

Abstract
It has been recognized that during chronic inflammatory systemic diseases (CIDs) maladaptations of the immune, nervous, endocrine and reproductive system occur. Maladaptation leads to disease sequelae in CIDs. The ultimate reason of disease sequelae in CIDs remained unclear because clinicians do not consider bodily energy trade-offs and evolutionary medicine. We review the evolution of physiological supersystems, fitness consequences of genes involved in CIDs during different life-history stages, environmental factors of CIDs, energy trade-offs during inflammatory episodes and the non-specificity of CIDs. Incorporating bodily energy regulation into evolutionary medicine builds a framework to better understand pathophysiology of CIDs by considering that genes and networks used are positively selected if they serve acute, highly energy-consuming inflammation. It is predicted that genes that protect energy stores are positively selected (as immune memory). This could explain why energy-demanding inflammatory episodes like infectious diseases must be terminated within 3–8 weeks to be adaptive, and otherwise become maladaptive. Considering energy regulation as an evolved adaptive trait explains why many known sequelae of different CIDs must be uniform. These are, e.g. sickness behavior/fatigue/depressive symptoms, sleep disturbance, anorexia, malnutrition, muscle wasting—cachexia, cachectic obesity, insulin resistance with hyperinsulinemia, dyslipidemia, alterations of steroid hormone axes, disturbances of the hypothalamic-pituitary-gonadal (HPG) axis, hypertension, bone loss and hypercoagulability. Considering evolved energy trade-offs helps us to understand how an energy imbalance can lead to the disease sequelae of CIDs. In the future, clinicians must translate this knowledge into early diagnosis and symptomatic treatment in CIDs.
Keywords: inflammatory systemic disease, neuroendocrine immunology, energy regulation, disease sequelae
INTRODUCTION
Chronic inflammatory systemic diseases (CIDs) like rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and many others are a burden to humans because of life-long debilitating illness, increased mortality and high costs for therapy and care. Other than CIDs, infectious disease like influenza or scarlet fever typically last only for a short period of time and they normally do not lead to chronic disease sequelae. The main difference between CIDs and acute infectious disease is span of time. While an acute infectious disease or inflammation during wound healing represents an adaptive response to overcome a disease and, thus, to increase life-time reproductive success, a CID is outside the adaptive reaction norm leading to maladaptive responses and a reduction of evolutionary fitness, because the stimulating trigger cannot be removed (for discussion of reaction norm see [1]).
CIDs are often discussed within the evolutionary medicine framework of the ‘hygiene hypothesis’ [2]. This model says that since the start of urbanization humans experienced a depletion of typical environmental infectious organisms such as helminths with which mammals co-evolved. The loss of these infectious agents provoked a change of normal background levels of immunoregulation during infancy, and this stimulated a more aggressive immune response in adulthood and old age and, thus, more frequent appearance of CIDs [2].
An approach of evolutionary medicine to CIDs is needed because of the fact that CIDs exist although they exert a negative effect on reproductive fitness [3–9]. So far, the persistence of CIDs was partly explained by the important roles a limited number of networked immune system genes play in pathogen defense and other functions that are under strong natural selection [10]. While these previous theories focused on the trigger of CIDs, here we extend this by focusing on common disease pathways and sequelae of CIDs.
Coordinated reactions of the supersystems (immune, nervous, endocrine and reproductive) that maintain homeostasis have been evolutionarily conserved to respond to and eliminate foreign agents over a period of days to a few weeks (e.g. influenza or scarlet fever) [11]. If the responses of these supersystems fail to return to normal such as in autoimmunity or other forms of systemic chronic inflammation (e.g. autoinflammatory syndromes), maladaptation of the supersystems perpetuates long-lasting CIDs [11, 12]. Since 2003 this theory has been refined, because new theoretical concepts from the field of bodily energy regulation were added [13–15]. This addition improved the model that is presented here in an updated version.
EVOLUTION OF THE SUPERSYSTEMS AND THEIR INTERACTIONS
Infectious and non-infectious hazards were present throughout the entire evolutionary history of all vertebrates including humans. With rats and mice, we share many similar inflammatory mechanisms, although the last common ancestor lived 65 million years ago [16]. Humans and chicken use immunoglobulin gene rearrangement and somatic hypermutation to shape the antibody response towards infectious agents [16]. Even sharks have a very similar immune system like ours with T cell receptors and immunoglobulin diversity based on V(D)J recombination and the RAG1/2 system [17]. Differences only become apparent when comparing humans with jawless fish (lampreys and hagfish; the last common ancestor with humans lived 460 million years ago), which use a more primitive immune system which, nevertheless, already consists of variable lymphocyte receptors [17].
Thus, when we study the activation of the immune system in the context of the most prevalent danger of infection, we have to consider that the evolutionary history of the human immune system spans more than 420 million years. The immunological pathways are conserved and are very similar between sharks, birds, rodents and humans. In sum, acute inflammation is an adaptive response present in nearly all vertebrates, which has evolved to cope with infections or short-lived inflammatory responses (e.g. wound healing or foreign body reactions).
In a similar way, fish, birds, rodents and humans share many neuroendocrine pathways responsible for bodily homeostasis such as the hypothalamic-pituitary-adrenal axis (HPA) axis or the sympathetic nervous system. For example, glucocorticoids of the HPA axis are present in mammals, birds, jawed fish and even in lamprey [18, 19].
The bilaterally organized sympathetic nervous system is present in jawed fish like sharks and rays and in all vertebrates, but it is absent in lampreys and hagfish [20]. Nevertheless, lampreys produce catecholamines in monoamine-containing neurons, which also happens in leukocytes in the human body [21, 22]. Even bacteria possess functional orthologs of tyrosine hydroxylase, which converts L-thyroxine to L-DOPA, the first catecholamine in the synthesis cascade towards noradrenaline and adrenaline and bacteria use catecholamines as growth factors [23, 24].
In addition, central nervous system responses such as sickness behavior [25] or anorexia [26] are similar in birds and rodents, indicating long evolutionary history. Rainbow trouts can become anorectic under stressful conditions [27]. Even crocodiles develop anorexia and lethargy during infection [28], which demonstrates the uniformity of the response. In sum, the immune, endocrine, and nervous systems have evolved over hundreds of millions of years, and to understand diseases in humans, including CIDs, the long evolutionary history of these supersystems has to be taken into account.
When we observe crosstalk between the supersystems, many similar pathways are used in humans and other vertebrates to maintain homeostasis. If homeostasis is disturbed by stressful events such as infection (leading to inflammation), the different supersystems need to cooperate in order to overcome the threat. The crosstalk of supersystems has co-evolved with infectious agents. Importantly, such a reaction to inflammatory stress is relatively uniform in many species. Genes enabling a coordinated response of the supersystems, allowing the organism to overcome infection or wounding, will increase survival probability and the potential for future reproduction and—as such—evolutionary fitness.
EVOLUTIONARY FITNESS AND CIDs
It can be expected that CIDs lead to reduced evolutionary fitness by indirect and direct effects. First, affected individuals would have reduced competitive abilities, reducing their access to resources such as food and sexual partners, and their social status in the group might be impaired. There might be even active repudiation in the group. In our ancestors, the risk of injury or even death in any competition including fights and warfare as well as encounters with predators would have significantly increased due to their handicap.
