prolonged the rise and reduced the amplitude the IPSCs and the effects were potentiated when uptake was inhibited. By examining the decay kinetics of IPSCs, we found that while spillover may allow for the activation of extrasynaptic receptors, efficient uptake by serotonin reuptake transporters (SERTs) prevented the pooling of serotonin from prolonging the duration of transmission when multiple inputs were active. Together the results suggest that the activation of 5‐HT1A receptors in the DRN results from the local release of serotonin rather than the extended diffusion throughout the extracellular space.
Key points
In the dorsal raphe nucleus, it is known that serotonin release activates metabotropic 5‐HT1A autoreceptors located on serotonin neurons that leads to an inhibition of firing through the activation of G‐protein‐coupled inwardly rectifying potassium channels.
We found that in mouse brain slices evoked serotonin release produced a 5‐HT1A receptor‐mediated inhibitory postsynaptic current (IPSC) that resulted in only a transient pause in firing.
While spillover activation of receptors contributed to evoked IPSCs, serotonin reuptake transporters prevented pooling of serotonin in the extrasynaptic space from activating 5‐HT1A‐IPSCs.
As a result, the decay of 5‐HT1A‐IPSCs was independent of the intensity of stimulation or the probability of transmitter release.
These results indicate that evoked serotonin transmission in the dorsal raphe nucleus mediated by metabotropic 5‐HT1A autoreceptors may occur via point‐to‐point synapses rather than by paracrine mechanisms.
Abbreviations
- 5‐HT
serotonin
- DRN
dorsal raphe nucleus
- GIRK
G‐protein coupled, inwardly rectifying potassium channel
- IPSC
inhibitory postsynaptic current
- SERT
serotonin reuptake transporter
- sIPSC
spontaneous inhibitory postsynaptic current
Introduction
The dorsal raphe nucleus (DRN) is a major source of ascending serotonergic innervation to the forebrain and limbic regions (Jacobs & Azmitia, 1992; Michelsen et al. 2008). Serotonin neurons of the DRN are implicated in multiple behavioural and cognitive functions (Jacobs & Azmitia, 1992; Lucki, 1998), and dysfunction in serotonin signalling is thought to underlie mood disorders and depression (Michelsen et al. 2008). In addition to release in projection regions, vesicular serotonin (5‐hydroxytryptamine; 5‐HT) release occurs locally in the DRN at somatic (Kaushalya et al. 2008; Colgan et al. 2009), dendritic (de Kock et al. 2006; Colgan et al. 2012) and axonal sites (Bruns et al. 2000) where it modulates the activity of DRN neurons (Pineyro & Blier, 1999; Adell et al. 2002; Michelsen et al. 2008; Andrade et al. 2015). Release of serotonin can activate inhibitory Gαi/o‐coupled 5‐HT1A autoreceptors that inhibit serotonergic neuron impulse activity through the opening of inwardly rectifying potassium (GIRK) channels (Aghajanian & Lakoski, 1984; De Vivo & Maayani, 1986; Pan & Williams, 1989; Pan et al. 1989; Bayliss et al. 1997; Katayama et al. 1997; Gantz et al. 2015) and inhibition of voltage‐dependent Ca2+ channels (Penington & Kelly, 1990; Penington et al. 1991). By suppressing pacemaker firing, 5‐HT1A autoreceptors regulate serotonin levels both locally in the dorsal raphe and in terminal projection regions (Aghajanian & Lakoski, 1984; Pan & Williams, 1989; Pan et al. 1989; Hjorth & Sharp, 1991; Portas et al. 1996; Adell et al. 2002; Michelsen et al. 2008), thereby influencing behaviours such as anxiety and stress (Richardson‐Jones et al. 2010).
Ultrastructural studies have found somatodendritic 5‐HT1A receptors at both synaptic (Kia et al. 1996) and extrasynaptic sites (Kia et al. 1996; Riad et al. 2000). As 5‐HT1A receptors can be located at extrasynaptic sites, it has been thought that serotonergic transmission through these receptors in the DRN occurs by their paracrine activation by low concentrations of transmitter that result from spillover of transmitter out from the synapse (Bunin & Wightman, 1999). In support of this extended form of spillover, known as volume transmission, electrochemical studies have found that evoked release in the DRN drives submicromolar increases in the concentration of 5‐HT throughout the extracellular space (Bunin & Wightman, 1998; Bunin et al. 1998; Jennings, 2013). While the increase in extracellular serotonin has been hypothesized to be sufficient to activate 5‐HT1A autoreceptors up to several micrometres away from release sites (Bunin & Wightman, 1999), the role of volume transmission in gating activation of these autoreceptors in the DRN has yet to be directly addressed.
While 5‐HT1A‐receptors in the DRN in vivo are thought to tonically modulate firing rates (Fornal et al. 1996; Mundey et al. 1996; Hajos et al. 2001; Haddjeri et al. 2004), electrophysiological studies using brain slices have found in contrast that stimulation evokes only 5‐HT1A receptor‐mediated hyperpolarizations in DRN neurons that cause only brief pauses in neuron firing (Williams et al. 1988; Pan et al. 1989; Morikawa et al. 2000; Levitt et al. 2013). To examine the mechanisms that underlie the synaptic activation of 5‐HT1A receptors, we recorded from identified serotonergic neurons in the dorsal raphe using ePET1‐eYFP mice. Through recording of 5‐HT1A receptor‐mediated IPSCs, we found that evoked serotonin release generated 5‐HT1A receptor‐mediated currents that rose and fell in a second. This resulted in transient pauses in firing without affecting overall firing rates. While spillover is likely to have allowed for the activation of some extrasynaptic receptors, serotonin reuptake transporters prevented transmitter pooling from adjacent release sites from extending the duration of transmission. These findings suggest that serotonergic transmission in the DRN occurs by the local activation of 5‐HT1A autoreceptors near the sites of transmitter release with limited crosstalk between synapses.
Methods
Ethical approval
All procedures were performed in accordance with United States law on the use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University, Cleveland, OH, USA.
Slice preparation and visualization
Mice were deeply anaesthetized with isoflurane and coronal brain slices (220 μm) containing the dorsal raphe nuclei were obtained from 4‐ to 8‐week‐old male and female ePET1‐eYFP mice (Scott et al. 2005). ePET1‐eYFP mice were donated by Dr Evan Deneris at Case Western Reserve University. Brain slices were cut using a vibrating blade microtome (Leica) in ice‐cold cutting solution containing (in mm): 75 NaCl, 2.5 KCl, 6 MgCl2, 0.1 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, 2.5 d‐glucose and 50 sucrose, continuously bubbled with 95% O2 and 5% CO2. After cutting, slices were transferred to artificial CSF (ACSF) containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 d‐glucose bubbled with 95% O2 and 5% CO2 and incubated at 35°C for at least 45 min prior to use. MK‐801 (10 μm) was included during the incubation to block NMDA receptors. After incubation, slices were transferred to a recording chamber and constantly perfused at 2 ml min−1 with oxygenated ACSF warmed to 34 ± 2°C. Slices were visualized with a BX51WI microscope (Olympus) with custom‐built infrared gradient contrast optics. Enhanced yellow fluorescent protein (eYFP) was visualized by fluorescence using a custom built cyan LED (505 nm, Luxeon Star, Brantford, Ontario, Canada) with a 515 nm long pass dicroic mirror and a 535/30 m emission filter (Chroma).
