

Abstract
Anaerosalibacter massiliensis sp. nov. strain ND1T (= CSUR P762 = DSM 27308) is the type strain of A. massiliensis sp. nov., a new species within the genus Anaerosalibacter. This strain, the genome of which is described here, was isolated from the faecal flora of a 49-year-old healthy Brazilian man. Anaerosalibacter massiliensis is a Gram-positive, obligate anaerobic rod and member of the family Clostridiaceae. With the complete genome sequence and annotation, we describe here the features of this organism. The 3 197 911 bp long genome (one chromosome but no plasmid) contains 3271 protein-coding and 62 RNA genes, including six rRNA genes.
Keywords: Anaerosalibacter massiliensis, Clostridiaceae, culturomics, genome, taxonogenomics
Introduction
We propose Anaerosalibacter massiliensis strain ND1T (= CSUR P762 = DSM 27308) as the type strain of A. massiliensis sp. nov., a new species within the genus Anaerosalibacter (Rezgui et al., 2012). Strain ND1T was isolated from the stool sample of a 49-year-old Brazilian man as part of a culturomics study aiming at cultivating individually all bacterial species of the human gut microbiota [1], [2]. Anaerosalibacter massiliensis is a Gram-positive, obligate anaerobic, moderately halophilic and motile rod-shaped bacillus. The genus Anaerosalibacter (Rezgui et al., 2012) was created in 2012 and contains, to date, only one species, A. bizertensis (Rezgui et al., 2012), an obligate anaerobic, Gram-positive and rod-shaped bacillus that was isolated from sludge in Bizerte, Tunisia [3].
We recently proposed a new taxonomic approach called taxonogenomics to describe new bacterial species [4]. This polyphasic strategy combines phenotypic characteristics that may be obtained by most clinical microbiology laboratories wordwide, the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF) spectrum, and the analysis and comparison of the complete genome sequence. To date, taxonogenomics has enabled us to validly publish 13 bacterial names [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20].
We current study assessed the characteristics of A. massiliensis sp. nov. strain ND1T (= CSUR P762 = DSM 27308), including its phenotype and genome sequence. On the basis of these characteristics, we found that strain ND1T is sufficiently different from A. bizertensis to be classified as a new Anaerosalibacter species, and we propose the creation of the species Anaerosalibacter massiliensis sp. nov.
Organism Information
A stool sample was collected from a healthy 49-year-old Brazilian volunteer living in Manaus, Brazil. The patient signed informed consent, and the agreement of the local ethics committee of the IFR48 (Marseille, France) was obtained under agreement 09-022. The patient had not received any antibiotics at the time of sampling. The faecal sample was frozen at −80°C after collection and shipped to Marseille, France. A. massiliensis strain ND1T (Table 1) was isolated in November 2013 by cultivation on 5% sheep's blood–enriched agar (bioMérieux, Marcy l’Étoile, France) in anaerobic conditions after 14 days of preincubation of the stool specimen in a blood bottle culture, with addition of 5 mL rumen sheep. Strain ND1T exhibited a 98.05% 16S rRNA sequence identity with A. bizertensis (GenBank accession no. HQ534365), the phylogenetically closest bacterial species with a validly published name (Figure 1). Its 16S rRNA sequence was deposited in GenBank under accession number HG315673. This value was lower than the 98.7% 16S rRNA gene sequence threshold recommended by Stackebrandt and Ebers [46] to delineate a new species without carrying out DNA-DNA hybridization.
Table 1.
Classification and general features of Anaerosalibacter massiliensis strain ND1T according to MIGS recommendations [21].
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain: Bacteria | TAS [22] | |
| Phylum: Firmicutes | TAS [23] | ||
| Class: Clostridia | TAS [24] | ||
| Order: Clostridiales | TAS [25] | ||
| Family: Clostridiaceae | TAS [22], [26] | ||
| Genus: Anaerosalibacter | TAS [3], [23], [25] | ||
| Species: Anaerosalibacter massiliensis | IDA | ||
| Type strain ND1T | IDA | ||
| Gram stain | Positive | IDA | |
| Cell shape | Rod-shaped | IDA | |
| Motility | Motile | IDA | |
| Sporulation | Sporulating | IDA | |
| Temperature range | Mesophile | IDA | |
| Optimum temperature | 37°C | IDA | |
| pH range; optimum | 7 | IDA | |
| MIGS-6.3 | Salinity | Moderately halophilic (5 g/L) | IDA |
| MIGS-22 | Oxygen requirement | Anaerobic | IDA |
| Carbon source | Unknown | IDA | |
| Energy source | Unknown | IDA | |
| MIGS-6 | Habitat | Human gut | IDA |
| MIGS-15 | Biotic relationship | Free-living | IDA |
| MIGS-14 | Pathogenicity | Unknown | |
| Biosafety level | 2 | ||
| Isolation | Human faeces | ||
| MIGS-4 | Geographic location | Brazil | IDA |
| MIGS-5 | Sample collection time | November 2012 | IDA |
| MIGS-4.1 | Latitude | −3.1190275 | IDA |
| MIGS-4.1 | Longitude | −60.0217314 | IDA |
| MIGS-4.3 | Depth | Surface | IDA |
| MIGS-4.4 | Altitude | 86 m above sea level | IDA |
The pH range is 7–7.5, with optimal pH at 7.
MIGS, minimum information about a genome sequence.
Evidence codes are as follows: IDA, inferred from direct assay; and TAS, traceable author statement (i.e. a direct report exists in the literature). These evidence codes are from the Gene Ontology project (http://www.geneontology.org/GO.evidence.shtml) [27]. If the evidence code is IDA, then the property should have been directly observed, for the purpose of this specific publication, for a live isolate by one of the authors, or by an expert or reputable institution mentioned in the acknowledgements.
