

Abstract
The p53-binding domains of Mdm2 and Mdmx, two negative regulators of the tumor suppressor, p53, are validated targets for cancer therapeutics, but correct binding poses of some proven inhibitors, particularly the nutlins, have been difficult to obtain with standard docking procedures. Virtual screening pipelines typically draw from a database of compounds represented with 1D or 2D structural information, from which one or more 3D conformations must be generated. These conformations are then passed to a docking algorithm that searches for optimal binding poses on the target protein. This work tests alternative pipelines using several commonly-used conformation generation programs (LigPrep, ConfGen, MacroModel and Corina/Rotate) and docking programs (GOLD, Glide, MOE-dock and AutoDock Vina) for their ability to reproduce known poses for a series of Mdmx and/or Mdm2 inhibitors, including several nutlins. Most combinations of these programs using default settings fail to find correct poses for the nutlins but succeed for all other compounds. Docking success for the nutlin class requires either computationally-intensive conformational exploration, or an “anchoring” procedure that incorporates knowledge of the orientation of the central imidazoline ring.
Keywords: Docking, Conformation generation, Mdm2, Mdmx, Mdm4, p53, Retinoblastoma, Nutlins, Re-docking, Cross-docking
INTRODUCTION
The tumor suppressor protein, p53, mediates cell-cycle arrest, senescence, or apoptosis in response to different types of cellular stresses such as oncogene activation, DNA damage, and hypoxia.1–4 Approximately 50% of human cancers have alterations in the p53 gene, resulting in inactivation or loss of p53, while in other cancers, p53 pathways are down-regulated or otherwise disabled. Among the most important p53-disabling mechanisms is overexpression of a negative regulator named murine double minute 2 (Mdm2; Hdm2 in humans).5,6 A related negative regulator of the p53 pathway is Mdmx (also called Mdm4). Mdm2 and Mdmx have homologous p53-binding domains, both of which interact with the same N-terminal transactivation domain (NTD) of p53. Mdmx overexpression has been observed in a number cancers,7,8 including pediatric retinoblastoma.9 Mdmx knockdown in breast carcinoma and retinoblastoma cell lines leads to p53-dependent growth-arrest or apoptosis.7,9,10 Although more effort has gone into targeting Mdm2, these studies suggest that Mdmx is a potential chemotherapeutic target for certain cancers, particularly retinoblastoma.11
Three dimensional structures of the p53NTD peptide with both Mdm2 and Mdmx have been determined.12,13 The structures show very similar protein folds and p53 binding modes, and together with mutagenesis studies, reveal the interactions important for binding. In particular, three hydrophobic residues on p53NTD — F19, W23 and L26 — occupy three hydrophobic pockets (Figure 1) on Mdm2/Mdmx (termed the Phe, Trp and Leu pockets) and make substantial contributions to binding.14
Figure 1.

Both Mdm2 (A) and Mdmx (B) interact with the NTD peptide of p53 via the same Phe, Trp and Leu sidechains which fit into three corresponding pockets on the protein. Structural data from PDB structures 1YCR [12] and 3DAB [13].
Development of drugs that rescue p53 function by disrupting the Mdm2/p53 interaction has been an important topic in cancer research.15,16 The first, and best characterized17–20 small molecule inhibitors of the Mdm2/p53 interaction were the nutlins, a series of tetra-substituted imidazolines discovered by high-throughput screening (HTS).21,22 Nutlin analogs are currently in early clinical trials.23,24 The “MI” series compounds, based on a spiro-oxindole scaffold, contain a 6-chlorooxindole unit that mimics p53NTD Trp23. These compounds were developed by a structure-based de novo design strategy targeting Mdm2.25–29 Benzodiazepinedione-based inhibitors were initially discovered by HTS against Mdm2, and were further optimized to improve pharmacokinetic properties.30–33 Amgen identified chromenotriazolopyrimidine derivatives as sub-micromolar Mdm2 inhibitors using HTS.34 A recently discovered class of Mdm2/Mdmx inhibitors is based on an imidazole-indole scaffold.35 All of the aforementioned chemical classes are well studied as Mdm2 inhibitors, but most of them are much weaker against Mdmx. WW298 (imidazole-indole derivative) is currently among one of the best reported inhibitors for Mdmx with Ki = 11 µM.35 Representatives of the five classes above have been co-crystallized with Mdm2. A co-crystal structure of the WW298/Mdmx complex is the only published Mdmx—small molecule structure available. Table 1 provides a summary of the available Mdm2/Mdmx-small molecule complex structures, all of which are included in the present study.
Table 1.
Compounds and structures used in this study.
| Compound | Cpd. Structure | Complex Structure |
Mdm2 IC50 (µM) |
Mdmx IC50 (µM) |
|---|---|---|---|---|
|
1 nutlin-2 |
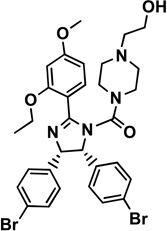 |
Mdm2: 1RV1 [18] |
0.14 [18] |
NDa |
|
2 imidazole-indole derivative |
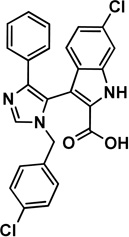 |
Mdm2: 3LBK [35] |
1.17 [35] |
44.5 [35] |
|
3 imidazole-indole, WW298 |
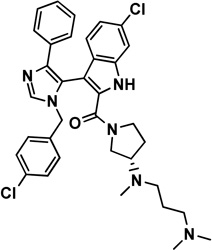 |
Mdmx: 3LBJ [35] |
0.19 [35] |
19.7 [35] |
|
4 MI63 analog |
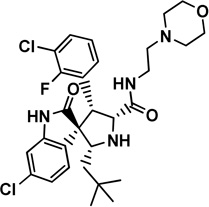 |
Mdm2: 3LBL [35] |
0.18 [35] |
NDa |
|
5 benzo- diazepineone |
 |
Mdm2: 1T4E [30] |
Kd=0.080 [30] |
NDa |
|
6 chromenotriazolo- pyrimidine |
 |
Mdm2: 3JZK [34] |
1.23 [34] |
NDa |
|
7 nutlin-3a |
 |
Mdm2: modeled via 1RV1 |
0.09 [18] |
Ki=9.38 [26] |
ND: not determined
Two classes of Mdmx inhibitors were not included in this study. Dimer inducing compounds developed by Roche revealed the possibility of dual inhibition of Mdm2 and Mdmx with similar affinity,36 but we consider this unique dimer-induction mechanism as outside the scope of this work. Recent pyrrolidine derivatives that were developed as specific Mdm2 inhibitors have shown moderate activity towards Mdmx (Ki = 2.11 µM),37 but no structures of this compound in complex with either Mdm2 or Mdmx are available. The difficulty of obtaining Mdmx co-structures could be due to several factors such as poor expression, poor Mdmx solubility, and weak binding of the inhibitors. Attempts to study the binding orientation of nutlin-3a with Mdmx did not produce definitive results due to weak binding affinity and the dynamic behavior of the protein.38 Although the nutlins are thought to bind Mdm2 and Mdmx in a similar orientation, some studies suggest that steric hindrance between nutlin-3 and Mdmx side chains M53 and Y99 may prevent nutlin-3 from binding to Mdmx in the same mode as it does to Mdm2.39 Differences in the binding cavity have led to several hypotheses regarding the binding mode of nutlins with Mdmx.38–40
A key component of structure-based drug discovery is computational docking, which seeks to position small molecules within the binding site of the target macromolecule in an energetically optimal fashion, and then score the resulting complexes to rank their binding affinity.41–43 Docking programs have been in development and use since the early 1980s,44 and over 50 different docking software packages such as GOLD, Glide, MOE, AutoDock, and Surflex-Dock are currently available to researchers.45–50 All docking programs require a 3D structure of the target protein, and even though modern docking programs search over 3D compound conformations, they also generally require an acceptable starting 3D structure for the compound. Therefore, a virtual screening pipeline that selects from a database of compounds represented in 1D (e.g., SMILES51) or 2D (e.g., connection table) must include a separate step for the generation of initial conformers. Programs for this purpose include LigPrep,52 ConfGen,53 MacroModel54 and Corina/Rotate.55 These programs may also be responsible for enumerating stereoisomers, tautomers, and ionization states. The overall pipeline is illustrated in Figure 2.