Immunohistochemistry
Eight‐week‐old ePet‐EYFP mice were deeply anaesthetized with avertin (44 mm tribromoethanol, 2.5% tert‐amyl alcohol), 20 ml kg−1. Mice were transcardially perfused with ice‐cold phosphate‐buffered saline (PBS; 137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.4), followed by a 20 min perfusion with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS. Brains were post‐fixed for 2 h in 4% paraformaldehyde. Fixed brains were sectioned coronally in 20 μm sections using a freezing microtome (American Optical Corp.) and mounted on Superfrost plus slides (Fisher), then air‐dried. Sections were permeabilized with 0.3% Triton X‐100 in PBS (PBS‐T) for 15 min and blocked in 5% normal goat serum for 1 h at room temperature. Slides were incubated with 1:1000 rabbit anti‐GFP antibodies (Invitrogen) in PBS‐T overnight at 4°C. Slides were incubated with 1:500 goat anti‐rabbit alexa fluor 488 (Invitrogen) in PBS‐T for 2 h at room temperature. Slides were coverslipped using prolong gold (Invitrogen). Fluorescent images were obtained using an Olympus BX51 compound microscope (Olympus) with a ×10 UPlanFl, NA 0.3 lens with a SPOT RT colour digital camera (Diagnostic Instruments, Sterling Heights, MI, USA). All images were processed using ImageJ.
Electrophysiology
Whole‐cell voltage‐clamp recordings (V h = −60 mV) were made using an Axopatch 200B amplifier (Molecular Devices). Patch pipettes (1.5–2.5 MΩ) were pulled from borosilicate glass (World Precision Instruments). The pipette intracellular solution contained (in mm): 115 potassium methylsulphate, 20 NaCl, 1.5 MgCl2, 10 K‐Hepes, 10 BAPTA‐tetrapotassium, 2 ATP, 0.3 GTP, and 6 sodium phosphocreatine, pH 7.4, 275 mosmol l−1. Data were acquired using an ITC‐18 interface (Instrutech) and Axograph X (Axograph Scientific) at 10 kHz and filtered to 2 kHz. Series resistance was not compensated and recordings were discarded if the access resistance rose above 15 MΩ. In the case of experiments with reduced extracellular calcium, MgCl2 was substituted to maintain a constant divalent ion concentration. All drugs were applied by bath perfusion. Recordings were made in the dorsomedial portion of the DRN. Serotonergic neurons within the DRN were fluorescently identified by eYFP expression driven by the PET1 enhancer (Scott et al. 2005). In some experiments, dextran (M r 35,000–50,000) was used to slow diffusion via macromolecular overcrowding. Dextran was added to ACSF (5% weight by volume) and bath perfused after achieving stable recordings. Bath temperature was monitored to ensure that it did not change by more than 1°C.
Cell attached recordings were made with internal solution containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4. Data were acquired in voltage‐clamp (0 mV) at 10 kHz and filtered to 5 kHz. Seals between the recording electrode and neuron ranged from 5 to 12 MΩ. Phenylephrine (3 μm) was included in the bath solution to mimic in vivo excitatory drive (Vandermaelen & Aghajanian, 1983).
Stimulation and iontophoresis
Serotonin release was evoked with an extracellular, ACSF‐filled monopolar glass electrode. The stimulating electrode was placed 50–100 μm away from the cell body of the recorded neuron. A single stimulation pulse (20–60 μA, 0.5 ms) was used to drive serotonin release. To prevent glutamate‐ and GABA‐mediated synaptic responses, recordings were made in the presence of 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX; 10 μm), MK801 (10 μm), picrotoxin (100 μm) and CGP55845 (200 nm). BAPTA (10 mm) in the internal pipette solution was used to prevent intracellular calcium signalling. In traces illustrating electric stimulation, the stimulation artifact was blanked for presentation. For the iontophoretic application, serotonin (100 mm) was ejected as a cation (160 nA) for 2000 ms using an Iontophoresis Generator (Dagan) from thin‐walled iontophoretic electrodes placed ∼ 30 μm from the cell. A negative retention current of 15–30 nA was applied to prevent leakage of serotonin.
Materials
Picrotoxin was from Abcam. DNQX, MK‐801, CGP 55845, SB 216641 hydrochloride, WAY 100635 maleate, citalopram hydrobromide, R‐(−)‐phenylephrine hydrochloride, forskolin, 8‐cyclopentyl‐1,3‐dipropylxanthine (DPCPX), and serotonin hydrochloride were from Tocris Bioscience. Potassium methylsulphate was from Acros Organics. BAPTA was from Invitrogen. d‐Glucose and HEPES sodium salt were from Sigma‐Aldrich. Dextran (M r 35,000–50,000) was from MP Biomedicals. All other chemicals were from Fisher Scientific.
Statistics and analysis
All data are shown as the mean ± SEM. Statistical significance (P < 0.05) was assessed by paired and unpaired Student's t tests, ANOVAs, and Pearson's correlations as noted (InStat 3.0). Decay kinetics of 5‐HT1A‐IPSCs were fitted with a single exponential using a Simplex algorithm optimized by the sum of squared errors in Axograph X (Axograph Scientific). Firing rates in cell‐attached experiments were calculated from the average inter‐spike intervals.
Results
Evoked 5‐HT1A‐IPSCs mediate a transient pause in DRN serotonergic neuron firing
Voltage clamp recordings were made from eYFP+ serotonergic neurons located in the dorsomedial DRN in brain slices obtained from ePET1‐eYFP mice (Fig. 1 A). A single stimulation was used to elicit 5‐HT1A receptor‐mediated inhibitory postsynaptic currents (IPSCs). In all neurons tested, IPSCs were abolished by the 5‐HT1A receptor antagonist WAY 100635 (200 nm, 97 ± 1% reduction in amplitude, n = 5, P < 0.01, Student's paired t test; Fig. 1 B), and IPSCs were reliably observed in > 95% of serotonin neurons (30/31 serotonergic neurons; 85 ± 10 pA, n = 30, Fig. 1 B). IPSCs were eliminated by TTX (200 nm, 98 ± 1% reduction, n = 3, P < 0.001, Student's paired t test), calcium‐free ACSF (n = 3, 97 ± 2% reduction, P < 0.001, Student's paired t test), and barium (200 μm, 81 ± 1% reduction, n = 4, P < 0.01, Student's paired t test)(Fig. 1 C). Thus, recorded IPSCs resulted from vesicular serotonin release acting on 5‐HT1A receptors to mediate a potassium conductance, likely to be via G‐protein‐coupled, inwardly rectifying potassium (GIRK) channels (Pan et al. 1989). Evoked IPSCs rose and fell within approximately 1 s. IPSCs activated in 168 ± 7 ms (10–90% rise time, n = 30) following a lag of 75 ± 3 ms (10% onset, n = 30), and decayed with a time constant of 0.44 ± 0.3 s (n = 30). This resulted in an overall duration of 0.58 ± 0.4 s measured at half‐maximal amplitude (n = 30, Fig. 1 D).
Figure 1. Electrical stimulation evokes 5‐HT1A‐IPSCs that drive pauses in DRN serotonin neuron firing .