Second, CIDs directly influence reproductive success by decreasing fecundity [3–9]. Even with optimum treatment in our days, fertility is reduced in both men and women with CIDs [29]. CIDs are accompanied by a severe decrease of androgen levels [30–32], which can be observed even after subcutaneous injection of the proinflammatory cytokine interleukin-6 (IL-6) into healthy volunteers. IL-6 blocks the release of testosterone by ∼50%, and it needs 7 days until restoration of normal serum levels [33]. In the acute phase of inflammatory diseases, IL-6 is nearly always increased and, thus, affects sexual function and sex hormone release. During a CID, this cytokine remains elevated if no appropriate treatment is applied.
There is other clear evidence that the proinflammatory TNF directly inhibits gonadal cells responsible for testosterone secretion [34]. Injection of another inflammatory cytokine, IL-1β, into the brain of experimental animals decreased testosterone production for a long time [35]. The same cytokines influence sperm motility resulting in reduced mucosal penetration properties and decreased male fertility [36]. Similarly, female reproduction is influenced by inflammatory cytokines such as IL-1 and TNF [37, 38], and infectious episodes reduce female fertility [39]. Inflammation has deleterious effects on the menstrual cycle, leading to amenorrhea [40]. Thus, the reproduction system is switched-off in both sexes during inflammation.
From a point of view of evolutionary history, it can be expected that the disease itself lead to early death in our ancestors, because no appropriate therapies were available until the 1950s. All conditions imply a high negative selection pressure for specific genetic factors leading to CIDs, especially, if these would have occurred in young reproducing adults.
GENETICS AND ENVIRONMENTAL INFLUENCE IN CIDs
Accumulation theory of CIDs
Natural selection cannot favor any chronic disease, because of the associated fitness costs and absence of any fitness gains. However, an individual that would suffer from a CID at a post-reproductive life history stage might have transmitted genes—increasing the risk to develop a chronic illness in late age—during an earlier life history stage when it was healthy and reproducing.
Several genes that play a role in CIDs have been identified. The best know association between CIDs and genes has been established for the HLA system, which is an important element of antigen presentation. Sometimes the risk to develop a disease in presence of a certain subtype of the HLA system can be high. Approximately 10–25% of humans carry risk HLA genotypes but the prevalence of any of the CIDs is 0.1–1% [41], and thus, far below the expected 10–25%. Thus, there have to be additional factors and a certain HLA protein alone is not necessary and not sufficient to elicit a certain CID.
Today we label such an alteration as a genetic disease-promoting risk factor. As the alteration of an HLA protein is not the single causative factor, we expect that other genetic factors and the environment (see below) are also relevant. Indeed, CIDs have a multifactorial genetic background as demonstrated, e.g. in rheumatoid arthritis [42]. Many additional genetic factors have been described that increase the risk to develop rheumatoid arthritis (Table 1). Therefore, negative selection for specific HLA gene variants most probably was small, because in most individuals carrying the gene the disease did not develop due to the lack of additional factors. In contrast, polymorphic HLA genes have been retained because they helped to overcome various infections [43], leading to positive selection. The genetic prerequisites for a CID can be retained over generations and generations via individuals that never express the disease because relevant co-factors may either not occur or are expressed following very irregular patterns.
Table 1.
Risk factors of rheumatoid arthritis
| HLA system (HLA DR4 [DRB1*04]) |
| tumor necrosis factor alpha-induced protein 3 (TNFAIP3) (negative regulator of the NF-kappaB pathway) |
| protein tyrosine phosphatase receptor type C (CD45) (receptor and protein tyrosine phosphatase) |
| interferon gamma receptor 2 (cytokine receptor) |
| CD40 (cell surface molecule and receptor) |
| tyrosine kinase 2 (TYK2) (signaling of cytokine receptors) |
| IL-6 receptor (cytokine receptor) |
| protein tyrosine phosphatase type 22 (PTPN22) (signaling of receptors) |
| Fc-gamma receptor II 2B (CD32, Fc fragment of IgG, low affinity IIb receptor) |
| IL-2 (cytokine) |
| IL-2 receptor alpha chain (cytokine receptor) |
| SH2B3 (member of the SH2B adaptor family of proteins, signaling of growth factor and cytokine receptors) |
| ICOSLG (inducible T-cell co-stimulator ligand) |
The respective single nucleotide polymorphism in the mentioned genes have been found in genome-wide association studies [42]. The list is ordered according to the significance level of the statistical test for relative risk. HLA, human leukocyte antigen; IL, interleukin; TNF, tumor necrosis factor.
Table 1 demonstrates that the same signaling factors can play a role in different CIDs such as multiple sclerosis (TYK2, CD40, TNFAIP3, PTPN22, IL-2, IL-2RA and others; ref. [44]), type 1 diabetes mellitus (TNFAIP3, PTPN22, IL-2, IL-2RA, SH2B3 and others, refs. [45–47]), systemic lupus erythematosus (TYK2, TNFAIP3, PTPN22, FCGR2B, reviewed in ref. [48]), and Crohn’s disease (TYK2, PTPN22, IL-2RA, ICOSLG and others, ref. [49]). This tells us that the same immunostimulatory or immunoinhibiting signaling pathways are used in the different diseases. These shared immune factors have been positively selected in the context of infectious diseases but not for CIDs (see next section ‘Pleiotropy theory of CIDs’, ref. [10]).
In addition, accumulation of different risk alleles does not only lead to additivity of risk but to synergistic effects as demonstrate for the R620W PTPN22 allele and ‘HLA-DRB1 shared epitope’ alleles in rheumatoid arthritis [50]. In addition, African Americans positive for ‘HLA-DRB1 shared epitope’ alleles have a higher risk of developing rheumatoid arthritis when they demonstrate a higher degree of European ancestry, which demonstrates genetic admixture [51].
From monozygotic twin studies, it is known that genes contribute to the appearance of CIDs (Table 2). In these twin studies, the concordance rate has been determined as the quantitative statistical expression for the concordance of a given genetic trait. The concordance rate varies between 0 and 50%, and is on average 25% for the mentioned CIDs in Table 2, indicating that the environment explains ∼75% of the variation. Thus, considering gene-environment interactions is of outstanding importance. The accumulation theory describes how the accumulation of several genetic and environmental risk factors increases the risk to develop a CID.
Table 2.
Concordance rate in twin studies in chronic inflammatory systemic diseases
An interesting example for an environmental factor affecting the risk to develop CIDs is smoking. Already in the 1970s, a link was demonstrated between smoking and increased serum levels of autoantibodies [58]. The first epidemiological studies on the link between smoking and the risk to develop rheumatoid arthritis appeared in the early 1990s [59–61]. One study on monozygotic twins showed that smoking increased the risk to develop rheumatoid arthritis by a factor of 12 [62]. The risk remains elevated for several years after smoking cessation [63]. This demonstrates that smoking is a very strong environmental risk factor, and Lars Klareskog et al. showed that smoking may trigger HLA-DR-restricted immune reactions to autoantigens modified by citrullination [64]. Importantly, smoking is also linked to an increased risk to develop other CIDs such as ankylosing spondylitis [65], multiple sclerosis [66], Crohn’s disease [67] and systemic lupus erythematosus [68, 69]. However, smoking can also reduce the risk of developing CIDs such as type 1 diabetes mellitus [70], ulcerative colitis [67] or pemphigus [71].