Figure 2.

Flow diagram for virtual screening. The components of the process tested here are the conformation generation and docking components. The specific programs tested are shown in italics. The normal information flow for virtual screening of a compound database is shown with solid arrows. Additional information flow for using a known target—compound complex to test the overall pipeline or its docking component is shown with dashed arrows and a dashed box.
Previous studies on the performance of currently available docking programs have found that most can reproduce experimental binding poses with deviations averaging from 1.5 to 2 Å.43,56 It has been shown that the initial input ligand conformation affects binding pose determination, and therefore multiple initial ligand conformations for each ligand are typically required.57,58 Retrieving the correct binding conformation for nutlins with Mdm2 has been particularly difficult,59 perhaps due to the complex stereochemistry60 or the high dimensionality of the ligand’s conformational space.
Here, we present systematic re-docking and cross-docking tests (dashed components of Figure 2) of the ability of four conformation-generation programs (LigPrep, ConfGen, Corina/Rotate and Macromodel) and four docking programs (GOLD, Glide, MOE-dock and AutoDock Vina) to correctly dock representatives of the five previously described Mdm2 inhibitor classes to Mdm2 and/or Mdmx. These programs typically have parameters that control the tradeoff between thoroughness of searching and computational cost, and our tests explore variations of these settings.
METHODS
Input structures and ligand preparation
Atomic structures of receptor—ligand complexes for ligand compounds 1—6 were taken from published structures deposited in the Protein Data Bank61 as indicated in Table 1. For compound 7 (nutlin-3a) bound to Mdm2, the complex structure was modeled from that of compound 1 (nutlin-2 bound to Mdm2). Hydrogen atoms were added to these heavy-atom ligand structures using the Protein Preparation Wizard in the Maestro suite from Schrodinger,62 and the results were manually cross checked for correctness of bond order and protonation of hetero atoms. This program also performs a short energy minimization of the complex to relieve small geometry problems and steric clashes. For the tests involving 2D-to-3D conversion of ligand structures, the 2D chemical structures in SDF format were taken either from our in-house chemical database or from the PubChem compound database.63 Care was taken that the chirality and ionization states were those of the known bound form, and this information was retained in subsequent 2D-to-3D conversion.
Conformation generation
The programs tested for the generation of multiple conformers for docking input were LigPrep,52 ConfGen,53 Corina/Rotate55 and MacroModel-MTLM (mixed torsional/low-mode); 54 the selection of options controlling the cost and comprehensiveness of searching is described in Results. For the nutlin compounds, 1 and 7, an anchor-based strategy was also used. This type of method is most useful when studying an analog series where the binding orientation of the scaffold is known. During library preparation, the three-dimensional coordinates of the scaffold are preserved and the ligand variations are enumerated in situ using virtual chemical reactions. Only the conformations of the new moieties are explored. For this study, anchor-based conformers were prepared in MOE using the QuaSAR-CombiGen module, and side-chain conformations for the resulting molecules were explored using low mode sampling with the scaffold fixed. The scaffold was defined as the central imidazoline ring oriented such that the two phenyl ring substitutions point into the Trp and Leu pockets.