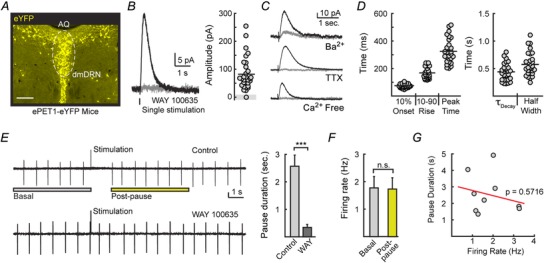
A, eYFP, driven by the PET1 enhancer, selectively labels serotonin neurons. Shown is eYFP immunoreactivity in a coronal brain slice obtained from ePET1‐eYFP mice containing the dorsal raphe nucleus (DRN). AQ, cerebral aqueduct; dmDRN, dorsomedial dorsal raphe nucleus. Scale bar represents 200 μm. B, left, averaged 5‐HT1A‐IPSCs from a dorsal raphe neuron evoked by a single stimulation in control conditions (black) and in the presence of the 5‐HT1A antagonist WAY 100635 (200 nm; grey). Whole‐cell recordings were voltage‐clamped to −60 mV. Right, IPSCs could be evoked in 30/31 DRN serotoninergic neurons examined. C, average traces of IPSCs recorded in 200 nm TTX, calcium‐free ASCF, and 200 μm barium. D, quantification of the rise and decay kinetics of evoked IPSCs. E, cell‐attached recordings made in 3 μm phenylephrine to mimic in vivo excitatory drive. Representative traces (left) and quantification (right) demonstrating that evoked serotonin release drives a pause in firing that is absent in the 5‐HT1A‐receptor antagonist WAY 100635 (200 nm). Vertical scale bar represents 25 pA. F, firing rates measured over 5 s before stimulation (basal) and over 5 s after firing had resumed (post‐pause) were identical. G, Pearson correlation demonstrating that the duration of the evoked pause was independent of the baseline firing rates (n = 9).
To examine the consequence of evoked serotonin release on excitability, cell‐attached recordings of DRN firing rates were made from serotonin neurons. In vivo, DRN neurons fire in a regular pattern at 0.5–3 Hz. To mimic the excitatory noradrenergic tone that facilitates firing (Baraban & Aghajanian, 1981; Haddjeri et al. 2004), phenylephrine (3 μm) was added to the bath to maintain regular pacemaker firing (Vandermaelen & Aghajanian, 1983). Single stimulation evoked serotonin release induced a 2.56 ± 0.41 s pause in firing (n = 10) that was blocked by the 5‐HT1A receptor antagonist WAY 100635 (200 nm; n = 5; Fig. 1 E). Evoked serotonin release did not cause a lasting depression of firing, as basal and post‐pause firing rates were similar (n = 5; P = 0.33; Fig. 1 E and F). Baseline‐firing rates did not correlate with the duration of the evoked pause (Fig. 1 G). Thus, serotonin release is capable of affecting DRN activity by generating transient pauses in serotonin neuron firing.
Receptor deactivation and not receptor desensitization terminated evoked 5‐HT1A‐receptor signalling
At many synapses, receptor desensitization can speed the decay of receptor‐mediated currents to shorten the duration of signalling (Trussell et al. 1993). To determine whether desensitization of 5‐HT1A receptors regulates the duration of the IPSC, a paired‐pulse protocol with an interstimulus interval of 4 s was used (Fig. 2 A). Evoking IPSCs 4 s apart resulted in a 73 ± 4% reduction in the amplitude of the second IPSC (n = 7; P < 0.001). The paired‐pulse depression was partially attenuated in the presence of the 5‐HT1B antagonist, SB 216641 (1 μm; 58 ± 8% reduction in amplitude of the 2nd IPSC; n = 4; P = 0.02 vs. 1st IPSC; P = 0.04 vs. reduction in control; Fig. 2 C), confirming that presynaptic activation by 5‐HT1B receptors by the first stimulation inhibited the subsequent release of serotonin (Morikawa et al. 2000). SB 216641 (1 μm) had no effect on the amplitude of unpaired IPSCs (P = 0.78 amplitude, n = 14), indicating that presynaptic 5‐HT1B receptors were not tonically active in slices (Morikawa et al. 2000). To examine whether some depression was postsynaptic in origin, 5‐HT was exogenously applied using paired iontophoretic applications (Fig. 2 B). The amplitude and decay kinetics of the two resulting currents generated 4 s apart were similar (amplitude: 1st iontophoretic current, 113 ± 14 pA; 2nd iontophoretic current, 123 ± 16 pA; n = 5, P = 0.69; Fig. 2C; τDecay: 1st iontophoretic current, 0.57 ± 0.06 s; 2nd iontophoretic current, 0.61 ± 0.06 s; n = 5, P = 0.78), suggesting that the paired‐pulse depression of IPSCs primarily occurred due to presynaptic mechanisms. When serotonin was iontophoretically applied during the decay phase of the evoked 5‐HT1A‐IPSC (1 s following stimulation) (Fig. 2 D), the amplitude and kinetics of the resulting current were also identical to currents that were not preceded by an IPSC (amplitude: during IPSC, 120 ± 21 pA; without IPSC, 124 ± 21 pA; n = 5, P = 0.89; τDecay: during IPSC, 0.80 ± 0.22 s; without IPSC, 0.78 ± 0.21 s; n = 5, P = 0.96; Fig. 2 E). The fact that exogenously serotonin evokes similar currents when applied on its own or during the decay of IPSCs suggests that limited desensitization of 5‐HT1A receptors occurred during the decay phase of the IPSC. Although iontophoresis and stimulation may recruit different receptor pools, these results suggest that receptor deactivation not desensitization controls the duration of serotonin transmission in the raphe.
Figure 2. 5‐HT1A‐receptor desensitization does not determine the decay time of IPSCs .
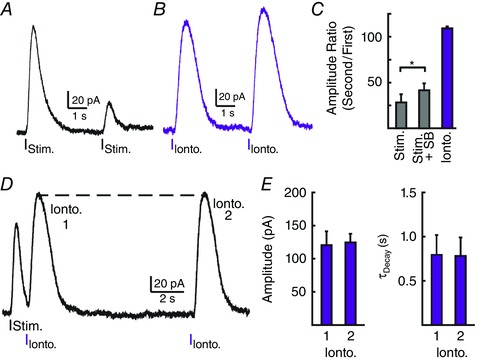
A, example trace demonstrating a paired‐pulse depression when stimulating at a 4 s interval. B, example trace demonstrating the lack of postsynaptic depression when serotonin was applied by iontophoresis twice at a 4 s interval (Ionto.). C, quantification of amplitude ratio of evoked IPSCs in the absence and presence of the 5‐HT1B presynaptic receptor antagonist SB 216641 (1 μm) or the current generated by the iontophoresis of serotonin at a 4 s interval. D, representative trace demonstrating the lack of receptor desensitization during evoked IPSCs. Serotonin was applied by iontophoresis during the decay of evoked IPSCs (1 s post‐stimulation; Ionto. 1) and without preceding IPSCs (12 s post‐stimulation; Ionto. 2). E, quantification demonstrating the similar amplitude (left) and time course (right) of currents generated by serotonin iontophoresis in D.