There are several other environmental factors that are known to affect the development pf CIDs. For example, alcohol can protect individuals from developing rheumatoid arthritis [72]. Intake of oily fish was associated with a modestly decreased risk of developing rheumatoid arthritis [73]. Another environmental risk factor for rheumatoid arthritis and other autoimmune diseases is silica exposure, which occurs in construction work (cement, demolition) [74, 75]. It was discussed that silica is a stimulator of the immune system, which may trigger the autoimmune process leading to chronic inflammation [74, 75]. These examples clearly demonstrate that environmental factors play an important role, supporting the accumulation theory.
Another environmental modulator is the microbiota in the gut and elsewhere on body surfaces. Over millennia of co-evolution, microbiota has been a strong stimulus for shaping the innate and adaptive immune system in vertebrates [76]. During evolution, responses towards microbes have established a fine-tuning of immune responses [43]. We expect that microbiota-reactive T and B lymphocytes support homeostasis of the body [76]. Thus, normal microbiota or changes in microbiota during disease states or continuous illness can modulate the expression of CIDs. The famous example of HLA-B27 transgenic rats clearly shows the phenomenon: these rats develop arthritis and colitis in the presence of microbiota but are protected in a germ-free environment [77]. Thus, microbiota can be a decisive environmental factor, and new studies suggest possible causal links between microbiota and CIDs [78].
Pleiotropy theory of CIDs
George C. Williams, an evolutionary biologist studying aging [79], stated in 1957:
It is necessary to postulate genes that have opposite effects on fitness at different ages, or, more accurately, in different somatic environments.
This theory is now widely accepted in aging research and elsewhere, and it is called ‘antagonistic pleiotropy theory’. The word pleiotropy comes from the Greek pleio, meaning ‘many’, and trepein, meaning ‘influencing’. Pleiotropic means that genes can influence different phenotypic traits at different life history points. We propose that the same theory applies to the situation in CIDs, with genes being adaptive at an early age, but maladaptive at older age (another somatic environment), leading to an overall increase in Darwinian fitness, even though causing disease and suffering at older age (Table 3).
Table 3.
Examples of antagonistic pleiotropy for genes that increase risk or severity of chronic inflammatory diseases [80]
| Genes | Chronic inflammatory disease | Pleiotropic meaning outside of chronic inflammatory diseases (with selection advantage) | Refs. |
|---|---|---|---|
| HLA DR4 (DRB1*04) | Rheumatoid arthritis and other autoimmune diseases | Decrease of risk of Dengue hemorrhagic fever (defense against infectious agents) | [81] |
| Fc-gamma receptor IIIA, 158 valine/valine | Rheumatoid arthritis and other autoimmune diseases | Decrease of poliomyelitis infection due to strong natural killer cell activity (defense against infectious agents) | [82] |
| HLA B27 | Ankylosing spondylitis and other axial forms of spondyloarthritis | Decrease of viral infection (defense against infectious agents) | [83, 84] |
| PTPN22 1858 C > T* | Many autoimmune diseases | Improved storage of energy-rich fuels (higher body mass index, higher waist-to-hip ratio in women) | [85] |
| CTLA4 49 A > G | Many autoimmune diseases | Better defense against hepatitis B virus and helicobacter pylori (defense against infectious agents) | [86, 87] |
| NOD2/CARD15 | Crohn’s disease | Hypertension (activation of the sympathetic nervous system and, thus, the fight-and-flight response) | [88] |
It can be expected that all genes associated with CIDs were positively selected because they confer fitness benefits by improving reproduction, stronger skeletal muscles, better storage of energy-rich fuels (in fat tissue), or better fight-and-flight responses (stronger activation of the sympathetic nervous system and HPA axis). However, a gene or allele increasing reproductive success in early adult life can play a deleterious role in an elderly person suffering from a CID, and still overall increase Darwinian fitness. For example, a gene may confer an excellent immune response against infectious agents because it leads to a stronger activity of immune cells or better bacteria/virus recognition. This gene is advantageous in younger ages because the gene carrier has less childhood infections and, thus, a higher chance to survive until reproductive age. However, the immune system of the gene carrier might be over-activated later in life, leading to the development of a CID. However, this cost at late age (often in post-reproductive life history stages) would be much lower than the benefit of increased chance of survival until reproduction.
Let us make another example from the endocrine world. It is widely accepted that the incidence rate of rheumatoid arthritis in women largely increases after menopause [89] (Fig. 1). Women with an early onset of menorrhoea, with shorter cycle lengths and higher numbers of ovulatory cycles have an increased risk of early menopause [90, 91]. In order to understand this phenomenon, one has to recall that ovaries are equipped with a limited number of oocytes, and, with every ovulation, oocytes are released and the remaining number decreases gradually. Under consideration of an early onset of menorrhoea and shorter ovulatory cycles, more oocytes will be released until age 40.
Figure 1.
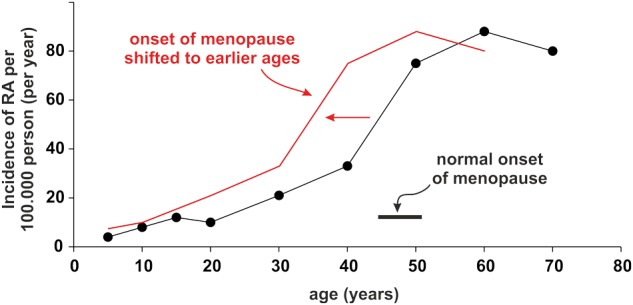
Incidence rate of rheumatoid arthritis (RA). The black line indicates the normal situation with a menopause starting at 45 years of age. The red line demonstrates a fictitious situation with accelerated menopause years before
The occurrence of menorrhoea and premature adrenarche has a strong genetic component [92]. Under some environmental conditions, the carrier of this trait should reproduce earlier and more often, thereby, increasing reproductive success. However, this trait also causes earlier menopause, increasing the risk to develop the CID rheumatoid arthritis [59, 89], but the cost of developing this CID might be low compared to the fitness benefits of early and frequent reproduction. Thus, a gene conferring an early onset of menorrhoea that might be favorable for sexual function and reproduction early in life, would lead to early menopause and an earlier onset of rheumatoid arthritis (red line in Fig. 1) [59]. In addition, RA patients with an earlier onset of menopause have a more severe disease with more pain and positive rheumatoid factors [93].