Docking
The docking programs used were GOLD, Glide, AutoDock Vina and MOE-dock. The program GOLD 5 (Genetic Optimization for Ligand Docking) from Cambridge Crystallographic Data Center, UK45 uses a genetic algorithm (GA) for docking flexible ligands into protein binding sites. The protein active sites were defined as extending 6 Å around the ligand positions observed in the crystal structures. For each of the GA runs, a maximum number of 100,000 operations were performed on a population of 100 individuals. GoldScore was used to rank-order the docked conformations, and the cutoff parameters for van der Waals and hydrogen-bond interactions were chosen as 4.0 and 2.5 Å, respectively. Glide v5.546,47 has three choices for default docking simulations: standard precision (SP), high-throughput virtual screening (HTVS), in which conformational sampling is significantly reduced relative to SP, and extra-precision (XP), which is designed to reduce the false positive rate. Sampling in XP is more extensive, using the results from SP docking as a starting point generating a more fine-grained set of conformers. In this study we have used Glide-SP except where use of Glide-XP is indicated. The Glide algorithm utilizes pre-computed grids generated using receptor sites defined by the centroids of the crystallographic ligands. The docking protocol starts with the systematic conformational expansion of the ligand, followed by placement in the receptor site. Minimization of the ligand in the field of the receptor is then carried out using the OPLS-AA force field with the default distance-dependent dielectric. The lowest energy poses are then subjected to a Monte Carlo procedure that samples nearby torsional minima. Different compounds can then be ranked using GlideScore, a modified version of the ChemScore function that includes terms for steric clashes and buried polar groups. Default van der Waal’s scaling was used (1.0 for the receptor and 0.8 for the ligand). MOE-Dock is a part of the Molecular Operating Environment software package from Chemical Computing Group.48 The active site was generated for each enzyme using the MOE alpha site finder. The ligand molecules were placed in the site with the Triangle Matcher method, and ranked with the London dG scoring function. The ten best poses (default is 30) were retained and further refined by energy minimization in the pocket, followed by rescoring with the GBVI/WSA dG scoring function. AutoDock Vina 1.149 is an open-source program for docking simulations. It uses the Iterated Local Search global optimizer algorithm64 in which a succession of steps consisting of a mutation and a local optimization are taken, with each step being accepted according to the Metropolis65 criterion. In the present study we have utilized the AutoDock plugin which can be incorporated in Pymol66 to analyze the binding sites and prepare the input parameters for AutoDock Vina runs. The grid box parameters were generated with the default selection around the crystallographic ligands and these parameters were utilized to generate the configuration file to run the AutoDock Vina. The receptor structural information required by the program (the pdbqt files) were generated using Pymol with the AutoDock plugin, and the ligand pdbqt files were generated by utilizing scripts included in the Molecular Graphics Laboratory (MGL) tools.67
Evaluation of dock poses
The straightforward use of root mean square deviation (RMSD) from the crystal structure as a measure of docking accuracy is subject to artifacts when equivalent, but differently indexed atoms are carried into one another by ring flips or other transformations performed in docking or conformation generation. We have therefore chosen, for each compound, a subset of heavy atoms not subject to such problems for RMSD comparisons (Figure S1). In evaluating cross-docking results, protein structures must first be superposed, and this was done so as to minimize the RMSD of Cα atoms using the superposition method included in Pymol and these aligned structures were utilized for all docking experiments in this study. RMSD calculations for ligand poses were done using the superposition module of Maestro v9. In addition to an RMSD criterion of docking success, we have also required that the ligand moieties that occupy the Trp, Leu and Phe pockets of the binding site are observed, by visual inspection, to occupy these sites (‘pocket occupancy test’).
RESULTS
Re-docking and cross-docking studies require compounds whose structure in complex with the target proteins are known, and we have based this study on the widest available set of such compounds in complex with Mdm2. Also included are a crystal structure of WW298 with Mdmx, and a model of a nutlin-3a—Mdmx complex based on the assumption that the binding mode is homologous to that of nutlin-2 with Mdm2. These compounds are shown in Table 1.
Comparison of available crystal structures
The Mdm2/Mdmx inhibitors considered in this study possess moieties that mimic the hydrophobic side chains of the p53-NTD that are essential for binding: F19, W23 and L26. Superposition of the crystal structures of Mdm2 bound with compounds 1, 2, 5, or 6, shows that the protein has negligible conformational differences at the binding sites when binding these diverse scaffolds (Figure 3A). The crystal structure of Mdm2 co-crystallized with the MI63-analog (compound 4) shows a significant reorientation of Y67, which makes up part of the Phe-binding pocket, suggesting some localized induced-fit phenomena in the MI63 case (Figure 3B). The sole crystal structure of Mdmx with a bound drug, the indole-imidazole derivative, WW298 (compound 3) has an overall binding-site shape very similar to those of the Mdm2 structures, but there are some differences at the Leu-binding pocket due to sequence differences (Figure 3C). The similarity of protein structure found in these comparisons implies that docking protocols that focus on ligand flexibility but not protein flexibility should have an acceptable success rate.
Figure 3.

Comparison of binding pockets from ligand bound co-crystal structures of Mdm2 and Mdmx. The structure of Mdm2 with nutlin-2 (compound 1) bound (1RV1) was taken as the reference and is shown in cyan. The nutlin-2 structure is shown as green sticks. (A) Superposition of protein structures of Mdm2 from co-crystal structures involving ligands 2, 5 and 6 on the 1 co-structure. These show very little variation in the backbone or residues lining the binding pocket. (B) Superposition of the 4-Mdm2 (3LBL) complex on the 1-complex shows changes in the phenylalanine binding pocket, particularly in residue Y67. (C) Superposition of the complex of Mdmx with 3 (1LBJ) on the 1-Mdm2 complex shows differences between the two targets, Mdmx and Mdm2, particularly near the leucine pocket.
Bridging water molecules are found in the complexes of compounds 2 and 5 with Mdm2 but not in the others. Because bridging water was not a consistent feature, we did not attempt to include them in the present docking tests.
Tests using ligand conformations from co-crystal structures
Ligand coordinates were taken from the six co-crystal structures and processed for use in docking algorithms as described in Methods. Correct bond order and ionization state were verified manually. These ligand conformers were taken as input coordinates to the four tested docking programs in re-docking (docking a ligand against the protein coordinates of the co-structure with that same ligand) and cross-docking (docking against protein coordinates of a co-structure with a different ligand). Because initial ligand coordinates were taken directly from the co-structures, these tests evaluate only the docking component of the pipeline (Figure 2) and exclude any effects from conformation generation. For each program, we took the 10 best scoring poses and determined the lowest RMSD relative to the crystallographic pose. A success was defined as RMSD ≤ 2.0Å and passing the pocket occupancy test as detailed in Methods.
Re-docking
All four docking programs performed well in re-docking experiments (diagonal elements of Figure 4). AutoDock Vina performed best by achieving an RMSD < 1.0 Å for all six re-docking tests, while the other programs had RMSD values between 1.0 and 2.0 Å for one of the six. Although Glide achieved a satisfactory RMSD (1.31 Å), it failed the pocket occupancy test for compound 1 (nutlin-2) due to a ring flip that left an ethoxy group protruding into solvent rather than placed in the Phe pocket. However, when the test was rerun using the extra-precision option (Glide-XP) instead of the standard-precision option (Glide-SP), the correct orientation was found and the RMSD fell to 0.39Å (Figure 5A vs. B).
Figure 4.

Re- and cross-docking experiments using compound conformers taken from protein— compound complex structures. The lowest-RMSD pose from the 10 best poses identified by each program is reported. Diagonal boxes (bold borders) indicate re-docking while the others represent cross docking. RMSD (Å) indicated by color: Green, 0—1; Yellow, 1—2; Orange, 2— 3; Red, >3. Failure to meet pocket-contact criteria is indicated by dual colors (first half of the box based on RMSD and second half red indicating failure to meet pocket criteria)
Figure 5.