Clearance via serotonin reuptake transporters limited serotonin spillover and was the primary mechanism driving 5‐HT1A‐receptor deactivation
Clearance of monoamines in the extracellular space is achieved by a combination of diffusion and uptake by transporters. The relative contributions of each vary widely across monoamine synapses (Courtney & Ford, 2014). In the DRN, it has been hypothesized that due to the limited role of uptake, diffusion of serotonin may extend the duration of serotonin transmission by allowing activation of distal extrasynaptic 5‐HT1A receptors through spillover transmission (Bunin & Wightman, 1999; Jennings, 2013). To determine the role of serotonin reuptake transporters (SERTs) in regulating 5‐HT1A activation, the selective serotonin reuptake inhibitor (SSRI) citalopram (Celexa, 200 nm) was bath applied while recording IPSCs. Citalopram significantly reduced the amplitude of 5‐HT1A‐IPSCs (39 ± 8% inhibition, n = 5, P < 0.05; Fig. 3 A–C). This was likely due to citalopram‐induced tonic activity of presynaptic 5‐HT1B receptors (Morikawa et al. 2000), as combining the 5‐HT1B antagonist, SB216641 (1 μm), with citalopram instead led to a stable increase in IPSC amplitude (ctrl, 134 ± 16 pA; citalopram + SB216641, 210 ± 33 pA; n = 5, P = 0.03; Fig. 3 A–C). Bath application of citalopram (200 nm) in both the absence and presence of SB 216641 (1 μm) greatly prolonged the duration of 5‐HT1A‐IPSCs. Compared to controls, IPSCs recorded in citalopram had delayed peak times (citalopram, 134 ± 22% increase vs. controls, n = 5, P = 0.003; citalopram + SB, 189 ± 29% increase, n = 5, P = 0.047; P = 0.292 citalopram vs. citalopram + SB; Fig. 3 D) and were significantly slower to decay (τDecay: citalopram, 963 ± 120% increase vs. controls; n = 5, P = 0.001; citalopram + SB, 830 ± 250% increase; n = 5, P = 0.005; P = 0.6334 citalopram vs. citalopram + SB; Fig. 3 D). As a result, citalopram induced a 5‐fold increase IPSC duration (half‐width) with or without SB 216641 (P = 0.98), indicating that clearance of serotonin clearance by reuptake transporters limits the duration of transmission.
Figure 3. Serotonin reuptake transporters limit the duration of IPSCs .
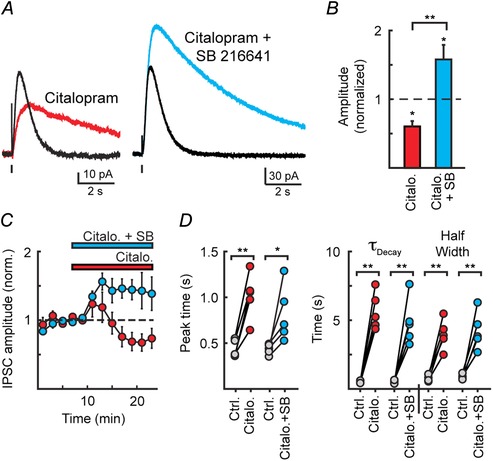
A, average 5‐HT1A‐IPSCs under control conditions and in the presence of citalopram (200 nm) and citalopram (200 nm) + SB216641 (1 μm), illustrating that blocking serotonin uptake increased the activation of 5‐HT1A receptors and led to the tonic activation of 5‐HT1B receptors. B, quantification of the change in amplitude of IPSCs induced by citalopram (200 nm) or citalopram (200 nm) + SB216641 (1 μm). C, time course of the relative change in IPSC amplitude quantified in B. D, quantification of the change in kinetics in either citalopram or citalopram + SB 216641. In both cases, inhibiting reuptake transporters prolongs the duration of 5‐HT1A‐IPSCs.
To examine the role of diffusion in receptor activation dextran (5%, 40 kDa) was applied to slow diffusion via macromolecular overcrowding. Stable IPSCs were recorded in control ACSF and then dextran (5 %, 40 kDa) was bath applied (Courtney & Ford, 2014). After 10 min of perfusion, dextran reduced the amplitude (54 ± 7% reduction; n = 6; P = 0.02) and slowed the rate of rise of 5‐HT1A‐IPSCs (38 ± 14% increase in peak time; n = 6, P = 0.02) (Fig. 4 A), but did not alter the decay kinetics of 5‐HT1A‐IPSCs (τDecay: control, 0.60 ± 0.13 s; 5% dextran, 0.79 ± 0.08 s; n = 6, P = 0.31, Student's paired t test; Fig. 4 B). As impairing diffusion limits the escape of serotonin from the synaptic cleft (Min et al. 1998), the reduction in amplitude and delay in time to peak in dextran indicates that spillover of serotonin is likely to have led to the activation of extrasynaptic 5‐HT1A receptors which contributed to the peak amplitude of IPSCs (Min et al. 1998; Nielsen et al. 2004; Szabadics et al. 2007; Markwardt et al. 2009; Ford et al. 2010; Courtney & Ford, 2014). The activation of these extrasynaptic receptors by spillover was likely to have been impaired in dextran, which may have resulted in the smaller amplitudes of IPSCs. In addition, dextran also hinders the diffusion‐mediated clearance of transmitter, which can prolong the activation of postsynaptic receptors (Min et al. 1998; Markwardt et al. 2009). The lack of a pronounced effect of dextran on IPSC decay suggests that clearance via reuptake, rather than diffusion, is the primary mechanism for receptor deactivation.
Figure 4. Slowing diffusion of serotonin alters the amplitude and kinetics of 5‐HT1A‐IPSCs .
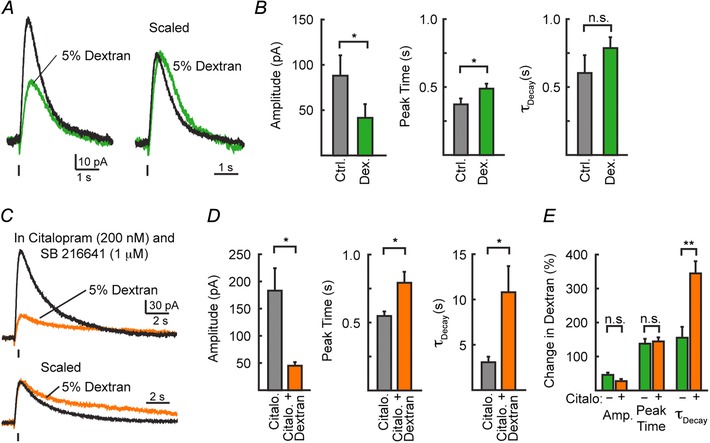
A, average traces of IPSCs recorded before (black) and during (green) the bath application of 5% dextran. B, quantification of the amplitude, peak time, and decay kinetics of IPSCs recorded in A. C, average traces of IPSCs before (black) and during (green) the bath application of 5% dextran recorded in the continuous presence of citalopram (200 nm) and SB 216641 (1 μm). When serotonin reuptake transporters and presynaptic autoreceptors were inhibited, slowing diffusion greatly prolonged the decay of 5‐HT1A‐IPSCs. D, quantification of the amplitude, peak time, and decay kinetics of IPSCs recorded in E. Relative change in IPSCs in the presence of dextran with and without citalopram (200 nm).
Next, we examined the ability of reuptake transporters to limit serotonin diffusion. When in the continuous presence of the selective serotonin reuptake inhibitor (SSRI) citalopram (Celexa, 200 nm) and the presynaptic 5‐HT1B autoreceptor antagonist SB 216641 (1 μm), bath application of 5% dextran resulted in similar changes in the amplitude (72 ± 6% reduction in 5% dextran, n = 4, P = 0.04; P = 0.10 vs. change in absence of citalopram) and peak time (44 ± 12% increase in 5% dextran, n = 4, P = 0.03; P = 0.75 vs. change in absence of citalopram) of 5‐HT1A‐IPSCs. However, slowing diffusion while reuptake was impaired now significantly extended the duration of IPSCs (τDecay: control, 3.1 ± 0.6 s; 5% dextran, 10.8 ± 2.9 s; n = 4, P = 0.04; P = 0.004 vs. change in absence of citalopram; Fig. 4 C–E). Thus when reuptake is inhibited, slowing diffusion now increased the duration of 5‐HT1A receptor activation, suggesting that diffusion became the primary clearance mechanism driving receptor deactivation.