The link between the HLA system, infection, and sets of secreted cytokines was nicely summarized by Chella David’s group of the Mayo Clinic [43]. This investigation demonstrated that cytokine sets are related to different types of infectious immune stimuli. A more clinical example is given in a Mexican population. It was demonstrated that the genetic risk factor HLA-DR4 (DRB1*04), known to be positively associated with rheumatoid arthritis and other CIDs, is highly negatively associated with the risk of dengue hemorrhagic fever [81]. HLA-DR4 (DRB1*04) homozygous individuals were 11.6 times less likely to develop dengue hemorrhagic fever in comparison to DR4(DRB1*04) negative persons, demonstrating that the genetic factor HLA-DR4 (DRB1*04) is protective against this infectious disease [81]. Since we are all descendants of African Homo sapiens, dengue was relevant to the entire human population some 100 000 years ago when humans lived in warmer climates. Thus, it seems that HLA-DR4 (DRB1*04) has been evolutionarily positively selected to fight off dengue hemorrhagic fever and other microbes in early life but later in old age supports the development of a CID. This leads to life-time reproductive success by its action in early life, even though imposing costs by increasing the risk to develop CIDs at late age.
Another example is the Fc-gamma receptor IIIA, which is responsible for the recognition of immunoglobulin G leading to immune cell activation. The recognition of immunoglobulin is important to detect bacteria and viruses, which are bound to immunoglobulin G (opsonization and phagocytosis). After binding to the cell surface Fc-gamma receptor IIIA, immunoglobulin G together with bacteria or viruses are taken up, and the cell gets stimulated to destroy the microbe. However, one form of the Fc-gamma receptor IIIA, with a genetic polymorphism in position 158, being homozygous for the amino acid valine (158 valine/valine), has a higher binding affinity for immunoglobulin G when compared to other Fc-gamma receptors [94], and this is associated with a higher risk to develop rheumatoid arthritis [95]. Importantly, the 158 valine/valine variant seems to protect against poliomyelitis [82], which indicates that this receptor variant confers protection against viral disease. Again, it seems that the genotype 158 valine/valine of the Fc-gamma receptor IIIA has been evolutionarily positively selected to fight off infectious agents by increasing microbe clearance. This trait is most advantageous in early ages (infants and young adults) by increasing the chances to reproduce, but the same surface molecule assists the development of a CID in older ages.
Pleiotropy occurs when a single gene influences multiple phenotypic traits. Such a gene can be largely advantageous for the carrier in earlier years in reproductive time, but it can also be highly disadvantageous in later ages in another somatic environment such as aging [79] or CIDs. These considerations might open our eyes when we study genetic risk factors in patients with CIDs. It also explains why a risky gene polymorphism is most probably not specific for a given CID, because the gene was evolutionarily positively selected for a different function.
IMMUNE RESPONSE, ENERGY CONSUMPTION VERSUS PROTECTION and CIDs
In a previous review, it was argued that a highly activated immune/repair system should not be switched on for a long time because this would be very energy-consuming [14]. Additionally, a highly activated immune system is accompanied by sickness behavior and anorexia, which prevents adequate food intake and necessitates life on stored reserves (inflammation-induced anorexia). Under systemic inflammatory conditions, breaking down all reserves takes 19–43 days [14]. An energy trade-off between immune system and other bodily systems explains this time-point, and this time-point also explains when inflammation becomes maladaptive and chronic (like in CIDs) [14].
A highly activated immune/repair system can need huge amounts of energy. For example, the energy consumption in the case of extensive burn wounds (up to 20 000 kJ/day [4777 kcal/day]) [14] is approximately the same as during military jungle training (also 20 000 kJ/day; ref. [96]), and more than during military arctic training (∼18 000 kJ/day; ref. [97]). Although burn wounds represent the extreme end of the spectrum, it demonstrates the high energy consumption of the immune system (another number on energy expenditure of the immune system is 15 000 kJ/day in sepsis). Energy is a limited resource for all vertebrates, a key determinant of survival and reproduction.
Energy consumption and energy storage are some of the most critical determinants in evolution [98]. If alterations of homeostasis lead to the selection of physiological mechanisms characterized by very high energy consumption, then the situation cannot be chronic—it must be acute. Such situations lead to allostatic overload type I, which means that energy consumption is higher than energy intake, which occurs during emergency life history stages [98]. Since time to total consumption of stored energy is ∼19–43 days [14], an acute energy-consuming change of homeostasis must be started and terminated within this time frame, as otherwise permanent damage and finally death occur.
A very good example for this time window is the germinal center reaction of B cell expansion and contraction that happens within ∼21–28 days [99]. Most acute disease states are terminated within this time frame such as infectious diseases, wound healing, and wound repair, but also strong mental activation in catastrophic stressful situations must be terminated because they are highly energy-consuming [100]. During evolution, respective homeostatic networks of supersystems were positively selected for short-lived acute energy-consuming responses but not for long-standing polygenic CIDs or chronic mental illness. These chronic situations would have generated a strong negative selection pressure if they occurred during early life and during the reproductive life history stage.
In contrast, if mutations were helpful to protect energy reserves, they were positively selected during evolution. This is true for memory responses because immediate reaction of an educated system can spare energy reserves. This is exemplified by the immune memory that leads to shorter, more effective and, finally, less energy-consuming reactions towards microbes. Importantly, acquisition of immune memory during the primary contact must fit into the above specified time frame of 19–43 days (and this happens as exemplified by the germinal center reaction in secondary lymphoid organs, ref. [99]). In this context, immunological tolerance versus harmless foreign antigens (e.g. of microbes on the skin) or harmless autoantigens is a memory function that spares energy reserves.
Another example of positively selected gene variants are genes for food intake and fat storage, both of which are important in determining above-mentioned total consumption time. Indeed, for a female Australopithecus afarensis it has been estimated that the consumption time was ∼19 days, while in a modern female Homo sapiens it is 43 days [14]. In human evolution, fat storage has been markedly increased over the last 6 million years [101]. Not surprisingly, the latest metaanalysis of genome-wide association studies of obesity and the metabolic syndrome found polymorphisms in genes relevant for food intake such as FTO (fat mass and obesity related), MC4R (melanocortin receptor type 4), POMC (proopiomelanocortin, the precursor of melanocortin) and genes relevant for fat storage such as the insulin-stimulating GIPR (gastric inhibitory polypeptide receptor) [102].
We explained these common signs and symptoms as a trade-off between energy allocation to the immune system versus distribution to the rest of the body. The immune system is selfish [103], because a fast and strong response is needed during infection to enable survival to future reproduction [104]. For example, inDrosophila melanogaster, which shares a common ancestor with us 530 million years ago, it was demonstrated that the immune system is selfish in the form that it requires a lot of energy which is then not available for other systems [104]. The authors described systemic changes in energy metabolism during an infectious immune challenge by extracellular adenosine. They found that extracellular adenosine, released from immune cells, induces a metabolic switch characterized by the suppression of nutrient storage and developmental growth in favor of the immune defense. This metabolic switch—a tradeoff between development and defense—is crucial for the resistance to infection. In Drosophila larvae lacking adenosine signaling, development is not suppressed and the resistance dramatically drops [104].
In sum, genes relevant for functioning of the supersystems are positively selected to protect energy stores under well-defined conditions (Table 4). Immune mechanisms were positively selected for either acute, highly energy-consuming responses terminated within 3–8 weeks or long-standing, energy-protective responses.