Effects of precision settings on re- and cross-docking performance of GOLD and Glide on nutlins with initial conformers from crystal structures. The protein from the 1RV1 crystal structure is shown as a surface and bound nutlin-2 is shown as gray sticks in all subpanels. The dock poses of compound 1 from Glide-SP (A) and Glide-XP (B) are shown with magenta and yellow sticks, respectively. The dock poses of compound 7 from GOLD with (C) and without (D) the early termination options are shown with magenta and yellow sticks, respectively.
Docking compound 7 (nutlin-3a) into the Mdm2 coordinates from the nutlin-2 co-structure, 1RV1, is not strictly a re-docking test, but it is germane given the structural similarity of the two compounds, 1 and 7. GOLD performed poorly on this test when using the default settings; the lowest RMSD found was 3.75Å, and the pose was clearly incorrect (Figures 4 & 5C). We explored various alternatives to the default settings and found that the problem lay in the “early termination” option. This feature stops the search when 3 docked solutions within 1.5 Å of each other are found. Upon switching off the early termination option, the RMSD was reduced to 1.73Å and the pose had correct contacts with all three hydrophobic pockets (Figure 5D). Other evaluations of GOLD docking have found that turning off early termination provides more robust results but can significantly increase computational costs.57
With respect to the re-docking tests, including compound 7 into 1RV1, the most successful programs using recommended settings were MOE-dock and Autodock Vina. In the six strict redocking tests, these programs achieved binding poses within 2.0 Å RMSD for all tests; Autodock Vina achieved poses within 1.0 Å for all six, and MOE dock achieved poses within 1.0 Å for all but compound 4. Both programs placed the correct moieties into the correct binding pockets in all six re-docking tests as well as the 7-into-1RV1 test. GOLD and Glide-SP performed nearly as well by RMSD in most tests, but failed to find correct pocket contacts in at least one case, and had to be re-run using more computationally intensive settings to correct these failures.
Cross-docking
In cross-docking runs, AutoDock Vina performed best by passing 32 out of the 35 cross-docking tests (Figure 4). The performance of MOE-dock was close (31/35), and GOLD and Glide-SP tied (23/35). For non-nutlins (compounds 2—6), the cross-docking success rates were: AutoDock Vina and Glide (22/25), MOE-dock (21/25), and GOLD (19/25). For the nutlins (1 and 7), both AutoDock Vina and MOE-dock succeeded in cross-docking tests with six protein structures, a 10/10 success rate, while GOLD and Glide-SP achieved only 4/10 and 1/10, respectively.
Aside from evaluating docking algorithms, re-/cross-docking studies can be used to select the single protein structure most likely to yield the highest docking success rate. Of the five structures of Mdm2 in complex with small molecules, the overall success of re-/cross-docking appears to be best with the 1RV1 structure (Mdm2 in complex with nutlin-2); AutoDock Vina achieved success with all 7 compounds, while GOLD, MOE-dock and GLIDE achieved success in 6 cases. There may be a slight pro-nutlin bias in this result; for the non-nutlins, the overall success rate is 19/20 with 1RV1, while the rate with 3JZK (compound 6 in complex with Mdm2) is 20/20. However, the one non-nutlin failure with 1RV1 occurs because of an RMSD of 2.0 Å, which is just on the success/failure threshold. Figure S2 compares the AutoDock Vina poses of all 7 compounds onto 1RV1, with those of the native crystal complexes.
In contrast, the overall failure rate was highest with the 3LBL structure (Mdm2 bound to compound 4, MI-63 analog). As noted above, this structure differs from the others by a rearrangement of Tyr67 causing the Phe-binding pocket to shrink. This is presumably related to the occupancy of this pocket by the neopentyl group of this compound, which is smaller than the corresponding groups of the other compounds. When compound 5, which all programs fail to cross dock into 3LBL, is superposed on the 3LBL structure based on the Mdm2/compound 5 complex, 1T4E, the iodo substituent at position 7, which in the native complex provides hydrophobic contacts in the Phe pocket, clashes with Tyr67.
Tests using computationally-generated ligand conformations
Testing the full workflow illustrated in Figure 2 requires dropping the use of crystallographic ligand conformations as inputs to docking and generating input conformers computationally. Typically, the conformation-generation programs can also explore alternative charge states, tautomeric states, and configurations at chiral centers. Here, we take bond structures from the literature and crystallographic studies, and use the programs for conformation generation only. The generated conformers are used as inputs to docking programs. Pose selection and success criteria are the same as those used in the tests with crystallographic ligand poses. In cases where multiple compound conformers are generated ahead of the docking component, 10 poses are selected from each docking run. Except where stated otherwise, both re-docking and cross-docking are considered equally, and the success percentages for a given compound or compound set include all possible re- and cross-dockings.
A single conformer (LigPrep)
LigPrep52 is a program that takes 2D chemical structures as input and, in the present usage, generates a single 3D structure as output. The structure is expected to be at or near a local energy minimum, but not necessarily either a global minimum or the optimal conformation for binding to the target. Indeed, the structure is calculated without reference to the target. Therefore, the task of conformational searching is left entirely to the docking algorithm. LigPrep-generated conformers of each of the 7 test compounds were docked against the six crystal structures. The results, summarized in Figure 6, are worse for all docking programs compared to docking using crystallographically-determined binding conformations. Failures are most acute for the nutlin compounds, 1 and 7 (Figure 6 vs. Figure 4) often due to a large error in overall orientation within the binding site (Figure S3). For the non-nutlins, 2—6, results of MOE-dock starting from the LigPrep conformer are significantly worse than those starting from the co-structure conformer, while for GOLD, Glide-SP (which is part of the same Schrodinger tool-chain as LigPrep) and AutoDock Vina, the results are only slightly worse. These results show that it is problematic to rely on docking algorithms alone to find binding-relevant ligand conformations for the Mdm2/Mdmx targets. The nutlin failures are noteworthy because they occur regardless of whether the target Mdm2 structure is the nutlin co-structure (1RV1) or another.
Figure 6.

Re-/cross-docking results of LigPrep-generated conformers against the Mdmx2 or Mdmx crystal structures. Color coding as in Figure 4.
Multiple conformations from faster algorithms
Two widely used programs for generating multiple conformations of compounds, ConfGen and Corina/Rotate, were tested with default settings which, in both cases, are for relatively fast calculations at the expense of thoroughness.