Fig. 1.

Phylogenetic tree highlighting position of Anaerosalibacter massiliensis sp. nov. strain ND1T relative to other type strains within Clostridiaceae. Strains and their corresponding GenBank accession numbers for 16S rRNA genes are (type = T): A. massiliensis strain ND1T, HG315673; A. bizertensis strain C5BELT, HQ534365[3]; A. bizertensis strain M3, HG964477; S. acetigenes strain Lup33T, NR_025151 [28]; C. ultunense strain BST, GQ461825[29]; T. creatinini strain BN11T, FR749955[30]; C. hastiforme strain ATCC 33268T, X80841[23], [31]; T. preacuta strain ATCC 25539T, GQ461814[32]; C. acidurici strain ATCC 7906T, M59084[23], [33]; C. aceticum strain ATCC 35044T, Y18183[34]; C. bifermentans strain ATCC 638T, AB075769[23], [35]; C. dakarense strain FF1T, KC517358[36]; C. saccharobutylicum strain ATCC BAA-117T, U16147[37]; C. butyricum strain ATCC 19398T, AJ458420[23], [38]; C. absonum strain ATCC 27555T, X77842[39]; C. senegalense strain JC122T, NR_125591 [7]; C. sporogenes strain ATCC 3584T, X68189[23], [35]; C. aciditolerans strain JW/YJL-B3T, DQ114945[40]; C. acidisoli strain CK74T, AJ237756[41]; Hungatella hatheweyi strain UB-B.2T, HE603919[42]; C. bolteae strain ATCC BAA-613T, AJ508452[43], [44]. 16S rRNA from A. massiliensis (1512 bp) was amplified and sequenced using fd1 (5′- AGAGTTTGATCCTGGCTCAG-3′) and rP2 (5′-ACGGCTACCTTGTTACGACTT-3′) primers; 16S rRNA sequences from all studied strains were aligned using CLUSTALW; total of 1182 nucleotide positions present in all studied sequences were used for phylogenetic inferences with maximum-likelihood method within MEGA6 software. Numbers at nodes are percentages of bootstrap values obtained by repeating analysis 500 times to generate majority consensus tree. Only bootstrap values greater than 70% are indicated. Acetobacterium bakii strain DSM 8239T, X96960[45], was used as outgroup. Scale bar = 2% nucleotide sequence divergence.
Different growth temperatures (30, 37, 45, 55°C) were tested. Growth occurred between 37 and 45°C, but optimal growth was observed at 37°C after 48 hours of incubation in anaerobic conditions. The colonies were 1.3 mm in diameter and moderately opaque on 5% sheep's blood–enriched agar (bioMérieux). Growth of the strain was tested under anaerobic and microaerophilic conditions using GENbag anaer and GENbag microaer systems respectively (bioMérieux) and under aerobic conditions with and without 5% CO2. Growth was observed only under anaerobic conditions and weakly with 5% CO2. No growth occurred under aerobic conditions. Gram staining showed rod-shaped, Gram-positive bacilli able to form spores (Figure 2). The motility test was positive. Cells grown on agar were moderately opaque and exhibited ranges in diameter and length of 0.5–1 and 2–5 μm respectively in electron microscopy (Figure 3). We also observed an oval and terminal spore of a 0.7 × 0.8 μm, causing a terminal swelling (Figure 3).
Fig. 2.
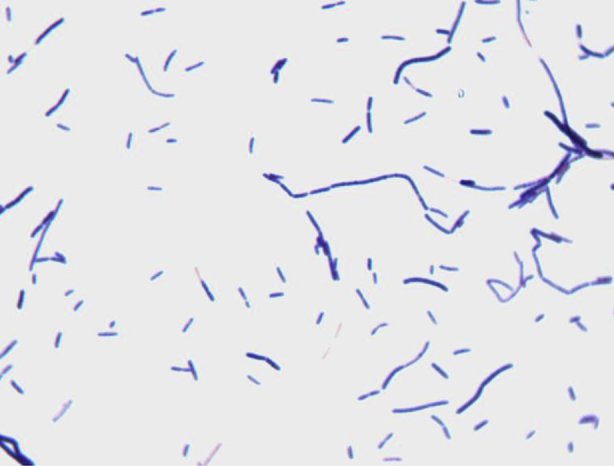
Gram staining of Anaerosalibacter massiliensis strain ND1T.
Fig. 3.

Transmission electron microscopy of Anaerosalibacter massiliensis strain ND1T using Morgani 268D (Philips, Amsterdam, The Netherlands) at operating voltage of 60 kV. Scale bar = 1 μm.
Strain ND1T exhibited neither catalase nor oxidase activities. Using API Rapid ID 32A (bioMérieux), positive reactions were observed for arginine dihydrolase, N-acetyl-β-glucosaminidase and pyroglutamic acid arylamidase. Negative reactions were observed for urease, indole, nitrate reduction, l-arabinose, ribose, mannose, d-lactose, d-fructose, d-maltose and sucrose activities. Using an API 50CH strip (bioMérieux), positive reactions were obtained for mannitol, arbutine, lactose and glycogen. Negative reactions were obtained for other constituents.
Anaerosalibacter massiliensis is susceptible to penicillin G, amoxicillin, gentamicin, amoxicillin/clavulanate, ciprofloxacine, metronidazole, ceftriaxone, imipenem, erythromycin, rifampicin and doxycycline but resistant to trimethoprim/sulfamethoxazole. Compared to A. bizertensis and representative species from other members of the genus Clostridium, A. massiliensis strain ND1T differed in a combination of nitrate reductase and β-galactosidase activities as well as arginine use (Table 2).
Table 2.