Table 4.
Positively selected immune mechanisms under defined conditions of A) acute, highly energy-consuming responses terminated within 3–8 weeks and B) long-standing, energy-protective responses
| Positively selected for acute, highly energy-consuming situations | Positively selected to protect energy stores |
|---|---|
| Immune response due to infection | Tolerogenic immune reactions |
| Immune response to foreign bodies | Control of inner and outer body surfaces |
| Clonal expansion and apoptosis | Memory of the immune system |
| Wound healing, burn wounds | Replacement of cells and tissue (physio logical regeneration and degeneration) |
| Implantation of stems cells into injured tissue | Implantation of a blastocyst into the uterine epithelium |
| Specific immunoglobulin production and affinity maturation | Immune phenomena facilitating semiallogenic pregnancy |
| High production rate of cytokines and chemokines | Allergic reactions (preformed response to clear or block threats on body surfaces) |
| Increased rate of phagocytosis | |
| Immune stimulated neoangiogenesis and wound healing |
The list is not complete.
MICROBIOTA, ENERGY REGULATION AND CIDs
Above, we mentioned that microbiota play an important role in HLA-B27 transgenic rats, which only develop CIDs when exposed to microbiota [77, 105]. Some discussed the influence of microbiota on development of CIDs simply as an unfortunate trigger of the innate and adpative immune systems leading to autoimmunity. However, microbiota also play an enormous role in metabolism and, thus, in energy regulation of the body [106, 107].
In experimental animals, there exist causal links between gut microbiota and obesity, which demonstrates an influence on energy regulation [107]. Conventionally raised mice develop diet-induced obesity, while germ-free animals do not [106]. Some bacterial divisions seem to play an important role for obesity [107, 108]. The obesity-triggering microbiome has an increased capacity to harvest energy from the diet [108], but it is also linked to more inflammation in the gut and liver [109, 110].
One may speculate that some bacterial divisions are important in establishing a more proinflammatory milieu supported by a higher degree of energy stores (obesity) to overcome acute infectious illness (free fatty acids as energy-rich fuels). Thus, the change in the microbiome can be positive for short-lived inflammatory diseases. However, the chronic change of the microbiome towards these proinflammatory bacterial divisions might be accompanied by chronic inflammatory illness. One has to keep in mind that bacterial divisions are also relevant for immunoregulatory function. Thus, it needs to be determined which groups play a causal role in the course of a CID.
The ‘NON-SPECIFICITY’ of SIGNS AND SYMPTOMS IN CIDs
In medicine, usually, the signs and symptoms in CIDs are thought to be an accident of the disease. The ultimate cause of CIDs and typical disease sequelae is not known. Symptomatology appears to be baffling and there is no common denominator to explain often common systemic disease sequelae in CIDs. In addition, physicians still believe that many symptoms are specific for one or the other CID, which most often is not the case.
At this point we recall that CIDs-related gene polymorphisms were not specifically selected for a given CID. In contrast, because of strong negative selection for these gene variants, natural selection would have outselected these genes. Thus, evolutionary medicine asks why these genes have not been outselected. In CIDs, which typically occur in a post-reproductive life history stage, uniform pathways and networks were positively selected as adaptive physiological mechanisms to deal with infections and wound healing in life history stages before or during reproduction (Table 4). We call this uniformity a non-specificity of CIDs.
This non-specificity is obvious in clinical medicine because symptoms in various infectious diseases including CIDs are surprisingly similar. Think of the erythrocyte sedimentation rate, which most often climbs independent of the type of acute inflammation or CID. Or think of fatigue, which often accompanies acute inflammation or CIDs. Many more common signs and symptoms have been recently summarized (Table 5) [13, 14, 103, 111–113].
Table 5.
Common signs and symptoms in chronic inflammatory systemic diseases
| Overt symptoms | Change |
|---|---|
| Amenorrhea | Increases |
| Avolition | Increases |
| Body temperature and sweating | Increases |
| Bone loss | Increases |
| Cachexia, cachectic obesity | Increases |
| Circadian rhythms of symptoms | Become apparent |
| Coagulation system | Activated |
| Disposition to pain (skeletal muscle, joints, other) | Increases |
| Erythrocyte sedimentation rate | Increases |
| Fatigue | Increases |
| Food intake, appetite (finally body weight), malnutrition | Decreases |
| Headache | Increases |
| Heart rate, sympathetic nervous tone | Increases |
| Hemoglobin per erythrocyte, inflammation-related anemia | Decreases |
| Hypertension and volume expansion/water retention | More often |
| Insulin resistance | Increases |
| Interleukin-6 serum levels (one example of a cytokine in the blood) | Increases |
| Libido, erectile dysfunction (loss of activity of the HPG axis) | Decreases |
| Numbness | Increases |
| Parasympathetic tone | Decreases |
| Physical activity | Decreases |
| Proinflammatory high density lipoproteins (HDL), dyslipidemia | Increases |
| Protein in the urine | Increases |
| Serum albumin | Decreases |
| Sleeping problems | Increases |
| Stress negatively influences inflammation | Becomes apparent |
| Symptoms of depression | Increases |
| Vertigo | Increases |
| Weakness | Increases |
This list is not complete. HPG axis, hypothalamic-pituitary-gonadal axis.
We explained these common signs and symptoms as a trade-off between energy allocation to the active immune system versus the rest of the body. While the presence of these phenomena is useful for short-lived inflammatory episodes in order to optimise recovery (it is not accident), long-term use in CIDs of the same programs is maladaptive.
CONCLUSIONS
CIDs lead to great and long-term suffering, imposing stress on people and high costs on society. During evolution, respective homeostatic networks of supersystems involved in CIDs were positively selected for short-lived acute energy-consuming responses but not for long-standing polygenic CIDs (Fig. 2). Several genetic factors of these supersystems that are favoring the development of CIDs have been identified. These factors typically increase fitness in young pre-reproductive and reproductive age, and resulting fitness benefits are expected to be higher than the fitness costs in post-reproductive age. Thus, life-time reproductive success will be increased, explaining why factors favoring CIDs have not been outselected but—under specific environmental conditions—have even been positively selected. Different CIDs result from uniform pathways and networks, which has been called the non-specificity of CIDs. For clinicians and the pharmaceutical industry to understand maladaptive responses in CIDs, they first need to comprehend adaptive functions in early life.
Figure 2.

There is a critical difference between typical inflammation and chronic inflammatory systemic diseases, which separates the asymptomatic phase from the symptomatic phase of a chronic inflammatory systemic disease, the asymptomatic-symptomatic-threshold (a-s-threshold). Importantly from an evolutionary point of view, reproduction is only impeded during the symptomatic, but not during the asymptomatic phase. Genes enabling an adaptive inflammatory response, allowing the organism to overcome an infection, will thus increase survival probability and the potential for future reproduction and as such evolutionary fitness in young age. After initiation of chronic systemic inflammation, however, reproduction is inhibited, and this for prolonged periods, often for years and until death, causing fitness costs. However, as these costs typically occur only at the end of the reproductive life-history stage or in post-reproductive age, these fitness costs are lower than the fitness benefits in early life leading to an overall increase in Darwinian fitness
FUNDING
For this work, the authors were funded by their institutions.