ConfGen (v2.1) is part of the Schrodinger software suite; in standard mode it has four different prepackaged protocols: Very Fast, Fast, Intermediate and Comprehensive. We first tested ConfGen Fast, which generates up to 5 conformers per degree of freedom and eliminates redundant conformers at a threshold of 1.0Å RMSD. In addition, conformers whose energies are more than 25 kcal/mol above that of the lowest-energy conformer are eliminated. We also selected the option to minimize structures by the truncated Newton conjugate gradient (TNCG) method68 using 100 iterations for input structures and 50 for output structures. The generated conformers were then utilized for re-/cross-docking in the same protocol as that used for the crystallographic ligand conformers discussed earlier. For the non-nutlin compounds, the success rate is significantly better than with the single LigPrep-generated conformer and very similar to that achieved using crystallographic compound conformers (Figure 7A). However, the performance is quite poor for the nutlins (Figure 7B).
Figure 7.

Percent success rates with various combinations of conformer generation and docking algorithm. A. Non-nutlin compounds 2—6. B. Nutlins 1 and 7
“Corina/Rotate Optimal” refers to the use of the Corina and Rotate programs with a set of search parameters designed for rapid generation of conformers for large scale virtual screening. Corina (v 3.44) is a rule- and data-based method for generating three-dimensional models from two-dimensional connection tables; it includes generation of alternative ring conformations.55 Models output from Corina are used as input to the Rotate program (v 1.4) which varies the torsion angles of the acyclic portions of molecules based on rules derived from a statistical analysis of small molecule crystal structures from the Cambridge Structural Database.69 The “optimal” parameter settings are, for Corina: rc (generate multiple ring conformations for rings less than 10 atoms), mc=10 (generate a maximum of 10 conformations per molecule), flapn (allow pyramidal ring nitrogens with one exocyclic neighbor to invert configuration when generating ring conformations), and sc (generate ring conformations simultaneously for efficiency); and for Rotate: rot=5 (process a maximum of the five most internal rotatable bonds), rms_ta=60 (cluster conformations in torsional space using an RMS cutoff of 60 degrees, and report only cluster heads). The results of applying Corina/Rotate Optimal with each of the docking programs are similar to those found with ConfGen Fast for the non-nutlins (compounds 2—6) results are comparable with those obtained with crystallographic conformers, while for the nutlins (compounds 1 and 7) the results are poor (Figure 7).
Multiple conformations from more comprehensive algorithms
We next ran ConfGen and Corina/Rotate by changing parameters to enable more extensive conformational exploration at the cost of increased computational time; these parameter settings are called “ConfGen Comprehensive” and “Corina/Rotate Best”, respectively. The ConfGen Comprehensive method generates up to 75 conformers per degree of freedom and uses an RMSD value of 0.5 Å to detect redundant conformers, and the threshold for eliminating higher-energy structures is 500 kJ/mol (119 kcal/mol). ConfGen Comprehensive also does more ring sampling (more conformers per ring, more ring combinations, and a higher energy limit for rings).53 The same minimization parameters for input and output structures were selected as with the ConfGen Fast method. The Corina/Rotate Best method uses identical parameters for the Corina part as in the Optimal method, but conformational sampling is increased in the Rotate part by changing the two parameter settings: rot=7 and rms_ta=30.
Another comprehensive search algorithm tested was mixed torsional/low-mode sampling (MTLM) which is implemented in MacroModel 9.0.54 It combines a Monte Carlo method of exploring torsional space that efficiently locates widely separated minima on the potential energy surface70 with a low-mode conformational search method that searches along the energetically “soft” degrees of freedom.71 The OPLS_2005 force field72 is used, and energy minimization is 500 steps of the TNCG method. The energy window for saving structures is set to 21 kJ/mol (default value). The redundancy threshold is 0.5Å RMSD and redundant conformers are removed. A maximum of 1000 steps is allowed for the Monte Carlo sampling and a maximum of 100 steps is allowed for the low-mode searching. No cutoff for the maximum number of conformers to be generated was set. Default parameters were used for the remaining options.
All 3 of these more comprehensive conformer-generation methods increase computational costs relative to their faster counterparts. The comprehensive methods themselves take more computer time, but more significantly, each generated conformer must be put through the subsequent docking algorithm, leading to an overall cost that scales approximately like the number of generated conformers. The ConfGen Fast and Corina/Rotate Optimal methods generated an average of 11.7 and 7.1 conformations per compound, respectively, while the ConfGen Comprehensive, Corina/Rotate Best and MTLM methods generated 74.7, 183 and 101, respectively (Table 2). Thus the more comprehensive methods’ costs were approximately an order of magnitude more than those of the fast methods. Within either the fast or comprehensive family of methods, there is significant variation in the numbers generated both per compound and by method. In most cases (as in the averages) MTLM generates a number of conformers intermediate between ConfGen Comprehensive and Corina/Rotate Best, which generated the largest number of conformers.
Table 2.
Numbers of conformers generated by method and compound
| Comp | MW | Rot Bonds |
ConfGen Fast |
Corina Optimal |
ConfGen Compr. |
Corina Best |
MTLM | Anchor |
|---|---|---|---|---|---|---|---|---|
| 1 | 686.4 | 10 | 8 | 11 | 131 | 527 | 217 | 6 |
| 2 | 461.3 | 5 | 9 | 2 | 55 | 12 | 35 | N.A. |
| 3 | 629.6 | 11 | 28 | 8 | 99 | 294 | 166 | N.A. |
| 4 | 577.5 | 8 | 22 | 10 | 197 | 196 | 163 | N.A. |
| 5 | 580.1 | 4 | 7 | 4 | 21 | 30 | 6 | N.A. |
| 6 | 536.2 | 2 | 1 | 8 | 1 | 32 | 2 | N.A. |
| 7 | 581.5 | 8 | 5 | 7 | 19 | 187 | 118 | 11 |
For non-nutlins, the docking performance of the overall pipeline when comprehensive methods are used for conformer generation remains similar to that when the fast methods are used (Figure 7A), which is in turn similar to that using the crystallographic conformations. For nutlins, however, there is a marked improvement in docking performance when using the comprehensive methods (Figure 7B). Of these, ConfGen Comprehensive (which generates the smallest number of conformers for the nutlins) has the lowest success rate. Corina/Rotate Best and MTLM both performed well for nutlins, giving results similar to each other and comparable to the success rates found with non-nutlins. A rather surprising result is that the comprehensive methods can “rescue” docking programs that fail when given only the crystallographic pose (lines for GOLD and Glide-SP in Figure 7B).