Differential characteristics of Anaerosalibacter massiliensis strain ND1T (data from this study); A. bizertensis strain C5BELT; Clostridium beijerinckii strain NCIMB 8052; C. dakarense strain FF1T; C. senegalense strain JC122T; C. ultunense strain BS T; and C. saccharobutylicum strain WM1T
| Property | A. massiliensis | A. bizertensis | C. beijerinckii | C. dakarense | C. senegalense | C. ultunense | C. saccharobutylicum |
|---|---|---|---|---|---|---|---|
| Cell diameter (μm) | 0.5–1 | 0.5–1 | 0.5–1.7 | 1.2 | 1.1 | 0.6 | 0.6 |
| Oxygen requirement | Anaerobic | Anaerobic | Anaerobic | Anaerobic | Anaerobic | Anaerobic | Anaerobic |
| Gram stain | + | + | + | + | + | + | − |
| Salt requirement | − | − | NA | − | − | NA | NA |
| Motility | + | + | + | + | + | + | − |
| Endospore formation | + | + | + | + | + | + | + |
| Production of: | |||||||
| Alkaline phosphatase | + | NA | NA | + | NA | NA | NA |
| Catalase | − | NA | − | − | − | − | NA |
| Oxidase | − | NA | NA | − | − | − | NA |
| Nitrate reductase | + | − | − | − | − | NA | + |
| Urease | − | NA | − | − | − | NA | NA |
| β-Galactosidase | + | − | NA | − | − | NA | NA |
| Acid from: | |||||||
| l-Arabinose | − | − | v | − | NA | − | + |
| Ribose | − | − | v | − | NA | − | w |
| Mannose | − | NA | + | − | NA | − | NA |
| Mannitol | + | NA | + | − | NA | − | w |
| Sucrose | − | NA | + | − | NA | − | w |
| d-Glucose | + | + | + | + | NA | − | |
| d-Fructose | − | − | NA | − | NA | − | + |
| d-Maltose | − | − | + | + | NA | − | w |
| d-Lactose | − | − | + | − | NA | − | w |
| Amino acid use: | |||||||
| Arginine | + | − | NA | + | − | NA | NA |
| Habitat | Human gut | Human gut | Human gut | Human gut | Human gut | Human gut | Human gut |
+, positive result; −, negative result; v, variable result; w, weakly positive result; NA, data not available.
MALDI-TOF protein analysis was carried out as previously described [47] using a Microflex spectrometer (Bruker Daltonics, Leipzig, Germany). Twelve individual colonies were deposited on a MTP 384 MALDI-TOF target plate (Bruker). The 12 ND1T spectra were imported into MALDI BioTyper 2.0 software (Bruker) and analysed by standard pattern matching (with default parameter settings) against the main spectra of 4706 bacteria, including one spectrum from Anaerosalibacter bizertensis, used as reference data in the BioTyper database. A score enabled the presumptive identification and discrimination of the tested species from those in the database; a score of >2 with a validated species enabled the identification at the species level and a score of <1.7 did not enable identification. For strain ND1T, no significant score was obtained, suggesting that our isolate was not a member of any known species (Fig. 4, Fig. 5). We added the spectrum from strain ND1T to our database (Figure 4). Finally, the gel view showed the spectral differences with other members of the family Clostridiaceae (Figure 5).
Fig. 4.

Reference mass spectrum from Anaerosalibacter massiliensis strain ND1T. This reference spectrum was generated by comparison of 12 individual colonies.
Fig. 5.

Gel view comparing Anaerosalibacter massiliensis strain ND1T to other members of family Clostridiaceae. Gel view displays raw spectra of loaded spectrum files arranged in pseudo-gel-like look. x-axis records m/z value. Left y-axis displays running spectrum number originating from subsequent spectra loading. Peak intensity is expressed as greyscale. The colour bar and right axis indicate the intensity each MALDI-TOF MS peak is displayed with and peak intensity in arbitrary units. Displayed species are detailed at left.
Fatty acid methyl ester (FAME) analysis by gas chromatography/mass spectrometry (GC/MS)
Approximately 40 mg of bacterial biomass was collected from three different culture plates. Cellular fatty acid methyl esters were prepared as described by Sasser [48]. GC/MS analyses were carried out on a Clarus 500 gas chromatograph equipped with a SQ8S MS detector (Perkin Elmer, Courtaboeuf, France). Two microlitres of FAME extracts were volatized at 250°C (split 20 mL/min) in a Focus liner with wool and separated on an Elite-5MS column (30 m, 0.25 mm i.d., 0.25 mm film thickness) using a linear temperature gradient (70 to 290°C at 6°C/min), enabling the detection of C4 to C24 fatty acid methyl esters. Helium flowing at 1.2 mL/min was used as carrier gas. The MS inlet line was set 250°C and EI source at 200°C. Full scan monitoring was performed from 45 to 500 m/z. All data were collected and processed using Turbomass 6.1 (Perkin Elmer). Fatty acid methyl esters were identified by spectral database search using MS Search 2.0 operated with the Standard Reference Database 1A (National Institute of Standards and Technology [NIST], Gaithersburg, MD, USA) and the FAMEs mass spectral database (Wiley, Chichester, UK). A 37-component FAME mix (Supelco; Sigma-Aldrich, Saint-Quentin Fallavier, France) was used for retention time correlations with estimated nonpolar retention indexes from the NIST database; FAME identifications were confirmed using this index. The major fatty acid detected was iso-C15:0 (80.3%). Small proportions of four other fatty acids were also detected (Table 3).
Table 3.