Conflict of interest: None declared.
REFERENCES
- 1.Stearns SC. The Evolution of Life Histories. Oxford: Oxford University Press, 1992. [Google Scholar]
- 2.Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol 2012;42:5–15. [DOI] [PubMed] [Google Scholar]
- 3.Soares PM, Borba EF, Bonfa E. et al. Gonad evaluation in male systemic lupus erythematosus. Arthritis Rheum 2007;56:2352–61. [DOI] [PubMed] [Google Scholar]
- 4.Suehiro RM, Borba EF, Bonfa E. et al. Testicular Sertoli cell function in male systemic lupus erythematosus. Rheumatology (Oxford) 2008;47:1692–7. [DOI] [PubMed] [Google Scholar]
- 5.Silva CA, Deen ME, Febronio MV. et al. Hormone profile in juvenile systemic lupus erythematosus with previous or current amenorrhea. Rheumatol Int 2011;31:1037–43. [DOI] [PubMed] [Google Scholar]
- 6.Villiger PM, Caliezi G, Cottin V. et al. Effects of TNF antagonists on sperm characteristics in patients with spondyloarthritis. Ann Rheum Dis 2010;69:1842–4. [DOI] [PubMed] [Google Scholar]
- 7.Richter JG, Becker A, Specker C. et al. Hypogonadism in Wegener's granulomatosis. Scand J Rheumatol 2008;37:365–9. [DOI] [PubMed] [Google Scholar]
- 8.Aikawa NE, Sallum AM, Leal MM. et al. Menstrual and hormonal alterations in juvenile dermatomyositis. Clin Exp Rheumatol 2010;28:571–5. [PubMed] [Google Scholar]
- 9.Wallenius M, Skomsvoll JF, Irgens LM. et al. Fertility in women with chronic inflammatory arthritides. Rheumatology (Oxford) 2011;50:1162–7. [DOI] [PubMed] [Google Scholar]
- 10.Brinkworth JF, Barreiro LB. The contribution of natural selection to present-day susceptibility to chronic inflammatory and autoimmune disease. Curr Opin Immunol 2014;31:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Straub RH, Besedovsky HO. Integrated evolutionary, immunological, and neuroendocrine framework for the pathogenesis of chronic disabling inflammatory diseases. Faseb J 2003;17:2176–83. [DOI] [PubMed] [Google Scholar]
- 12.Straub RH, Pongratz G, Cutolo M. et al. Increased cortisol relative to adrenocorticotropic hormone predicts improvement during anti-tumor necrosis factor therapy in rheumatoid arthritis. Arthritis Rheum 2008;58:976–84. [DOI] [PubMed] [Google Scholar]
- 13.Straub RH, Cutolo M, Buttgereit F. et al. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med 2010;267:543–60. [DOI] [PubMed] [Google Scholar]
- 14.Straub RH. Evolutionary medicine and chronic inflammatory state – known and new concepts in pathophysiology. J Mol Med 2012;90:523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Straub RH. Interaction of the endocrine system with inflammation: a function of energy and volume regulation. Arthritis Res Ther 2014;16:203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cortez D, Marin R, Toledo-Flores D. et al. Origins and functional evolution of Y chromosomes across mammals. Nature 2014;508:488–93. [DOI] [PubMed] [Google Scholar]
- 17.Rast JP, Buckley KM. Lamprey immunity is far from primitive. Proc Natl Acad Sci USA 2013;110:5746–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ritchie M. Neuroanatomy and physiology of the avian hypothalamic/pituitary axis: clinical aspects. Vet Clin North Am Exot Anim Pract 2014;17:13–22. [DOI] [PubMed] [Google Scholar]
- 19.Rai S, Szeitz A, Roberts BW. et al. A putative corticosteroid hormone in Pacific lamprey, Entosphenus tridentatus. Gen Comp Endocrinol 2014;10. [DOI] [PubMed] [Google Scholar]
- 20.Funakoshi K, Nakano M. The sympathetic nervous system of anamniotes. Brain Behav Evol 2007;69:105–13. [DOI] [PubMed] [Google Scholar]
- 21.Bergquist J, Tarkowski A, Ekman R. et al. Discovery of endogenous catecholamines in lymphocytes and evidence for catecholamine regulation of lymphocyte function via an autocrine loop. Proc Natl Acad Sci USA 1994;91:12912–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller LE, Jüsten HP, Schölmerich J. et al. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. Faseb J 2000;14:2097–107. [DOI] [PubMed] [Google Scholar]
- 23.Connor KL, Colabroy KL, Gerratana B. A heme peroxidase with a functional role as an L-tyrosine hydroxylase in the biosynthesis of anthramycin. Biochemistry 2011;50:8926–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyte M. Microbial endocrinology: Host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes 2014;5:381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dantzer R, O'Connor JC, Freund GG. et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 2008;9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter ME, Soden ME, Zweifel LS. et al. Genetic identification of a neural circuit that suppresses appetite. Nature 2013;503:111–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoglund E, Sorensen C, Bakke MJ. et al. Attenuation of stress-induced anorexia in brown trout (Salmo trutta) by pre-treatment with dietary l-tryptophan. Br J Nutr 2007;97:786–9. [DOI] [PubMed] [Google Scholar]
- 28.Clippinger TL, Bennett RA, Johnson CM. et al. Morbidity and mortality associated with a new mycoplasma species from captive American alligators (Alligator mississippiensis). J Zoo Wildl Med 2000;31:303–14. [DOI] [PubMed] [Google Scholar]
- 29.Ostensen M, Almberg K, Koksvik HS. Sex, reproduction, and gynecological disease in young adults with a history of juvenile chronic arthritis. J Rheumatol 2000;27:1783–7. [PubMed] [Google Scholar]
- 30.Masi AT, Josipovic DB, Jefferson WE. Low adrenal androgenic-anabolic steroids in women with rheumatoid arthritis (RA): gas-liquid chromatographic studies of RA patients and matched normal control women indicating decreased 11-deoxy-17-ketosteroid excretion. Semin Arthritis Rheum 1984;14:1–23. [DOI] [PubMed] [Google Scholar]
- 31.Cutolo M, Balleari E, Giusti M. et al. Sex hormone status of male patients with rheumatoid arthritis: evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Arthritis Rheum 1988;31:1314–7. [DOI] [PubMed] [Google Scholar]
- 32.Lahita RG, Bradlow HL, Ginzler E. et al. Low plasma androgens in women with systemic lupus erythematosus. Arthritis Rheum 1987;30:241–8. [DOI] [PubMed] [Google Scholar]
- 33.Tsigos C, Papanicolaou DA, Kyrou I. et al. Dose-dependent effects of recombinant human interleukin-6 on the pituitary-testicular axis. J Interferon Cytokine Res 1999;19:1271–6. [DOI] [PubMed] [Google Scholar]
- 34.Watson ME, Newman RJ, Payne AM. et al. The effect of macrophage conditioned media on Leydig cell function. Ann Clin Lab Sci 1994;24:84–95. [PubMed] [Google Scholar]
- 35.Turnbull AV, Rivier C. Inhibition of gonadotropin-induced testosterone secretion by the intracerebroventricular injection of interleukin-1 beta in the male rat. Endocrinology 1997;138:1008–13. [DOI] [PubMed] [Google Scholar]
- 36.Gruschwitz MS, Brezinschek R, Brezinschek HP. Cytokine levels in the seminal plasma of infertile males. J Androl 1996;17:158–63. [PubMed] [Google Scholar]