Reuptake transporters prevent pooling and crosstalk from prolonging the duration of IPSCs
Spillover from synaptic sites can result in extrasynaptic pooling and synaptic crosstalk (Otis & Mody, 1992; Isaacson et al. 1993; Balakrishnan et al. 2009; Courtney & Ford, 2014). As pooling allows for the duration of signalling to become dependent on the number of active release sites, greater amounts of transmitter release result in longer durations of receptor activation. While pooling is likely to account for the increases in extracellular levels of serotonin that can be detected by electrochemical approaches (Bunin & Wightman, 1998; Jennings, 2013), it is unclear if it plays a physiological role in the activation of postsynaptic 5‐HT1A receptors.
To determine the contribution of transmitter pooling to evoked serotonin transmission, we initially compared the amplitude and duration of IPSCs recorded from serotonin neurons across the DRN (Fig. 5 A). We found that across neurons, decay time did not correlate with the amplitude of evoked IPSCs (n = 30, R 2 = 0.017, P = 0.54, Pearson's correlation; Fig. 5 B), suggesting that variations in the amount of transmitter release did not alter the duration of 5‐HT1A receptor signalling. For a given cell, we next varied the intensity of stimulation used to evoke IPSCs. Decreasing the stimulation intensity reduced the amplitude of events (65 ± 2% reduction, n = 7; P < 0.001; Student's paired t test) but again did not alter the rate of decay (n = 7, P = 0.15, Student's paired t test; Fig. 5 C). Likewise, decreasing the probability of transmitter release by lowering the concentration of extracellular calcium from 2.5 mm to 1.0 mm similarly reduced the amplitude of synaptic currents (n = 8, P < 0.001; Student's paired t test) with no effect on the decay time (n = 8, P = 0.99, Student's paired t test; Fig. 5 D). Because decreases in serotonin release did not result in shorter duration events, these results suggest that the time course of IPSCs under control conditions is not shaped by pooling of serotonin in the extracellular space.
Figure 5. Reuptake transporters prevent transmitter pooling and synaptic crosstalk from prolonging the decay time of 5‐HT1A‐IPSCs .

A, example IPSCs recorded from 4 neurons demonstrating the variation in amplitude and duration. B, lack of correlation between decay kinetics and amplitude of 5‐HT1A‐IPSCs in 30 DRN neurons from Fig. 1. C, example IPSCs evoked with different stimulation intensities (low: 10–20 μA; high: 60–80 μA). Inset shows the traces normalized and aligned to their peak, illustrating that responses to lower stimuli had similar rates of decay. Right, quantification of amplitudes and decay times between conditions. D, example IPSCs evoked in 2.5 mm and 1.0 mm extracellular Ca2+. Inset shows the traces normalized and aligned to their peak, illustrating that responses in reduced calcium had similar rates of decay. Right, quantification of amplitudes and decay times between conditions. Scale bar (for D and E): 30 pA, 500 ms. n.s. represents P > 0.05. E, example traces (left) and quantification (right) of IPSCs in the presence of SB216641 (1 μm) + citalopram (200 nm) evoked with low (10–20 μA) and high (60–80 μA) stimulation intensities. Lower trace shows the recordings normalized, illustrating that in the presence of citalopram, larger stimuli prolong the decay time of 5‐HT1A‐IPSCs. F, cartoon schematic of the proposed mechanisms underlying 5‐HT1A receptor‐mediated transmission in the DRN. Reuptake transporters limit the extent of transmitter spillover to prevent synaptic crosstalk. This may result in functionally independent sites of transmission.
In other regions, spillover of transmitter between synapses can be facilitated by blocking uptake (Barbour et al. 1994; Otis et al. 1996; Silver et al. 1996; Overstreet and Westbrook, 2003; Balakrishnan et al. 2009; Courtney & Ford, 2014). As blocking reuptake transporters enabled extended diffusion to prolong the decay of 5‐HT1A‐IPSCs (Fig. 3), we next examined whether blocking reuptake enhanced transmitter crosstalk and pooling. In the presence of citalopram (200 nm) and SB216641 (1 μm), increasing the strength of stimulation not only increased the amplitude of IPSCs (n = 4, P = 0.01) but also prolonged the time course of decay (80 ± 26% increase, n = 4, P = 0.006; Fig. 5 E). Thus efficient uptake of serotonin by transporters limited pooling in the extracellular space from extending the duration of IPSCs. This suggests that the postsynaptic activation of 5‐HT1A receptors may result from the local release of serotonin with limited spillover transmission, rather than extended diffusion away from sites of release resulting in transmitter pooling (Fig. 5 F).
Spontaneous IPSCs occur with similar kinetics to evoked
While making long recordings from serotonergic neurons in the DRN, we observed spontaneous IPSCs (sIPSCs) that occurred in 16/38 neurons (42%; Fig. 6 A). The kinetics of sIPSCs were similar to evoked 5‐HT1A‐IPSCs (peak time: 346 ± 23 ms; τdecay: 0.37 ± 0.03 s; n = 27; Fig. 6 B and D) and were prolonged in the presence of citalopram (200 nm, n = 10; P < 0.001; Fig. 6 C and D). The low frequency of events that could be detected (0.2 ± 0.1 events per 10 min, n = 38 neurons) limited quantitative analysis of the underlying mechanisms of individual events. To increase the ability to detect events, phenylephrine (3 μm), forskolin (1 μm), DPCPX (500 nm), and citalopram (50 nm) were included in the bath solution. Activation of adenylyl cyclase by forskolin is known to increase evoked and spontaneous catecholamine release (Beckstead & Williams, 2007; Gantz et al. 2013) and DPCPX was used to prevent adenylyl cyclase from inducing adenosine A1‐receptor activity. This pharmacological cocktail increased the frequency of spontaneous events (1.2 ± 0.3 events per 10 min, n = 15 neurons) with spontaneous events being observed in 13/15 neurons (87%; Fig. 6 E). The apparent increase in spontaneous event frequency may be in part due to increased amplitude of resolvable events (28 ± 3% increase, n = 21 sIPSCs, P < 0.05; Fig. 6 F). Bath application of the 5‐HT1A antagonist WAY 100635 (200 nm) eliminated sIPSCs (8/8 cells) and sIPSCs were never observed in recordings made in the continuous presence of WAY 100635 (200 nm, 9 cells; Fig. 6 E), indicating that spontaneous events were driven by 5‐HT1A receptor activation. While these observations cannot determine whether evoked and spontaneous IPSCs arose from similar or separate pools of presynaptic release sites, the existence of sIPSCs implies that serotonin release from some presynaptic sites was sufficient to independently activate postsynaptic receptors without requiring broad stimulation.
Figure 6. Spontaneous 5‐HT1A receptor‐mediated currents have similar kinetics to evoked IPSCs .