Total cellular fatty acid composition of Anaerosalibacter massiliensis strain ND1T
| Fatty acid | IUPAC name | Mean relative %a |
|---|---|---|
| Iso15:0 | 13-Methyl-tetradecanoic acid | 80.3 ± 0.3 |
| 16:0 | Hexadecanoic acid | 6.5 ± 0.1 |
| 18:1n9 | 9-Octadecenoic acid | 4.8 ± 0.1 |
| Iso5:0 | 3-Methyl-butanoic acid | 3.9 ± 0.6 |
| 18:0 | Octadecanoic acid | 2.4 ± 0.1 |
| 18:2n6 | 9,12-Octadecadienoic acid | tr |
| 18:1n7 | 11-Octadecenoic acid | tr |
| 14:0 | Tetradecanoic acid | tr |
| 4:0 | Butanoic acid | ND |
| 18:1n6 | 12-Octadecenoic acid | ND |
IUPAC, International Union of Pure and Applied Chemistry; ND, not detected, tr, trace amounts (<1%).
Mean peak area percentage calculated from analysis of FAMEs in three sample preparations ± standard deviation (n = 3).
Genome Sequencing Information
Genome project history
The organism was selected for sequencing on the basis of its phylogenetic position and 16S rRNA similarity to A. bizertensis and other members of the family Clostridiaceae. It is part of a culturomics study of human digestive flora that aims to isolate all bacterial species within human faeces [2]. It was the first genome of Anaerosalibacter species and the first genome of A. massiliensis sp. nov. A summary of the project information is shown in Table 4. The GenBank accession number is CCEZ01000001 and consists of 82 contigs. Table 4 shows the project information and its association with minimum information about a genome sequence (MIGS) version 2.0 compliance [21].
Table 4.
Genome sequencing information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | High-quality draft |
| MIGS-28 | Libraries used | Paired end and Mate pair |
| MIGS-29 | Sequencing platform | Illumina MiSeq |
| MIGS-31.2 | Fold coverage | 94.9× |
| MIGS-30 | Assemblers | Newbler version 2.5.3 |
| MIGS-32 | Gene calling method | Prodigal |
| GenBank date of release | 31 July 2014 | |
| NCBI project ID | CCEZ01000001 | |
| MIGS-13 | Source material identifier | DSM 27308 |
| Project relevance | Study of human gut microbiome |
MIGS, minimum information about a genome sequence.
Growth conditions and DNA isolation
A. massiliensis sp. nov., strain ND1T (= CSUR P762 = DSM 27308), was grown on 5% sheep's blood–enriched Columbia agar (bioMérieux) at 37°C in anaerobic atmosphere. Bacteria grown on three petri dishes were collected and resuspended in 4× 100 μL Tris-EDTA (TE) buffer. Then 200 μL of this suspension was diluted in 1 mL TE buffer for lysis treatment that included a 30-minute incubation with 2.5 μg/μL lysozyme at 37°C, followed by an overnight incubation with 20 μg/μL proteinase K at 37°C. Extracted DNA was then purified using three successive phenol–chloroform extractions and ethanol precipitations at −20°C overnight. After centrifugation, the DNA was resuspended in 160 μL TE buffer.
Genome sequencing and assembly
Genomic DNA of Anaerosalibacter massiliensis was sequenced on the MiSeq sequencer (Illumina, San Diego, CA, USA) using two sequencing strategies: paired end and mate pair. The paired end and the mate pair strategies were barcoded in order to be mixed respectively with 11 other genomic projects prepared with the Nextera XT DNA sample prep kit (Illumina) and 11 other projects with the Nextera Mate Pair sample prep kit (Illumina).
Genomic DNA was quantified at 35.3 ng/μL using the Qubit assay (Life Technologies, Carlsbad, CA, USA) and diluted to 1 ng/μL as input to prepare the paired-end library. After tagmentation to fragment and tag the DNA, limited cycle PCR amplification (12 cycles) completed the tag adapters and introduced dual-index barcodes. After purification using AMPure XP beads (Beckman Coulter, Fullerton, CA, USA), the library was normalized using specific beads according to the Nextera XT protocol (Illumina). The library was loaded onto the reagent cartridge and then onto the instrument along with the flow cell. Automated cluster generation and paired-end sequencing with dual index reads were performed in a single 39-hour run at a 2 × 250 bp read length. The sequencing generated 5.7 Gb, of which 732 000 reads were from A. massiliensis.
The mate pair library was prepared with 1 μg of genomic DNA using the Nextera mate pair Illumina guide. The genomic DNA sample was simultaneously fragmented and tagged with a mate pair junction adapter. The profile of the fragmentation was validated on an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA) with a DNA 7500 lab chip. The DNA fragments ranged in size from 1 to 11 kb, with an optimal size of 5 kb. No size selection was performed, and 600 ng of tagmented fragments were circularized. The circularized DNA was mechanically sheared to small fragments with optimum at 692 bp on the Covaris S2 device in microtubes (Covaris, Woburn, MA, USA). The library profile was visualized on a High Sensitivity Bioanalyzer LabChip (Agilent Technologies). The libraries were normalized at 2 nM and pooled. After a denaturation step and dilution at 10 pM, the pool of libraries was loaded onto the reagent cartridge and then onto the MiSeq instrument along with the flow cell. Automated cluster generation and the sequencing run were performed in a single 42-hour run at a 2 × 250 bp read length.
The mate pair sequencing generated 3.2 Gb, 925 000 reads of which were from A. massiliensis. The reads obtained from both applications were trimmed, and the optimal assembly was obtained through the Spades software with eight scaffolds, which generated a genome size of 3.28 Mb. The GC% was calculated at 29%.
Genome annotation
Open reading frames (ORFs) were predicted using Prodigal [49] with default parameters, but the predicted ORFs were excluded if they spanned a sequencing gap region. The predicted bacterial protein sequences were searched against the GenBank database [50] and the Clusters of Orthologous Groups (COGs) databases using BLASTP. The tRNAScanSE tool [51] was used to find tRNA genes, whereas ribosomal RNAs were found by using RNAmmer [52] and BLASTn against the GenBank database. Lipoprotein signal peptides and the number of transmembrane helices were predicted using SignalP [53] and TMHMM [54] respectively. ORFans were identified if their BLASTP E value was lower than 1e-03 for alignment length greater than 80 amino acids. If alignment lengths were smaller than 80 amino acids, we used an E value of 1e-05. Such parameter thresholds have already been used in previous works to define ORFans.