Anchored conformer generation for nutlins
In anticipation of a project (to be reported elsewhere) that would require the docking of a large number of nutlin derivatives, we developed a conformation generation method that would use existing knowledge regarding the nutlin pose in Mdm2. The idea was to “anchor” the conformations of the central imidazoline ring and the two moieties that bind in the Leu and Trp pockets, while allowing other degrees of freedom to be searched (see Methods for details). This anchoring strategy was applied only at the conformation-generation stage; in the docking stage no anchoring restraints were applied.
The Anchor-based method generated 17 conformers for the two nutlin derivatives, compared to 13 and 18 total conformers generated by ConfGen Fast and Corina/Rotate optimal, respectively. MOE-dock and AutoDock Vina yielded 100% success with anchor based conformers of both nutlin derivatives (compounds 1 and 7) while Glide-SP and GOLD provided correct poses in 9 and 7 instances out of 12 re-/cross-docking cases, respectively.
DISCUSSION
Mdm2 and Mdmx are targets of much current interest16,73 but have proven to be difficult targets for docking in virtual screening campaigns, particularly in the case of the nutlin class of compounds, which remain perhaps the best characterized and most promising class.38,59,74,75 We have therefore made a systematic study, using available structural data, of the ability of various conformation-generating and docking protocols to correctly predict the binding modes of known inhibitors. Our tests involved various combinations of four different docking programs and four different conformation-generating tools, all of which are widely used.
A test that one might expect any docking program to pass is re-docking, in which a known ligand/receptor complex structure is used as input and the docking program is asked to reproduce the known pose from that input structure. Re-docking success demonstrates a docking algorithm’s ability to sample pose space and find the correct poses among its few top-scoring poses. For the available complexes of Mdm2 or Mdmx with a small-molecule ligand, all docking programs could successfully re-dock compounds in the test set although for one of them an extra-precision option had to be invoked to achieve success with nutlin-2. A somewhat more difficult test is cross-docking, in which a ligand conformation from one complex is taken as the starting point for docking against a receptor structure bound to a different ligand. Cross-docking probes a docking algorithm’s ability to account for perturbations of the receptor conformations resulting from complexes with different ligands. (The docking algorithms tested here incorporate ligand flexibility, while receptor flexibility has not been included.) However, in the case of Mdm2 in complex with small-molecule inhibitors, we observed these receptor differences to be relatively small, implying that cross-docking should not be a major challenge. The relative lack of receptor variation from ligand to ligand has enabled us to focus the present study on issues of ligand flexibility without addressing receptor flexibility. It should be noted that cases of flexible receptors add an additional level of complexity that the methods explored here do not address. As expected, the cross-docking tests were mostly successful, although there was more variation from program to program, and the programs that had difficulty had their most acute problems with the nutlin class (Figure 4). The re- and cross-docking tests involving compounds 2 and 5 and their corresponding Mdm2 protein structures were done without the bridging water molecules seen in crystal structures of the complexes. In spite of this, the re-docking tests succeeded with all docking programs, as did the cross-docking tests for compound 2. Somewhat more difficulty was encountered in cross-docking compound 5, but not as much difficulty as for the nutlins (Figure 4) and much of this difficulty was apparently associated with the rearrangement of Y67 noted above. Missing bridging water molecules do not seem to be a factor in difficulties with nutlin docking as the complex structure, 1RV1,18 does not show any.
In typical, large-scale virtual screening campaigns, the correct docked pose must be identified with no foreknowledge of the bound ligand conformation. In such cases, it is critical that relevant 3D conformations be explored. Our tests of docking with the single LigPrep-generated conformers showed relatively poor overall performance, and complete failure in the case of the nutlins. The docking programs alone could not adequately search the compounds’ internal degrees of freedom starting from an arbitrary (but sterically reasonable) point in conformational space. The limitations of docking algorithms in handling ligand flexibility have been observed previously, and methods to pre-generate a diverse set of starting conformers have been developed.53,55,57,58 For the non-nutlin compounds in our test set, these methods provided satisfactory results with standard parameter settings using our panel of docking algorithms. However, for the nutlins, the default settings of the multi-conformer generation programs were not sufficient. Success in docking nutlins with no foreknowledge of the correct pose was achieved only when much more comprehensive conformer generation was enabled. This more comprehensive search had little effect on the non-nutlin results apart from increasing compute time.
The use of more comprehensive conformation generation prior to docking as described in Figure 2 greatly increases computational cost, primarily because it increases the number of docking runs proportional to the number of conformations generated (Table 2). If these more comprehensive calculations are only needed for a subset of the candidate compounds, it would be useful to identify in advance which compounds require additional sampling. It might be expected that larger and more flexible compounds require greater conformational exploration. However, the nutlins do not particularly stand out from the other compounds in terms of either molecular weight, number of rotatable bonds, radius of gyration, or flexibility as calculated by MOE’s descriptors method (Table S1).76 One possible explanation is that the nutlins have what might be called ‘four hands for three pockets’: in addition to the three moieties that fit in the Trp, Leu and Phe pockets, they have a piperazine moiety that the docking programs sometimes erroneously fit into one of these three pockets.
The apparent need for a more fine-grained search for starting conformations of nutlins for the docking programs suggests that docking success requires an input conformation quite close to the correct final conformation. To put it in a strong sense, it suggests that the docking programs have radii of convergence that are unusually small in the nutlin case. One might therefore expect that from the ensemble generated by the conformation-generation step of Figure 2 the conformer closest (in the RMSD sense) to the crystal structure conformer would be the one that, when submitted to docking, would lead to the correct pose. However, our findings show otherwise. A detailed examination of our results shows that while the successful inputs are indeed within 1.0 Å of the correct conformation, they may only be the 10th or 20th best in RMSD terms, compared to inputs that do not lead to docking success. In other words, the domain of input conformers that map, via docking, to correct output poses may lie within a region of conformation space expressible by a simple metric such as RMSD. Yet, within this region, the true domain for success is more scattered and complex. The chaotic behavior of docking simulations has been analyzed recently.77 This complexity may explain our finding that sufficiently comprehensive conformation generation can “rescue” cross-docking failures.
For the particular problem of the nutlins docking against Mdm2 or Mdmx, we have found a practical solution by exploiting our knowledge of the complexes using an anchoring-based method. This allows conformational searching to be focused on those moieties that are to be varied in compound optimization efforts going forward. The computational costs are similar to those of the standard conformation-generation parameter settings of ConfGen Fast and Corina/Rotate Optimal.