- 37.Gerard N, Caillaud M, Martoriati A. et al. The interleukin-1 system and female reproduction. J Endocrinol 2004;180:203–12. [DOI] [PubMed] [Google Scholar]
- 38.Adashi EY, Resnick CE, Croft CS. et al. Tumor necrosis factor alpha inhibits gonadotropin hormonal action in nontransformed ovarian granulosa cells. A modulatory noncytotoxic property. J Biol Chem 1989;264:11591–7. [PubMed] [Google Scholar]
- 39.Fray MD, Mann GE, Bleach EC. et al. Modulation of sex hormone secretion in cows by acute infection with bovine viral diarrhoea virus. Reproduction 2002;123:281–9. [PubMed] [Google Scholar]
- 40.Xiao E, Ferin M. Stress-related disturbances of the menstrual cycle. Ann Med 1997;29:215–9. [DOI] [PubMed] [Google Scholar]
- 41.Cooper GS, Miller FW, Pandey JP. The role of genetic factors in autoimmune disease: implications for environmental research. Environ Health Perspect 1999;107(Suppl 5):693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada Y, Wu D, Trynka G. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol 2013;190:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beecham AH, Patsopoulos NA, Xifara DK. et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013;45:1353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plagnol V, Howson JM, Smyth DJ. et al. Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PLoS Genet 2011;7:e1002216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bradfield JP, Qu HQ, Wang K. et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet 2011;7:e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrett JC, Clayton DG, Concannon P. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis 2013;72(Suppl 2):ii56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franke A, McGovern DP, Barrett JC. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010;42:1118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kallberg H, Padyukov L, Plenge RM. et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet 2007;80:867–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hughes LB, Morrison D, Kelley JM. et al. The HLA-DRB1 shared epitope is associated with susceptibility to rheumatoid arthritis in African Americans through European genetic admixture. Arthritis Rheum 2008;58:349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Svendsen AJ, Holm NV, Kyvik K. et al. Relative importance of genetic effects in rheumatoid arthritis: historical cohort study of Danish nationwide twin population. BMJ 2002;324:264–6. [PMC free article] [PubMed] [Google Scholar]
- 53.Silman AJ, MacGregor AJ, Thomson W. et al. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol 1993;32:903–7. [DOI] [PubMed] [Google Scholar]
- 54.Ringold DA, Nicoloff JT, Kesler M. et al. Further evidence for a strong genetic influence on the development of autoimmune thyroid disease: the California twin study. Thyroid 2002;12:647–53. [DOI] [PubMed] [Google Scholar]
- 55.Deapen D, Escalante A, Weinrib L. et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum 1992;35:311–8. [DOI] [PubMed] [Google Scholar]
- 56.Duffy DL, Spelman LS, Martin NG. Psoriasis in Australian twins. J Am Acad Dermatol 1993;29:428–34. [DOI] [PubMed] [Google Scholar]
- 57.Jarvinen P. Occurrence of ankylosing spondylitis in a nationwide series of twins. Arthritis Rheum 1995;38:381–3. [DOI] [PubMed] [Google Scholar]
- 58.Mathews JD, Whittingham S, Hooper BM. et al. Association of autoantibodies with smoking, cardiovascular morbidity, and death in the Busselton population. Lancet 1973;2:754–8. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez AM, Liang MH, Willett WC. et al. Reproductive factors, smoking, and the risk for rheumatoid arthritis. Epidemiology 1990;1:285–91. [DOI] [PubMed] [Google Scholar]
- 60.Heliovaara M, Aho K, Aromaa A. et al. Smoking and risk of rheumatoid arthritis. J Rheumatol 1993;20:1830–5. [PubMed] [Google Scholar]
- 61.Voigt LF, Koepsell TD, Nelson JL. et al. Smoking, obesity, alcohol consumption, and the risk of rheumatoid arthritis. Epidemiology 1994;5:525–32. [PubMed] [Google Scholar]
- 62.Silman AJ, Newman J, MacGregor AJ. Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins. Arthritis Rheum 1996;39:732–5. [DOI] [PubMed] [Google Scholar]
- 63.Stolt P, Bengtsson C, Nordmark B. et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis 2003;62:835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klareskog L, Stolt P, Lundberg K. et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006;54:38–46. [DOI] [PubMed] [Google Scholar]
- 65.Videm V, Cortes A, Thomas R. et al. Current Smoking is Associated with Incident Ankylosing Spondylitis - The HUNT Population-based Norwegian Health Study. J Rheumatol 2014;41:2041–8. [DOI] [PubMed] [Google Scholar]
- 66.Hernan MA, Olek MJ, Ascherio A. Cigarette smoking and incidence of multiple sclerosis. Am J Epidemiol 2001;154:69–74. [DOI] [PubMed] [Google Scholar]
- 67.Tobin MV, Logan RF, Langman MJ. et al. Cigarette smoking and inflammatory bowel disease. Gastroenterology 1987;93:316–21. [DOI] [PubMed] [Google Scholar]
- 68.Ghaussy NO, Sibbitt WL, Jr., Qualls CR. Cigarette smoking, alcohol consumption, and the risk of systemic lupus erythematosus: a case-control study. J Rheumatol 2001;28:2449–53. [PubMed] [Google Scholar]
- 69.Hardy CJ, Palmer BP, Muir KR. et al. Smoking history, alcohol consumption, and systemic lupus erythematosus: a case-control study. Ann Rheum Dis 1998;57:451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carlsson S, Midthjell K, Grill V. Smoking is associated with an increased risk of type 2 diabetes but a decreased risk of autoimmune diabetes in adults: an 11-year follow-up of incidence of diabetes in the Nord-Trondelag study. Diabetologia 2004;47:1953–6. [DOI] [PubMed] [Google Scholar]
- 71.Brenner S, Tur E, Shapiro J. et al. Pemphigus vulgaris: environmental factors. Occupational, behavioral, medical, and qualitative food frequency questionnaire. Int J Dermatol 2001;40:562–9. [DOI] [PubMed] [Google Scholar]
- 72.Kallberg H, Jacobsen S, Bengtsson C. et al. Alcohol consumption is associated with decreased risk of rheumatoid arthritis: results from two Scandinavian case-control studies. Ann Rheum Dis 2009;68:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosell M, Wesley AM, Rydin K. et al. Dietary fish and fish oil and the risk of rheumatoid arthritis. Epidemiology 2009;20:896–901. [DOI] [PubMed] [Google Scholar]
- 74.Parks CG, Conrad K, Cooper GS. Occupational exposure to crystalline silica and autoimmune disease. Environ Health Perspect 1999;107(Suppl 5):793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khuder SA, Peshimam AZ, Agraharam S. Environmental risk factors for rheumatoid arthritis. Rev Environ Health 2002;17:307–15. [DOI] [PubMed] [Google Scholar]