Because no genome was available for A. bizertensis [3], the only Anaerosalibacter species with standing in nomenclature, we compared the genome from A. massiliensis strain ND1T to those of other members of the family Clostridiaceae, including Clostridium ultunense strain Esp (CARA00000000) [29], C. acidurici strain 9a (CP003326) [23], [33] C. dakarense strain FF1 (GenBank accession no. CBTZ00000000) [36] and C. senegalense strain JC122 (CAEV00000000) [7]. The last two species have as yet no standing in nomenclature but were proposed as new species [7], [36] and are phylogenetically close to A. massiliensis. Ortholog sets composed of one gene from each of these five genomes were identified using Proteinortho 1.4 software [55] using threshold values of 30% protein identity and a 1e-05 E value. The average percentages of nucleotide sequence identity between corresponding orthologous sets were determined using the Needleman-Wunsch algorithm global alignment technique. Artemis [56] was used for data management, and DNAPlotter [57] was used for visualization of genomic features. The Mauve alignment tool was used for multiple genomic sequence alignment and visualization [58].
Genome properties
The genome of A. massiliensis strain ND1T is 3 197 911 bp long with a 29.70% G+C content (Figure 6, Table 5). Of the 3333 predicted genes, 3271 were protein-coding genes and 62 were RNAs. Six rRNA genes (two identical 16S rRNAs, two identical 23S rRNAs and two 5S rRNAs) and 56 predicted tRNA genes were identified in the genome. A total of 2376 genes (71.37%) were assigned a putative function. Fifty-three genes were identified as ORFans (1.59%). The remaining genes were annotated as hypothetical proteins. The properties and statistics of the genome are summarized in Table 5. The distribution of genes into COGs functional categories is presented in Table 6 and Figure 7.
Fig. 6.

Graphical circular map of chromosome. From outside in, outer two circles show ORFs oriented in forward (coloured by COGs categories) and reverse (coloured by COGs categories) direction respectively. Third circle marks rRNA gene operon (red) and tRNA genes (green). Fourth circle shows G+C% content plot. Innermost circle shows GC skew, with purple indicating negative values and olive positive values.
Table 5.
Nucleotide content and gene count levels of genome
| Attribute | Genome (total) |
|
|---|---|---|
| Value | % of totala | |
| Size (bp) | 3 197 911 | 100 |
| G+C content (bp) | 949 779 | 29.70 |
| Coding region (bp) | 2 785 986 | 87.12 |
| Total genes | 3333 | 100 |
| RNA genes | 62 | 1.86 |
| Pseudo genes | 53 | 1.59 |
| Protein-coding genes | 3271 | 98.13 |
| Genes with function prediction | 2376 | 71.28 |
| Genes assigned to COGs | 2102 | 63.06 |
| Genes with Pfam domains | 1660 | 49.80 |
| Genes with peptide signals | 68 | 2.04 |
| Genes with transmembrane helices | 824 | 24.72 |
COGs, Clusters of Orthologous Groups database.
Total is based on either size of genome in base pairs or total number of protein-coding genes in annotated genome.
Table 6.
Number of genes associated with 25 general COGs functional categories
| Code | Value | % of totala | Description |
|---|---|---|---|
| J | 164 | 5.02 | Translation |
| A | 0 | 0 | RNA processing and modification |
| K | 229 | 7.00 | Transcription |
| L | 164 | 5.02 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 30 | 0.92 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0 | Nuclear structure |
| V | 91 | 2.78 | Defense mechanisms |
| T | 156 | 4.77 | Signal transduction mechanisms |
| M | 117 | 3.58 | Cell wall/membrane biogenesis |
| N | 52 | 1.59 | Cell motility |
| Z | 0 | 0 | Cytoskeleton |
| W | 0 | 0 | Extracellular structures |
| U | 37 | 1.13 | Intracellular trafficking and secretion |
| O | 87 | 2.66 | Post-translational modification, protein turnover, chaperones |
| C | 195 | 5.96 | Energy production and conversion |
| G | 189 | 5.78 | Carbohydrate transport and metabolism |
| E | 223 | 6.82 | Amino acid transport and metabolism |
| F | 75 | 2.29 | Nucleotide transport and metabolism |
| H | 94 | 2.87 | Coenzyme transport and metabolism |
| I | 64 | 1.96 | Lipid transport and metabolism |
| P | 115 | 3.52 | Inorganic ion transport and metabolism |
| Q | 69 | 2.11 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 324 | 9.91 | General function prediction only |
| S | 203 | 6.21 | Function unknown |
| — | 1169 | 35.73 | Not in COGs |
COGs, Clusters of Orthologous Groups database.
Total is based on total number of protein-coding genes in annotated genome.
Fig. 7.

Distribution of functional classes of predicted genes in genomes from Anaerosalibacter massiliensis (AM), Clostridium acidurici (CA), C. dakarense (CD), C. senegalense (CS) and C. ultunense (CU) chromosomes according to clusters of orthologous groups of proteins.