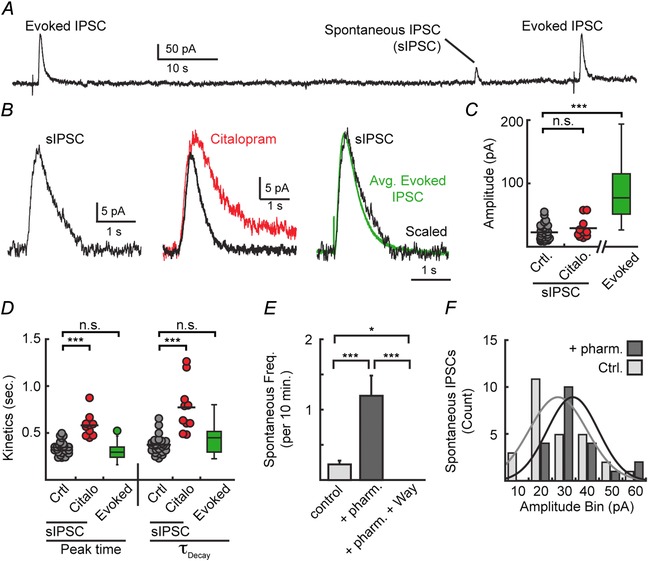
A, representative trace of evoked and spontaneous IPSCs. B, average spontaneous IPSCs (sIPSC, left) overlaid with average sIPSCs recorded in citalopram (200 nm, centre) and average evoked 5‐HT1A‐IPSCs (right). C, quantification of the amplitudes of sIPSCs recorded in control bath and citalopram vs. evoked IPSCs (ANOVA, P < 0.001). D, quantification of the kinetics of the spontaneous IPSCs recorded in control bath and citalopram (200 nm) compared to evoked 5‐HT1A‐IPSCs (ANOVA: peak time P < 0.001, τDecay P < 0.001). sIPSCs recorded in control bath had similar kinetics to evoked IPSCs. E, inclusion of phenylephrine (3 μm), forskolin (1 μm), DPCPX (500 nm), and citalopram (50 nm) in the bath increased the frequency of sIPSCs (pharm.). Further addition of WAY 100635 (200 nm) abolished sIPSCs (ANOVA P < 0.001). F, histogram demonstrating that sIPSCs had significantly greater amplitudes (P = 0.04; n = 21 pharm.; n = 27 ctrl.) in the pharmacological bath (from E) compared to control bath. *P < 0.05; ***P < 0.001; n.s., P > 0.05.
Discussion
In the present study, we examined the mechanisms regulating serotonergic transmission by 5‐HT1A receptors in the dorsal raphe. We found that stimulation evoked serotonin release‐generated 5‐HT1A receptor‐mediated inhibitory currents that rose and fell within a second and paused basal firing without causing long‐lasting depressions to firing rates. The transient nature of 5‐HT1A receptor‐mediated transmission was primarily driven by transmitter clearance via serotonin reuptake transporters and the ensuing deactivation of 5‐HT1A receptors. Reuptake transporters limited the spillover activation of distal, extrasynaptic receptors and prevented serotonin pooling in the extracellular space from prolonging the duration of 5‐HT1A receptor activity via synaptic crosstalk. Taken together, these results suggest that 5‐HT1A receptor‐mediated serotonin transmission in the DRN occurs through functionally independent synapses despite the observation of a limited amount of spillover.
Local feedback inhibition by inhibitory autoreceptors regulates cellular excitability at multiple monoamine synapses. In midbrain dopamine neurons, evoked and spontaneous vesicular dopamine release activates D2 receptor‐mediated IPSCs through GIRK channels (Beckstead et al. 2004; Gantz et al. 2013). Like axonal synapses in the striatum (Marcott et al. 2014), somatodendritic transmission in the midbrain is mediated by a high concentration of dopamine (Ford et al. 2009) that occurs in the absence of transmitter pooling and spillover (Ford et al. 2010; Courtney & Ford, 2014). This differs from somatodendritic transmission of noradrenaline in the locus coeruleus where less efficient uptake allows for low concentrations of noradrenaline to pool in the extracellular space when multiple inputs are synchronously active (Courtney & Ford, 2014). The lack of dopamine pooling in the ventral tegmental prevents crosstalk between synaptic sites to maintain independence between release sites (Courtney & Ford, 2014). In this study, we also found that the local release of serotonin in the DRN can activate synaptic 5‐HT1A receptors in the absence of pooling. This was likely to be true for both evoked and spontaneous release due to the similarity in the rise and decay kinetics. Unlike dopamine transmission, however, spillover allowed for the activation of some pool of extrasynaptic receptors. Serotonergic 5‐HT1A‐IPSCs were more variable cell to cell in their duration, and thus time course of inhibition, than was previously observed for midbrain dopamine transmission (Courtney & Ford, 2014). This increased variability may be due to the recruitment of extrasynaptic receptors in serotonin signalling. The lack of pooling at synaptic sites was attributed to efficient uptake by SERT. Like somatodendritic dopamine transmission (Courtney & Ford, 2014), blocking uptake extended the time course of IPSCs such that the duration of transmission was now dependent upon the amount of serotonin released. How serotonin reuptake transporters both allowed for the spillover activation of extrasynaptic receptors yet prevented pooling and synaptic crosstalk remains unclear. One possibility may be that a non‐uniform distribution of SERTs in DRN neurons around different release sites (Colgan et al. 2012) allows serotonin to spill over into the extrasynaptic space yet limits pooling between synapses.
Throughout these recordings we observed spontaneous IPSCs in roughly half of the neurons examined. The similarity in kinetics between these IPSCs and the finding that no events could be detected in the presence of 5‐HT1A antagonists suggests that these IPSCs could be the result of spontaneous release of serotonin leading to the activation of 5‐HT1A autoreceptors. Similar spontaneous release of dopamine has been recently described at synapses in the substantia nigra (Gantz et al. 2013). It is unclear if the same pool of serotonergic vesicles mediates both evoked and spontaneous events. At ionotropic synapses, different populations of vesicles have been proposed to underlie spontaneous and evoked release (Ramirez & Kavalali, 2011; Kavalali, 2015). In the presence of phenylephrine, which increases DRN firing to 2–3 Hz, the frequency of observable events was only once per ∼10 min. While many events might be below the level of detection in our experiments, the low frequency of events suggests that these events did not result from background firing of DRN neurons. One possibility may be that these events could arise from dendritic sites, where serotonin release occurs in an action potential‐independent manner (Colgan et al. 2012).
Vesicular serotonin release in the DRN occurs at axonal (Bruns et al. 2000), somatic (Kaushalya et al. 2008; Colgan et al. 2009), and dendritic sites (de Kock et al. 2006; Colgan et al. 2012). As evoked serotonin release underlying 5‐HT1A‐IPSCs is modulated by 5‐HT1B receptors (Fig. 2) (Morikawa et al. 2000), which are located only on axon terminals (Sari, 2004), axonal release is likely to contribute to the activation of the synaptic receptors that underlie the IPSC. It is not clear whether these axons originate locally from within the DRN or arise from other brain serotoninergic nuclei (Bang et al. 2012; Andrade et al. 2015). Cell body release sites lack defined presynaptic active zones (Colgan et al. 2009) making it unclear the extent to which somatic release participates in evoked IPSCs. While axonal and somatic transmission are dependent on action potentials (Bruns et al. 2000; Colgan et al. 2012), dendritic release is instead impulse independent, relying on local NMDA receptors and L‐type calcium channels (de Kock et al. 2006; Colgan et al. 2012). Glutamate receptors were blocked in our experiments, making it less likely that dendritic release sites contributed to evoked 5‐HT1A‐IPSCs.
Recent studies have linked both tonic and transient inhibitory regulation of DRN serotonergic neurons to behaviours such as reward encoding (Ranade & Mainen, 2009; Cohen et al. 2015). During these behaviours, tonic and transient inhibition independently influenced DRN neurons and could be used to identify neuronal subpopulations (Cohen et al. 2015). There is a growing consensus that DRN serotonergic neurons are non‐homogeneous, as heterogeneity of these neurons has already been suggested by anatomical, biochemical and electrophysiological properties (Abrams et al. 2004; Marinelli et al. 2004; Calizo et al. 2011; Vasudeva et al. 2011; Andrade & Haj‐Dahmane, 2013). Subpopulations of serotonin neurons, either within the DRN or between various raphe nuclei, are hypothesized to be interconnected, and form complex microcircuits (Bang et al. 2012; Gaspar & Lillesaar, 2012; Altieri et al. 2013). Serotonergic neurons send rich networks of recurrent axon collaterals that can span long distances inside of the DRN, often skipping their nearest neighbours to innervate more distant targets (Altieri et al. 2013). By signalling through independent, 5‐HT1A receptor‐mediated synapses, serotonergic innervation within the DRN could selectively inhibit targeted microcircuits to dampen terminal serotonin release only in their associated projection regions. Thus, independent synapses with these DRN networks may allow spatial and temporal precision in the encoding of mood and behaviour by serotonin throughout the brain.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
N.A.C. and C.P.F. designed the research, analysed the data, prepared the figures and wrote the manuscript. N.A.C. performed the research. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH grants DA035821 (C.P.F.) and T32‐NS077888 (N.A.C.).