We have tested four different docking programs, AutoDock Vina, Glide, GOLD and MOE-dock. In several specific settings, such as cross docking from X-ray structures, there were significant differences in program performance (Figure 4) but these differences were not consistent throughout the study. In general, a program that did best in one setting, did not do best in another, and no very clear overall “best” program emerged. These results support the “consensus” approach to the use of docking programs.78,79
Supplementary Material
Acknowledgements
This work was supported by the American Lebanese Syrian Associated Charities (ALSAC). N.B and D.B were also supported by National Institutes of Health (GM57513).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Details related to atoms considered in the ligand RMSD calculations (Figure S1), dock poses orientations in case of re-/cross-docking of crystal ligand conformations (Figure S2), LigPrep generated conformations (Figure S3) as well as important descriptor information of test set ligands (Table S1) are provided as supplementary information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 2.Teodoro JG, Evans SK, Green MR. Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. J. Mol. Med. (Heidelberg, Ger.) 2007;85:1175–1186. doi: 10.1007/s00109-007-0221-2. [DOI] [PubMed] [Google Scholar]
- 3.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 4.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 6.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 7.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, Marine JC. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 2004;24:5835–5843. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valentin-Vega YA, Barboza JA, Chau GP, El-Naggar AK, Lozano G. High levels of the p53 inhibitor MDM4 in head and neck squamous carcinomas. Hum. Pathol. 2007;38:1553–1562. doi: 10.1016/j.humpath.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, Guy RK, Marine JC, Jochemsen AG, Dyer MA. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 10.Bartel F, Schulz J, Bohnke A, Blumke K, Kappler M, Bache M, Schmidt H, Wurl P, Taubert H, Hauptmann S. Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int. J. Cancer. 2005;117:469–475. doi: 10.1002/ijc.21206. [DOI] [PubMed] [Google Scholar]
- 11.Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J. Cell Sci. 2007;120:371–378. doi: 10.1242/jcs.03362. [DOI] [PubMed] [Google Scholar]
- 12.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 13.Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–2443. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 14.Li C, Pazgier M, Li C, Yuan W, Liu M, Wei G, Lu WY, Lu W. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J. Mol. Biol. 2010;398:200–213. doi: 10.1016/j.jmb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millard M, Pathania D, Grande F, Xu S, Neamati N. Small-molecule inhibitors of p53-MDM2 interaction: the 2006–2010 update. Curr. Pharm. Des. 2011;17:536–559. doi: 10.2174/138161211795222649. [DOI] [PubMed] [Google Scholar]
- 16.Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011;8:25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- 17.Fry DC, Emerson SD, Palme S, Vu BT, Liu CM, Podlaski F. NMR structure of a complex between MDM2 and a small molecule inhibitor. J. Biomol. NMR. 2004;30:163–173. doi: 10.1023/B:JNMR.0000048856.84603.9b. [DOI] [PubMed] [Google Scholar]
- 18.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Vassilev LT. p53 Activation by small molecules: application in oncology. J. Med. Chem. 2005;48:4491–4499. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 21.Dickens MP, Fitzgerald R, Fischer PM. Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin. Cancer Biol. 2010;20:10–18. doi: 10.1016/j.semcancer.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 23.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, Nichols GL, Bui BN. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 24.Carter BZ, Mak DH, Schober WD, Koller E, Pinilla C, Vassilev LT, Reed JC, Andreeff M. Simultaneous activation of p53 and inhibition of XIAP enhance the activation of apoptosis signaling pathways in AML. Blood. 2010;115:306–314. doi: 10.1182/blood-2009-03-212563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 26.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu S, Qin D, Shangary S, Chen J, Wang G, Ding K, McEachern D, Qiu S, Nikolovska-Coleska Z, Miller R, Kang S, Yang D, Wang S. Potent and orally active small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2009;52:7970–7973. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008;14:5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, Cummings MD, LaFrance LV, Milkiewicz KL, Calvo RR, Maguire D, Lattanze J, Franks CF, Zhao S, Ramachandren K, Bylebyl GR, Zhang M, Manthey CL, Petrella EC, Pantoliano MW, Deckman IC, Spurlino JC, Maroney AC, Tomczuk BE, Molloy CJ, Bone RF. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 31.Parks DJ, Lafrance LV, Calvo RR, Milkiewicz KL, Gupta V, Lattanze J, Ramachandren K, Carver TE, Petrella EC, Cummings MD, Maguire D, Grasberger BL, Lu T. 1,4-Benzodiazepine-2,5-diones as small molecule antagonists of the HDM2-p53 interaction: discovery and SAR. Bioorg. Med. Chem. Lett. 2005;15:765–770. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Marugan JJ, Raboisson P, Schubert C, Koblish HK, Zhao S, Franks CF, Lattanze J, Carver TE, Cummings MD, Maguire D, Grasberger BL, Maroney AC, Lu T. Enhanced pharmacokinetic properties of 1,4-benzodiazepine-2,5-dione antagonists of the HDM2-p53 protein-protein interaction through structure-based drug design. Bioorg. Med. Chem. Lett. 2006;16:3310–3314. doi: 10.1016/j.bmcl.2006.03.055. [DOI] [PubMed] [Google Scholar]
- 33.Cummings MD, Schubert C, Parks DJ, Calvo RR, LaFrance LV, Lattanze J, Milkiewicz KL, Lu T. Substituted 1,4-benzodiazepine-2,5-diones as alpha-helix mimetic antagonists of the HDM2-p53 protein-protein interaction. Chem. Biol. Drug Des. 2006;67:201–205. doi: 10.1111/j.1747-0285.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 34.Allen JG, Bourbeau MP, Wohlhieter GE, Bartberger MD, Michelsen K, Hungate R, Gadwood RC, Gaston RD, Evans B, Mann LW, Matison ME, Schneider S, Huang X, Yu D, Andrews PS, Reichelt A, Long AM, Yakowec P, Yang EY, Lee TA, Oliner JD. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J. Med. Chem. 2009;52:7044–7053. doi: 10.1021/jm900681h. [DOI] [PubMed] [Google Scholar]
- 35.Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, Domling A, Holak TA. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle. 2010;9:1104–1111. doi: 10.4161/cc.9.6.10956. [DOI] [PubMed] [Google Scholar]