Genomic comparison with other members of the family Clostridiaceae
We compared the genome of A. massiliensis strain ND1T those of C. acidurici strain 9a (GenBank accession no. CP003326) [23], [33], C. dakarense strain FF1T (CBTZ00000000) [36], C. senegalense strain JC122T (CAEV00000000) [7] and C. ultunense strain Esp (CARA00000000) [29]. The draft genome of A. massiliensis has a larger size than that of C. acidurici (3.19 and 3.11 Mb respectively) but is smaller than those of C. dakarense, C. senegalense and C. ultunense (3.73, 3.89 and 6.13 Mb respectively). The G+C content of A. massiliensis is higher than those of C. dakarense and C. senegalense (29.70, 27.98 and 26.8% respectively) but lower than those of C. acidurici and C. ultunense (29.9 and 40.9% respectively). The gene content of A. massiliensis is larger than those of C. acidurici (3330 and 2957 genes respectively) and smaller than those of C. dakarense, C. senegalense and C. ultunense (3916, 3761 and 6744 genes respectively). In addition, A. massiliensis shared 3271, 2839, 3808, 3704 and 5711 orthologous genes with C. acidurici, C. dakarense, C. senegalense and C. ultunense respectively. The average nucleotide sequence identity ranged from 71.49% to 66.45% between A. massiliensis and other members of the family Clostridiaceae (Table 7).
Table 7.
Numbers of orthologous protein shared between genomes (upper right)a
| Anaerosalibacter massiliensis | Clostridium acidurici | Clostridium dakarense | Clostridium senegalense | Clostridium ultunense | |
|---|---|---|---|---|---|
| A. massiliensis | 3271b | 1116 | 1071 | 1036 | 1323 |
| C. acidurici | 71.49 | 2839b | 974 | 941 | 1166 |
| C. dakarense | 69.38 | 69.51 | 3808b | 1045 | 1127 |
| C. senegalense | 69.42 | 69.24 | 70.12 | 3704b | 1077 |
| C. ultunense | 72.40 | 68.55 | 66.48 | 66.45 | 5711b |
Average percentage similarity of nucleotides corresponding to orthologous protein shared between genomes (lower left).
Numbers of proteins per genome.
Conclusions
On the basis of phenotypic, phylogenetic and genomic analyses, we formally propose the creation of Anaerosalibacter massiliensis sp. nov. that contains the strain ND1T. This bacterial strain was isolated from the faecal flora of a 49-year-old Brazilian man in good health.
Description of Anaerosalibacter massiliensis strain ND1T sp. nov.
Anaerosalibacter massiliensis (ma.si.li.en'.sis., L. gen. masc. n., from massiliensis, Massilia, the Latin name for Marseille, where A. massiliensis was first isolated).
Colonies were 1.3 mm in diameter and moderately opaque on 5% sheep's blood–enriched agar. Cells are Gram positive, rod shaped, motile and obligate anaerobic, with a mean diameter and length of 0.97 and 2.71 μm respectively. Optimal growth was observed at 37°C. The major fatty acid is iso-C15:0.
A. massiliensis is negative for catalase, oxidase, urease, indole, nitrate reduction, l-arabinose, ribose, mannose, d-lactose, d-fructose, d-maltose and sucrose activities but positive for arginine dihydrolase, N-acetyl-β-glucosaminidase, pyroglutamic acid arylamidase, mannitol, arbutine, lactose and glycogen activities. Cells were susceptible to penicillin G, amoxicillin, amoxicillin/clavulanate, ceftriaxone, imipenem, gentamicin, ciprofloxacine, metronidazole, erythromycin, rifampicin and doxycycline but resistant to trimethoprim/sulfamethoxazole.
The G+C content of the genome is 29.70%. The 16S rRNA and genome sequences are deposited in GenBank under accession numbers HG315673 and CCEZ01000001- CCEZ010000082 respectively. The type strain ND1T (= CSUR P762 = DSM 27308) was isolated from the faecal flora of a 49-year-old healthy Brazilian man.
Acknowledgements
The authors thank the Xegen Company (http://www.xegen.fr/) for automating the genomic annotation process. This study was funded by the Fondation Méditerranée Infection.
Conflict of Interest
None declared.
References
- 1.Dubourg G., Lagier J.C., Armougom F., Robert C., Hamad I., Brouqui P. The gut microbiota of a patient with resistant tuberculosis is more comprehensively studied by culturomics than by metagenomics. Eur J Clin Microbiol Infect. 2013;32:637–645. doi: 10.1007/s10096-012-1787-3. [DOI] [PubMed] [Google Scholar]
- 2.Lagier J.C., Armougom F., Million M., Hugon P., Pagnier I., Robert C. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–1193. doi: 10.1111/1469-0691.12023. [DOI] [PubMed] [Google Scholar]
- 3.Rezgui R., Maaroufi A., Fardeau M.L., Ben Ali Gam Z., Cayol J.L., Ben Hamed S. Anaerosalibacter bizertensis gen. nov., sp. nov., a halotolerant bacterium isolated from sludge. Int J Syst Evol Microbiol. 2014;62:2469–2474. doi: 10.1099/ijs.0.036566-0. [DOI] [PubMed] [Google Scholar]
- 4.Ramasamy D., Mishra A.K., Lagier J.C., Padhmanabhan R., Rossi M., Sentausa E. A polyphasic strategy incorporating genomic data for the taxonomic description of new bacterial species. Int J Syst Evol Microbiol. 2014;64:384–391. doi: 10.1099/ijs.0.057091-0. [DOI] [PubMed] [Google Scholar]