Acknowledgements
We thank Evan Deneris and William Clay Spencer for providing ePET1‐YFP mice and confocal raphe images and Michael Beckstead and John Williams for helpful discussions and a critical reading of the manuscript.
References
- Abrams JK, Johnson PL, Hollis JH & Lowry CA (2004). Anatomic and functional topography of the dorsal raphe nucleus. Ann N Y Acad Sci 1018, 46–57. [DOI] [PubMed] [Google Scholar]
- Adell A, Celada P, Abellan MT & Artigas F (2002). Origin and functional role of the extracellular serotonin in the midbrain raphe nuclei. Brain Res Brain Res Rev 39, 154–180. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK & Lakoski JM (1984). Hyperpolarization of serotonergic neurons by serotonin and LSD: studies in brain slices showing increased K+ conductance. Brain Res 305, 181–185. [DOI] [PubMed] [Google Scholar]
- Altieri SC, Garcia‐Garcia AL, Leonardo ED & Andrews AM (2013). Rethinking 5‐HT1A receptors: emerging modes of inhibitory feedback of relevance to emotion‐related behavior. ACS Chem Neurosci 4, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R & Haj‐Dahmane S (2013). Serotonin neuron diversity in the dorsal raphe. ACS Chem Neurosci 4, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R, Huereca D, Lyons JG, Andrade EM & McGregor KM (2015). 5‐HT receptor‐mediated autoinhibition and the control of serotonergic cell firing. ACS Chem Neurosci 6, 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD & Trussell LO (2009). Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci 12, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang SJ, Jensen P, Dymecki SM & Commons KG (2012). Projections and interconnections of genetically defined serotonin neurons in mice. Eur J Neurosci 35, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban JM & Aghajanian GK (1981). Noradrenergic innervation of serotonergic neurons in the dorsal raphe: demonstration by electron microscopic autoradiography. Brain Res 204, 1–11. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I & Marty A (1994). Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron 12, 1331–1343. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Li YW & Talley EM (1997). Effects of serotonin on caudal raphe neurons: activation of an inwardly rectifying potassium conductance. J Neurophysiol 77, 1349–1361. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K & Williams JT (2004). Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42, 939–946. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ & Williams JT (2007). Long‐term depression of a dopamine IPSC. J Neurosci 27, 2074–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D, Riedel D, Klingauf J & Jahn R (2000). Quantal release of serotonin. Neuron 28, 205–220. [DOI] [PubMed] [Google Scholar]
- Bunin MA, Prioleau C, Mailman RB & Wightman RM (1998). Release and uptake rates of 5‐hydroxytryptamine in the dorsal raphe and substantia nigra reticulata of the rat brain. J Neurochem 70, 1077–1087. [DOI] [PubMed] [Google Scholar]
- Bunin MA & Wightman RM (1998). Quantitative evaluation of 5‐hydroxytryptamine (serotonin) neuronal release and uptake: an investigation of extrasynaptic transmission. J Neurosci 18, 4854–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunin MA & Wightman RM (1999). Paracrine neurotransmission in the CNS: involvement of 5‐HT. Trends Neurosci 22, 377–382. [DOI] [PubMed] [Google Scholar]
- Calizo LH, Akanwa A, Ma X, Pan YZ, Lemos JC, Craige C, Heemstra LA & Beck SG (2011). Raphe serotonin neurons are not homogenous: electrophysiological, morphological and neurochemical evidence. Neuropharmacology 61, 524–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JY, Amoroso MW & Uchida N (2015). Serotonergic neurons signal reward and punishment on multiple timescales. eLife 4, e06346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Cavolo SL, Commons KG & Levitan ES (2012). Action potential‐independent and pharmacologically unique vesicular serotonin release from dendrites. J Neurosci 32, 15737–15746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Putzier I & Levitan ES (2009). Activity‐dependent vesicular monoamine transporter‐mediated depletion of the nucleus supports somatic release by serotonin neurons. J Neurosci 29, 15878–15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney NA & Ford CP (2014). The timing of dopamine‐ and noradrenaline‐mediated transmission reflects underlying differences in the extent of spillover and pooling. J Neurosci 34, 7645–7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Cornelisse LN, Burnashev N, Lodder JC, Timmerman AJ, Couey JJ, Mansvelder HD & Brussaard AB (2006). NMDA receptors trigger neurosecretion of 5‐HT within dorsal raphe nucleus of the rat in the absence of action potential firing. J Physiol 577, 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vivo M & Maayani S (1986). Characterization of the 5‐hydroxytryptamine1a receptor‐mediated inhibition of forskolin‐stimulated adenylate cyclase activity in guinea pig and rat hippocampal membranes. J Pharmacol Exp Ther 238, 248–253. [PubMed] [Google Scholar]
- Ford CP, Gantz SC, Phillips PE & Williams JT (2010). Control of extracellular dopamine at dendrite and axon terminals. J Neurosci 30, 6975–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE & Williams JT (2009). The time course of dopamine transmission in the ventral tegmental area. J Neurosci 29, 13344–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornal CA, Metzler CW, Gallegos RA, Veasey SC, McCreary AC & Jacobs BL (1996). WAY‐100635, a potent and selective 5‐hydroxytryptamine1A antagonist, increases serotonergic neuronal activity in behaving cats: comparison with (S)‐WAY‐100135. J Pharmacol Exp Ther 278, 752–762. [PubMed] [Google Scholar]
- Gantz SC, Bunzow JR & Williams JT (2013). Spontaneous inhibitory synaptic currents mediated by a G protein‐coupled receptor. Neuron 78, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Levitt ES, Llamosas N, Neve KA & Williams JT (2015). Depression of serotonin synaptic transmission by the dopamine precursor L‐DOPA. Cell Rep 12, 944–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P & Lillesaar C (2012). Probing the diversity of serotonin neurons. Philos Trans R Soc Lond B Biol Sci 367, 2382–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddjeri N, Lavoie N & Blier P (2004). Electrophysiological evidence for the tonic activation of 5‐HT1A autoreceptors in the rat dorsal raphe nucleus. Neuropsychopharmacology 29, 1800–1806. [DOI] [PubMed] [Google Scholar]
- Hajos M, Hoffmann WE, Tetko IV, Hyland B, Sharp T & Villa AE (2001). Different tonic regulation of neuronal activity in the rat dorsal raphe and medial prefrontal cortex via 5‐HT1A receptors. Neurosci Lett 304, 129–132. [DOI] [PubMed] [Google Scholar]
- Hjorth S & Sharp T (1991). Effect of the 5‐HT1A receptor agonist 8‐OH‐DPAT on the release of 5‐HT in dorsal and median raphe‐innervated rat brain regions as measured by in vivo microdialysis. Life Sci 48, 1779–1786. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM & Nicoll RA (1993). Local and diffuse synaptic actions of GABA in the hippocampus. Neuron 10, 165–175. [DOI] [PubMed] [Google Scholar]
- Jacobs BL & Azmitia EC (1992). Structure and function of the brain serotonin system. Physiol Rev 72, 165–229. [DOI] [PubMed] [Google Scholar]