- 76.Peterson DA, Cardona RA. Specificity of the adaptive immune response to the gut microbiota. Adv Immunol 2010;107:71–107. [DOI] [PubMed] [Google Scholar]
- 77.Taurog JD, Richardson JA, Croft JT. et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med 1994;180:2359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Zhang D, Jia H. et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 2015;21:895–905. [DOI] [PubMed] [Google Scholar]
- 79.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution 1957;11:398–411. [Google Scholar]
- 80.Straub RH. Neuroendocrine immunology: new pathogenetic aspects and clinical application. Z Rheumatol 2011;70:767–74. [DOI] [PubMed] [Google Scholar]
- 81.LaFleur C, Granados J, Vargas-Alarcon G. et al. HLA-DR antigen frequencies in Mexican patients with dengue virus infection: HLA-DR4 as a possible genetic resistance factor for dengue hemorrhagic fever. Hum Immunol 2002;63:1039–44. [DOI] [PubMed] [Google Scholar]
- 82.Rekand T, Langeland N, Aarli JA. et al. Fcgamma receptor IIIA polymorphism as a risk factor for acute poliomyelitis. J Infect Dis 2002;186:1840–3. [DOI] [PubMed] [Google Scholar]
- 83.McKiernan SM, Hagan R, Curry M. et al. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology 2004;40:108–14. [DOI] [PubMed] [Google Scholar]
- 84.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol 2008;8:619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pertovaara M, Raitala A, Juonala M. et al. Autoimmunity and atherosclerosis: functional polymorphism of PTPN22 is associated with phenotypes related to the risk of atherosclerosis. The Cardiovascular Risk in Young Finns Study. Clin Exp Immunol 2007;147:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thio CL, Mosbruger TL, Kaslow RA. et al. Cytotoxic T-lymphocyte antigen 4 gene and recovery from hepatitis B virus infection. J Virol 2004;78:11258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Raitala A, Karjalainen J, Oja SS. et al. Helicobacter pylori-induced indoleamine 2,3-dioxygenase activity in vivo is regulated by TGFB1 and CTLA4 polymorphisms. Mol Immunol 2007;44:1011–4. [DOI] [PubMed] [Google Scholar]
- 88.Gu CC, Hunt SC, Kardia S. et al. An investigation of genome-wide associations of hypertension with microsatellite markers in the family blood pressure program (FBPP). Hum Genet 2007;121:577–90. [DOI] [PubMed] [Google Scholar]
- 89.Masi AT. Incidence of rheumatoid arthritis: do the observed age-sex interaction patterns support a role of androgenic-anabolic steroid deficiency in its pathogenesis? Br J Rheumatol 1994;33:697–9. [DOI] [PubMed] [Google Scholar]
- 90.Cramer DW, Xu H, Harlow BL. Does “incessant” ovulation increase risk for early menopause? Am J Obstet Gynecol 1995;172:568–73. [DOI] [PubMed] [Google Scholar]
- 91.Chang SH, Kim CS, Lee KS. et al. Premenopausal factors influencing premature ovarian failure and early menopause. Maturitas 2007;58:19–30. [DOI] [PubMed] [Google Scholar]
- 92.Roldan MB, White C, Witchel SF. Association of the GAA1013–>GAG polymorphism of the insulin-like growth factor-1 receptor (IGF1R) gene with premature pubarche. Fertil Steril 2007;88:410–7. [DOI] [PubMed] [Google Scholar]
- 93.Wong LE, Huang WT, Pope JE. et al. Effect of age at menopause on disease presentation in early rheumatoid arthritis: results from the canadian early arthritis cohort. Arthritis Care Res (Hoboken) 2015;67:616–23. [DOI] [PubMed] [Google Scholar]
- 94.van de Winkel JG, Anderson CL. Biology of human immunoglobulin G Fc receptors. J Leukoc Biol 1991;49:511–24. [DOI] [PubMed] [Google Scholar]
- 95.Morgan AW, Keyte VH, Babbage SJ. et al. FcgammaRIIIA-158V and rheumatoid arthritis: a confirmation study. Rheumatology (Oxford) 2003;42:528–33. [DOI] [PubMed] [Google Scholar]
- 96.Forbes-Ewan CH, Morrissey BL, Gregg GC. et al. Use of doubly labeled water technique in soldiers training for jungle warfare. J Appl Physiol (1985) 1989;67:14–8. [DOI] [PubMed] [Google Scholar]
- 97.Jones PJ, Jacobs I, Morris A. et al. Adequacy of food rations in soldiers during an arctic exercise measured by doubly labeled water. J Appl Physiol (1985) 1993;75:1790–7. [DOI] [PubMed] [Google Scholar]
- 98.McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav 2003;43:2–15. [DOI] [PubMed] [Google Scholar]
- 99.Meyer-Hermann ME, Maini PK. Cutting edge: back to “one-way” germinal centers. J Immunol 2005;174:2489–93. [DOI] [PubMed] [Google Scholar]
- 100.Hitze B, Hubold C, van DR. et al. How the selfish brain organizes its supply and demand. Front Neuroenergetics 2010;2:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zihlman AL, Bolter DR. Body composition in Pan paniscus compared with Homo sapiens has implications for changes during human evolution. Proc Natl Acad Sci USA 2015;112:7466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fall T, Ingelsson E. Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol 2014;382:740–57. [DOI] [PubMed] [Google Scholar]
- 103.Straub RH. Insulin resistance, selfish brain, and selfish immune system: an evolutionarily positively selected program used in chronic inflammatory diseases. Arthritis Res Ther 2014;16(Suppl 2):S4 pages 1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bajgar A, Kucerova K, Jonatova L. et al. Extracellular adenosine mediates a systemic metabolic switch during immune response. PLoS Biol 2015;13:e1002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rath HC, Herfarth HH, Ikeda JS. et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest 1996;98:945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Janssen AW, Kersten S. The role of the gut microbiota in metabolic health. Faseb J 2015;29:3111–23. [DOI] [PubMed] [Google Scholar]
- 107.Cox LM, Yamanishi S, Sohn J. et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014;158:705–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turnbaugh PJ, Ley RE, Mahowald MA. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–31. [DOI] [PubMed] [Google Scholar]
- 109.Lam YY, Ha CW, Campbell CR. et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS One 2012;7:e34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Henao-Mejia J, Elinav E, Jin C. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Straub RH. Concepts of evolutionary medicine and energy regulation contribute to the etiology of systemic chronic inflammatory diseases. Brain Behav Immun 2011;25:1–5. [DOI] [PubMed] [Google Scholar]
- 112.Straub RH. The origin of chronic inflammatory systemic diseases and their sequelae. San Diego: Academic Press, 2015. [Google Scholar]
- 113.Straub RH, Cutolo M, Pacifici M. Evolutionary medicine and bone loss in chronic inflammatory diseases – a theory of inflammation-related osteopenia. Semin Arthritis Rheum 2015;45:220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]