- 5.Lagier J.C., El Karkouri K., Nguyen T.T., Armougom F., Raoult D., Fournier P.E. Non-contiguous finished genome sequence and description of Anaerococcus senegalensis sp. nov. Stand Genomic Sci. 2012;6:116–125. doi: 10.4056/sigs.2415480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lagier J.C., Armougom F., Mishra A.K., Nguyen T.T., Raoult D., Fournier P.E. Non-contiguous finished genome sequence and description of Alistipes timonensis sp. nov. Stand Genomic Sci. 2012;6:315–324. doi: 10.4056/sigs.2685971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra A.K., Lagier J.C., Robert C., Raoult D., Fournier P.E. Non-contiguous finished genome sequence and description of Clostridium senegalense sp. nov. Stand Genomic Sci. 2012;6:386–395. doi: 10.4056/sigs.2766062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishra A.K., Lagier J.C., Robert C., Raoult D., Fournier P.E. Non contiguous-finished genome sequence and description of Peptoniphilus timonensis sp. nov. Stand Genomic Sci. 2012;7:1–11. doi: 10.4056/sigs.2956294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra A.K., Lagier J.C., Rivet R., Raoult D., Fournier P.E. Non-contiguous finished genome sequence and description of Paenibacillus senegalensis sp. nov. Stand Genomic Sci. 2012;7:70–81. doi: 10.4056/sigs.3056450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagier J.C., Gimenez G., Robert C., Raoult D., Fournier P.E. Non-contiguous finished genome sequence and description of Herbaspirillum massiliense sp. nov. Stand Genomic Sci. 2012;7:200–209. doi: 10.4056/sigs.3086474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roux V., El Karkouri K., Lagier J.C., Robert C., Raoult D. Non-contiguous finished genome sequence and description of Kurthia massiliensis sp. nov. Stand Genomic Sci. 2012;7:221–232. doi: 10.4056/sigs.3206554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokcha S., Ramasamy D., Lagier J.C., Robert C., Raoult D., Fournier P.E. Non-contiguous finished genome sequence and description of Brevibacterium senegalense sp. nov. Stand Genomic Sci. 2012;7:233–245. doi: 10.4056/sigs.3256677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramasamy D., Kokcha S., Lagier J.C., Nguyen T.T., Raoult D., Fournier P.E. Genome sequence and description of Aeromicrobium massiliense sp. nov. Stand Genomic Sci. 2012;7:246–257. doi: 10.4056/sigs.3306717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lagier J.C., Ramasamy D., Rivet R., Raoult D., Fournier P.E. Non contiguous-finished genome sequence and description of Cellulomonas massiliensis sp. nov. Stand Genomic Sci. 2012;7:258–270. doi: 10.4056/sigs.3316719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagier J.C., Elkarkouri K., Rivet R., Couderc C., Raoult D., Fournier P.E. Non contiguous-finished genome sequence and description of Senegalemassilia anaerobia gen. nov., sp. nov. Stand Genomic Sci. 2013;7:343–356. doi: 10.4056/sigs.3246665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagier J.C., El Karkouri K., Mishra A.K., Robert C., Raoult D., Fournier P.E. Non contiguous-finished genome sequence and description of Enterobacter massiliensis sp. nov. Stand Genomic Sci. 2013;7:399–412. doi: 10.4056/sigs.3396830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hugon P., Mishra A.K., Lagier J.C. Non-contiguous finished genome sequence and description of Brevibacillus massiliensis sp. nov. Stand Genomic Sci. 2013;8:1–14. doi: 10.4056/sigs.3466975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oren A., Garrity G.M. Validation list no. 154. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2013;63:3931–3934. doi: 10.1099/ijsem.0.001181. [DOI] [PubMed] [Google Scholar]
- 19.Oren A., Garrity G.M. Validation list no. 155. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2014;64:1–5. doi: 10.1099/ijsem.0.003881. [DOI] [PubMed] [Google Scholar]
- 20.Oren A., Garrity G.M. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2015;65:3763–3767. doi: 10.1099/ijsem.0.000632. [DOI] [PubMed] [Google Scholar]
- 21.Field D., Garrity G., Gray T., Morrison N., Selengut J., Sterk P. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woese C.R., Kandler O., Wheelis M.L. Towards a natural system of organisms: proposal for the domains Archae, Bacteria, and Eukarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skerman V.B.D., McGowan V., Sneath P.H.A. Approved list of bacterial names. Int J Syst Bacteriol. 1980;30:225–420. [Google Scholar]
- 24.Rainey F.A. Class II. Clostridia class nov. In: De Vos P., Garrity G.M., Jones D., Krieg N.R., Ludwig W., Rainey F.A., editors. Bergey’s manual of systematic bacteriology. 2nd ed. vol. 3. Springer; New York: 2009. p. 736. [Google Scholar]
- 25.Prévot A.R. Dictionnaire des bactéries pathogens. In: Hauduroy P., Ehringer G., Guillot G., Magrou J., Prevot A.R., Rosset A., editors. Masson; Paris: 1953. pp. 142–165. [Google Scholar]
- 26.Garrity G.M., Holt J.G. The road map to the manual. In: Garrity G.M., Boone D.R., Castenholz R.W., editors. Bergey’s manual of systematic bacteriology. 2nd ed. vol. 1. Springer; New York: 2001. pp. 119–169. [Google Scholar]
- 27.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez-Eugenio G., Fardeau M.L., Cayol J.L., Patel B.K., Thomas P., Macarie H. Sporanaerobacter acetigenes gen. nov., sp. nov., a novel acetogenic, facultatively sulfur-reducing bacterium. Int J Syst Evol Microbiol. 2002;52:1217–1223. doi: 10.1099/00207713-52-4-1217. [DOI] [PubMed] [Google Scholar]
- 29.Schnürer A., Schink B., Svensson B.H. Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int J Syst Bacteriol. 1996;46:1145–1152. doi: 10.1099/00207713-46-4-1145. [DOI] [PubMed] [Google Scholar]