- 36.Graves B, Thompson T, Xia M, Janson C, Lukacs C, Deo D, Di Lello P, Fry D, Garvie C, Huang KS, Gao L, Tovar C, Lovey A, Wanner J, Vassilev LT. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11788–11793. doi: 10.1073/pnas.1203789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhuang C, Miao Z, Zhu L, Dong G, Guo Z, Wang S, Zhang Y, Wu Y, Yao J, Sheng C, Zhang W. Discovery, synthesis, and biological evaluation of orally active pyrrolidone derivatives as novel inhibitors of p53-MDM2 protein-protein interaction. J. Med. Chem. 2012;55:9630–9642. doi: 10.1021/jm300969t. [DOI] [PubMed] [Google Scholar]
- 38.Sanchez MC, Renshaw JG, Davies G, Barlow PN, Vogtherr M. MDM4 binds ligands via a mechanism in which disordered regions become structured. FEBS Lett. 2010;584:3035–3041. doi: 10.1016/j.febslet.2010.05.058. [DOI] [PubMed] [Google Scholar]
- 39.Phan J, Li Z, Kasprzak A, Li B, Sebti S, Guida W, Schonbrunn E, Chen J. Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem. 2010;285:2174–2183. doi: 10.1074/jbc.M109.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joseph TL, Madhumalar A, Brown CJ, Lane DP, Verma CS. Differential binding of p53 and nutlin to MDM2 and MDMX: computational studies. Cell Cycle. 2010;9:1167–1181. doi: 10.4161/cc.9.6.11067. [DOI] [PubMed] [Google Scholar]
- 41.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 42.Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem., Int. Ed. Engl. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 43.Kontoyianni M, McClellan LM, Sokol GS. Evaluation of docking performance: comparative data on docking algorithms. J. Med. Chem. 2004;47:558–565. doi: 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- 44.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 45.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 46.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 47.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 48.Molecular Operating Environment, version 2010.10. Quebec, Canada: Chemical Computing Group Inc; 2010. [Google Scholar]
- 49.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain AN. Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput.-Aided Mol. Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 51.Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988;28:31–36. [Google Scholar]
- 52.Chen IJ, Foloppe N. Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: comparison to programs MOE and catalyst. J. Chem. Inf. Model. 2010;50:822–839. doi: 10.1021/ci100026x. [DOI] [PubMed] [Google Scholar]
- 53.Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC. ConfGen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010;50:534–546. doi: 10.1021/ci100015j. [DOI] [PubMed] [Google Scholar]
- 54.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel—an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
- 55.Sadowski J, Gasteiger J, Klebe G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Comput. Sci. 1994;34:1000–1008. [Google Scholar]
- 56.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 57.Corbeil CR, Moitessier N. Docking ligands into flexible and solvated macromolecules. 3. Impact of input ligand conformation, protein flexibility, and water molecules on the accuracy of docking programs. J. Chem. Inf. Model. 2009;49:997–1009. doi: 10.1021/ci8004176. [DOI] [PubMed] [Google Scholar]
- 58.Feher M, Williams CI. Effect of input differences on the results of docking calculations. J. Chem. Inf. Model. 2009;49:1704–1714. doi: 10.1021/ci9000629. [DOI] [PubMed] [Google Scholar]
- 59.Lu Y, Nikolovska-Coleska Z, Fang X, Gao W, Shangary S, Qiu S, Qin D, Wang S. Discovery of a nanomolar inhibitor of the human murine double minute 2 (MDM2)-p53 interaction through an integrated, virtual database screening strategy. J. Med. Chem. 2006;49:3759–3762. doi: 10.1021/jm060023+. [DOI] [PubMed] [Google Scholar]
- 60.Brooks WH, Daniel KG, Sung SS, Guida WC. Computational validation of the importance of absolute stereochemistry in virtual screening. J. Chem. Inf. Model. 2008;48:639–645. doi: 10.1021/ci700358r. [DOI] [PubMed] [Google Scholar]
- 61.Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 62.Suite 2011: Maestro, version 9.2. New York, NY: Schrödinger, LLC; 2011. [Google Scholar]
- 63.Bolton EWY, Thiessen PA, Bryant SH. Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem. 2008;4:217–241. [Google Scholar]
- 64.Baxter J. Local Optima Avoidance in Depot Location. J. Oper. Res. Soc. 1981;32:815–819. [Google Scholar]
- 65.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953;21:1087–1092. [Google Scholar]
- 66.The PyMOL Molecular Graphics System, Version 1.5.0.4. New York, NY: Schrödinger, LLC; 2012. [Google Scholar]
- 67.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schlick T, Overton M. A powerful truncated Newton method for potential energy minimization. J. Comput. Chem. 1987;8:1025–1039. [Google Scholar]
- 69.Renner S, Schwab CH, Gasteiger J, Schneider G. Impact of Conformational Flexibility on Three-Dimensional Similarity Searching Using Correlation Vectors. J. Chem. Inf. Model. 2006;46:2324–2332. doi: 10.1021/ci050075s. [DOI] [PubMed] [Google Scholar]
- 70.Li Z, Scheraga HA. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. U. S. A. 1987;84:6611–6615. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kolossváry I, Guida WC. Low Mode Search. An Efficient, Automated Computational Method for Conformational Analysis: Application to Cyclic and Acyclic Alkanes and Cyclic Peptides. J. Am. Chem. Soc. 1996;118:5011–5019. [Google Scholar]
- 72.Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM. Integrated Modeling Program, Applied Chemical Theory (IMPACT) J. Comput. Chem. 2005;26:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 74.Secchiero P, Bosco R, Celeghini C, Zauli G. Recent advances in the therapeutic perspectives of Nutlin-3. Curr. Pharm. Des. 2011;17:569–577. doi: 10.2174/138161211795222586. [DOI] [PubMed] [Google Scholar]
- 75.Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr. Pharm. Des. 2008;14:2100–2110. doi: 10.2174/138161208785294663. [DOI] [PubMed] [Google Scholar]
- 76.Labute P. A widely applicable set of descriptors. J. Mol. Graphics Modell. 2000;18:464–477. doi: 10.1016/s1093-3263(00)00068-1. [DOI] [PubMed] [Google Scholar]
- 77.Feher M, Williams CL. Numerical errors and chaotic behavior in docking simulations. J. Chem. Inf. Model. 2012;52:724–738. doi: 10.1021/ci200598m. [DOI] [PubMed] [Google Scholar]
- 78.Houston DR, Walkinshaw MD. Consensus docking: improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013;53:384–390. doi: 10.1021/ci300399w. [DOI] [PubMed] [Google Scholar]
- 79.Plewczynski D, Lazniewski M, von Grotthuss M, Rychlewski L, Ginalski K. VoteDock: consensus docking method for prediction of protein-ligand interactions. J. Comput. Chem. 2011;32:568–581. doi: 10.1002/jcc.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.