- Jennings KA (2013). A comparison of the subsecond dynamics of neurotransmission of dopamine and serotonin. ACS Chem Neurosci 4, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama J, Yakushiji T & Akaike N (1997). Characterization of the K+ current mediated by 5‐HT1A receptor in the acutely dissociated rat dorsal raphe neurons. Brain Res 745, 283–292. [DOI] [PubMed] [Google Scholar]
- Kaushalya SK, Desai R, Arumugam S, Ghosh H, Balaji J & Maiti S (2008). Three‐photon microscopy shows that somatic release can be a quantitatively significant component of serotonergic neurotransmission in the mammalian brain. J Neurosci Res 86, 3469–3480. [DOI] [PubMed] [Google Scholar]
- Kavalali ET (2015). The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16, 5–16. [DOI] [PubMed] [Google Scholar]
- Kia HK, Brisorgueil MJ, Hamon M, Calas A & Verge D (1996). Ultrastructural localization of 5‐hydroxytryptamine1A receptors in the rat brain. J Neurosci Res 46, 697–708. [DOI] [PubMed] [Google Scholar]
- Levitt ES, Hunnicutt BJ, Knopp SJ, Williams JT & Bissonnette JM (2013). A selective 5‐HT1a receptor agonist improves respiration in a mouse model of Rett syndrome. J Appl Physiol (1985) 115, 1626–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki I (1998). The spectrum of behaviors influenced by serotonin. Biol Psychiatry 44, 151–162. [DOI] [PubMed] [Google Scholar]
- Marcott PF, Mamaligas AA & Ford CP (2014). Phasic dopamine release drives rapid activation of striatal D2‐receptors. Neuron 84, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Schnell SA, Hack SP, Christie MJ, Wessendorf MW & Vaughan CW (2004). Serotonergic and nonserotonergic dorsal raphe neurons are pharmacologically and electrophysiologically heterogeneous. J Neurophysiol 92, 3532–3537. [DOI] [PubMed] [Google Scholar]
- Markwardt SJ, Wadiche JI & Overstreet‐Wadiche LS (2009). Input‐specific GABAergic signaling to newborn neurons in adult dentate gyrus. J Neurosci 29, 15063–15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelsen KA, Prickaerts J & Steinbusch HW (2008). The dorsal raphe nucleus and serotonin: implications for neuroplasticity linked to major depression and Alzheimer's disease. Prog Brain Res 172, 233–264. [DOI] [PubMed] [Google Scholar]
- Min MY, Rusakov DA & Kullmann DM (1998). Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron 21, 561–570. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Manzoni OJ, Crabbe JC & Williams JT (2000). Regulation of central synaptic transmission by 5‐HT1B auto‐ and heteroreceptors. Mol Pharmacol 58, 1271–1278. [DOI] [PubMed] [Google Scholar]
- Mundey MK, Fletcher A & Marsden CA (1996). Effects of 8‐OHDPAT and 5‐HT1A antagonists WAY100135 and WAY100635, on guinea‐pig behaviour and dorsal raphe 5‐HT neurone firing. Br J Pharmacol 117, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TA, DiGregorio DA & Silver RA (2004). Modulation of glutamate mobility reveals the mechanism underlying slow‐rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron 42, 757–771. [DOI] [PubMed] [Google Scholar]
- Otis TS & Mody I (1992). Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J Neurophysiol 67, 227–235. [DOI] [PubMed] [Google Scholar]
- Otis TS, Wu YC & Trussell LO (1996). Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci 16, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS & Westbrook GL (2003). Synapse density regulates independence at unitary inhibitory synapses. J Neurosci 23, 2618–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Colmers WF & Williams JT (1989). 5‐HT‐mediated synaptic potentials in the dorsal raphe nucleus: interactions with excitatory amino acid and GABA neurotransmission. J Neurophysiol 62, 481–486. [DOI] [PubMed] [Google Scholar]
- Pan ZZ & Williams JT (1989). Differential actions of cocaine and amphetamine on dorsal raphe neurons in vitro. J Pharmacol Exp Ther 251, 56–62. [PubMed] [Google Scholar]
- Penington NJ & Kelly JS (1990). Serotonin receptor activation reduces calcium current in an acutely dissociated adult central neuron. Neuron 4, 751–758. [DOI] [PubMed] [Google Scholar]
- Penington NJ, Kelly JS & Fox AP (1991). A study of the mechanism of Ca2+ current inhibition produced by serotonin in rat dorsal raphe neurons. J Neurosci 11, 3594–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineyro G & Blier P (1999). Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev 51, 533–591. [PubMed] [Google Scholar]
- Portas CM, Thakkar M, Rainnie D & McCarley RW (1996). Microdialysis perfusion of 8‐hydroxy‐2‐(di‐n‐propylamino)tetralin (8‐OH‐DPAT) in the dorsal raphe nucleus decreases serotonin release and increases rapid eye movement sleep in the freely moving cat. J Neurosci 16, 2820–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM & Kavalali ET (2011). Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol 21, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade SP & Mainen ZF (2009). Transient firing of dorsal raphe neurons encodes diverse and specific sensory, motor, and reward events. J Neurophysiol 102, 3026–3037. [DOI] [PubMed] [Google Scholar]
- Riad M, Garcia S, Watkins KC, Jodoin N, Doucet E, Langlois X, el Mestikawy S, Hamon M & Descarries L (2000). Somatodendritic localization of 5‐HT1A and preterminal axonal localization of 5‐HT1B serotonin receptors in adult rat brain. J Comp Neurol 417, 181–194. [PubMed] [Google Scholar]
- Richardson‐Jones JW, Craige CP, Guiard BP, Stephen A, Metzger KL, Kung HF, Gardier AM, Dranovsky A, David DJ, Beck SG, Hen R & Leonardo ED (2010). 5‐HT1A autoreceptor levels determine vulnerability to stress and response to antidepressants. Neuron 65, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y (2004). Serotonin1B receptors: from protein to physiological function and behavior. Neurosci Biobehav Rev 28, 565–582. [DOI] [PubMed] [Google Scholar]
- Scott MM, Wylie CJ, Lerch JK, Murphy R, Lobur K, Herlitze S, Jiang W, Conlon RA, Strowbridge BW & Deneris ES (2005). A genetic approach to access serotonin neurons for in vivo and in vitro studies. Proc Natl Acad Sci USA 102, 16472–16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA, Cull‐Candy SG & Takahashi T (1996). Non‐NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol 494, 231–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Tamas G & Soltesz I (2007). Different transmitter transients underlie presynaptic cell type specificity of GABAA,slow and GABAA,fast . Proc Natl Acad Sci USA 104, 14831–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Zhang S & Raman IM (1993). Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron 10, 1185–1196. [DOI] [PubMed] [Google Scholar]
- Vandermaelen CP & Aghajanian GK (1983). Electrophysiological and pharmacological characterization of serotonergic dorsal raphe neurons recorded extracellularly and intracellularly in rat brain slices. Brain Res 289, 109–119. [DOI] [PubMed] [Google Scholar]
- Vasudeva RK, Lin RC, Simpson KL & Waterhouse BD (2011). Functional organization of the dorsal raphe efferent system with special consideration of nitrergic cell groups. J Chem Neuroanat 41, 281–293. [DOI] [PubMed] [Google Scholar]
- Williams JT, Colmers WF & Pan ZZ (1988). Voltage‐ and ligand‐activated inwardly rectifying currents in dorsal raphe neurons in vitro. J Neurosci 8, 3499–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]