- 30.Farrow J.A.E., Lawson P.A., Hippe H., Gauglitz U., Collins M.D. Phylogenetic evidence that the Gram-negative nonsporulating bacterium Tissierella (Bacteroides) praeacuta is a member of the Clostridium subphylum of the Gram-positive bacteria and description of Tissierella creatinini sp. nov. Int J Syst Bacteriol. 1995;45:436–440. doi: 10.1099/00207713-45-3-436. [DOI] [PubMed] [Google Scholar]
- 31.Maclennan J.D. The nonsaccharolytic plectridial anaerobes. J Pathol Bacteriol. 1939;49:535–548. [Google Scholar]
- 32.Collins M.D., Shah H.N. Reclassification of Bacteroides praeacutus Tissier (Holdeman and Moore) in a new genus, Tissierella, as Tissierella praeacuta comb. nov. Int J Syst Bacteriol. 1986;36:461–463. [Google Scholar]
- 33.Barker H.A. The fermentation of definite nitrogenous compounds by members of the genus Clostridium. J Bacteriol. 1938;36:322–323. [Google Scholar]
- 34.Gottschalk G., Braun M. Revival of the name Clostridium aceticum. Int J Syst Bacteriol. 1981;31:476. [Google Scholar]
- 35.Bergey D.H., Harrison F.C., Breed R.S., Hammer B.W., Huntoon F.M., editors. Bergey’s manual of determinative bacteriology. Williams & Wilkins; Baltimore: 1923. [Google Scholar]
- 36.Lo C.I., Mishra A.K., Padhmanabhan R., Samb B., Sow A.G., Robert C. Non-contiguous finished genome sequence and description of Clostridium dakarense sp. nov. Stand Genomic Sci. 2013;9:14–27. doi: 10.4056/sigs.4097825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keis S., Shaheen R., Jones D.T. Emended descriptions of Clostridium acetobutylicum and Clostridium beijerinckii, and descriptions of Clostridium saccharoperbutylacetonicum sp. nov. and Clostridium saccharobutylicum sp. nov. Int J Syst Evol Microbiol. 2001;51:2095–2103. doi: 10.1099/00207713-51-6-2095. [DOI] [PubMed] [Google Scholar]
- 38.Prazmowski A. Hugo Voigt; Leipzig: 1880. Untersuchung über die Entwickelungsgeschichte und Fermentwirking einiger Bacterien-Arten. Inaugural dissertation. [Google Scholar]
- 39.Nakamura S., Shimamura T., Hayase M., Nishida S. Numerical taxonomy of saccharolytic clostridia, particularly Clostridium perfringens–like strains: descriptions of Clostridium absonum sp. n. and Clostridium paraperfringens. Int J Syst Bacteriol. 1973;23:419–429. [Google Scholar]
- 40.Lee Y.J., Romanek C.S., Wiegel J. Clostridium aciditolerans sp. nov., an acid-tolerant spore-forming anaerobic bacterium from constructed wetland sediment. Int J Syst Evol Microbiol. 2007;57:311–315. doi: 10.1099/ijs.0.64583-0. [DOI] [PubMed] [Google Scholar]
- 41.Kuhner C.H., Matthies C., Acker G., Schmittroth M., Göβner A.S., Drake H.L. Clostridium akagii sp. nov. and Clostridium acidisoli sp. nov., acid-tolerant, N2-fixing clostridia isolated from acidic forest soil and litter. Int J Syst Evol Microbiol. 2000;50:873–881. doi: 10.1099/00207713-50-2-873. [DOI] [PubMed] [Google Scholar]
- 42.Kaur S., Yawar M., Kumar P.A., Suresh K. Hungatella effluvii gen. nov., sp. nov., an obligately anaerobic bacterium isolated from an effluent treatment plant, and reclassification of Clostridium hathewayi as Hungatella hathewayi gen. nov., comb. nov. Int J Syst Evol Microbiol. 2014;64:710–718. doi: 10.1099/ijs.0.056986-0. [DOI] [PubMed] [Google Scholar]
- 43.Song Y., Liu C., Molitoris D.R., Tomzynski T.J., Lawson P.A., Collins M.D., Finegold S.M. Clostridium bolteae sp. nov., isolated from human sources. Syst Appl Microbiol. 2003;26:84–89. doi: 10.1078/072320203322337353. [DOI] [PubMed] [Google Scholar]
- 44.Validation list no 92. Validation of publication of new names and new combinations previously. Int J Syst Evol Microbiol. 2003;53:935–937. doi: 10.1099/ijs.0.02853-0. [DOI] [PubMed] [Google Scholar]
- 45.Johnson J.L., Moore W.E.C., Moore L.V.H. Bacteroides caccae sp. nov., Bacteroides merdae sp. nov., and Bacteroides stercoris sp. nov., isolated from human feces. Int J Syst Bacteriol. 1986;36:499–501. [Google Scholar]
- 46.Stackebrandt E., Ebers J. Taxonomic parameters revisited: tarnished gold standards. Microbiol Today. 2006;33:152–155. [Google Scholar]
- 47.Seng P., Drancourt M., Gouriet F., La Scola B., Fournier P.E., Rolain J.M. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin Infect Dis. 2009;49:543–551. doi: 10.1086/600885. [DOI] [PubMed] [Google Scholar]
- 48.Sasser M. MIDI Inc.; Newark, DE: 2006. Bacterial identification by gas chromatographic analysis of fatty acids methyl esters (GC-FAME) Technical note 101. [Google Scholar]
- 49.Hyatt D., Chen G.L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benson D.A., Karsch-Mizrachi I., Clark K., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2012;40:D48–D53. doi: 10.1093/nar/gkr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lagesen K., Hallin P., Rodland E.A., Staerfeldt H.H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bendtsen J.D., Nielsen H., von Heijne G., Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 54.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 55.Lechner M., Findeib S., Steiner L., Marz M., Stadler P.F., Prohaska S.J. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rutherford K., Parkhill J., Crook J., Horsnell T., Rice P., Rajandream M.A. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 57.Carver T., Thomson N., Bleasby A., Berriman M., Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25:119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Darling A.C., Mau B., Blattner F.R., Perna N.T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]