

Abstract
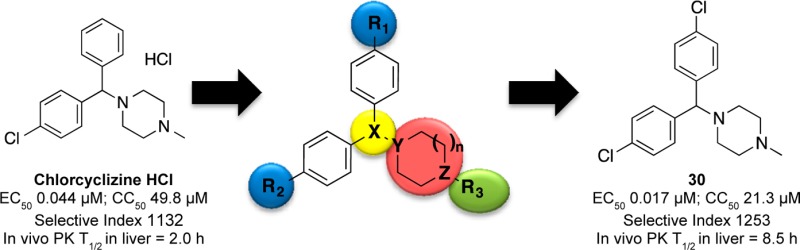
Recently, we reported that chlorcyclizine (CCZ, Rac-2), an over-the-counter antihistamine piperazine drug, possesses in vitro and in vivo activity against hepatitis C virus. Here, we describe structure–activity relationship (SAR) efforts that resulted in the optimization of novel chlorcyclizine derivatives as anti-HCV agents. Several compounds exhibited EC50 values below 10 nM against HCV infection, cytotoxicity selectivity indices above 2000, and showed improved in vivo pharmacokinetic properties. The optimized molecules can serve as lead preclinical candidates for the treatment of hepatitis C virus infection and as probes to study hepatitis C virus pathogenesis and host–virus interaction.
Introduction
Hepatitis C virus (HCV) is an enveloped positive-stranded RNA virus of the Flaviviridae family. The HCV replication cycle is initiated by virions entering the host cell via interaction with cell surface receptors. Following pH-dependent fusion and uncoating, the HCV RNA genome is translated. The resulting polyprotein undergoes proteolytic cleavage into structural (core, E1, and E2) and nonstructural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins. After RNA replication, HCV undergoes assembly, maturation, and secretion processes.1,2
HCV leads to acute and chronic inflammatory hepatic infections that often progress toward chronic liver diseases, including cirrhosis, with an elevated risk of developing hepatocellular carcinoma. There is no vaccine for HCV to date.3 Besides there being around 180 million people chronically infected worldwide with HCV, the standard of care for many years has been limited to interferon α (IFN-α) in combination with ribavirin (RBV).4,5 This combination therapy is partially effective with serious adverse effects. Drug discovery efforts during the past 20 years led to a new paradigm for HCV treatment, which is marked by the recent approval of multiple direct-acting antivirals (DAAs) for interferon-free regimens.2,6,7 The DAAs inhibit certain replication steps in the HCV replication cycle by directly targeting viral proteins, such as NS3/4A proteases, NS5A or NS5B polymerase.8 Viral rebounds were often observed during monotherapy of these DAAs due to the selection of drug-resistant viral mutants. Thus, the clinical application of DAAs is mostly limited to combination regimens, which face challenges including additional side effects, complex administration, and drug–drug interactions.2 Sofosbuvir,9 a DAA that has been approved for interferon-free regimen, costs more than $80,000 during a typical 12-week course of treatment alone, raising concerns about the affordability of DAAs in order to globally impact the burden of HCV disease.10,11 Host-targeting agents (HTAs), however, inhibit host factors that are essential in the viral replication cycle. Promising host-factor targets of HTAs include entry factors, components of viral replication complex cyclophilin A, and miR122 that bind to viral RNA to facilitate replication.1 There is a higher genetic barrier to develop resistance to HTAs.12 Moreover, HTAs can also be used as chemical probes to elucidate anti-HCV mechanisms and host–virus interactions. However, few HTA candidates are in the anti-HCV drug discovery pipeline, possibly due to the nature of primary drug screen assays that were mostly based on certain viral proteins or HCV replicons. Overall, there is still need to improve the current therapeutic regimens by exploring novel anti-HCV targets and small molecules.
Recently, we reported the anti-HCV activity of chlorcyclizine (CCZ, Rac-2), discovered through the screening of the NCGC Pharmaceutical Collection (NPC), in a cell-based anti-HCV quantitative high-throughput screening (qHTS) platform.13−15 The hit compound chlorcylizine HCl (CCZ (Rac-2), Figure 1) showed potent in vitro anti-HCV activity and preferable liver distribution in mouse models, as well as in vivo efficacy against HCV infection in Alb-uPA/SCID chimeric mouse model engrafted with primary human hepatocytes.14 CCZ (Rac-2) was also reported by Chamoun-Emanuelli et al. to block HCV entry, possibly via a cholesterol-dependent pathway.16 However, the target and precise mechanism of action of CCZ (Rac-2) in inhibiting HCV entry remains unknown. Here, we present a SAR study aiming to optimize CCZ (Rac-2) for an anti-HCV application focusing on the following features: generation of a nonchiral lead, improvement of its anti-HCV potency, modulation of its physicochemical properties to potentially reduce CNS exposure, reduction or elimination of its antihistamine activity, and improvement of pharmacokinetic properties. The resulting lead compounds in this series, represented by nonchiral compound 30, exhibited increased anti-HCV activity and selectivity (up to 19-fold and 8-fold, respectively) and improved in vivo pharmacokinetics properties. The optimized lead compounds merit further preclinical development for the treatment of hepatitis C.
Figure 1.

Chlorcylizine HCl identified from qHTS. (A) Chemical structure of chlorcylizine HCl (CCZ, Rac-2). (B) Anti-HCV activity and selectivity of chlorcyclizine HCl.
Results
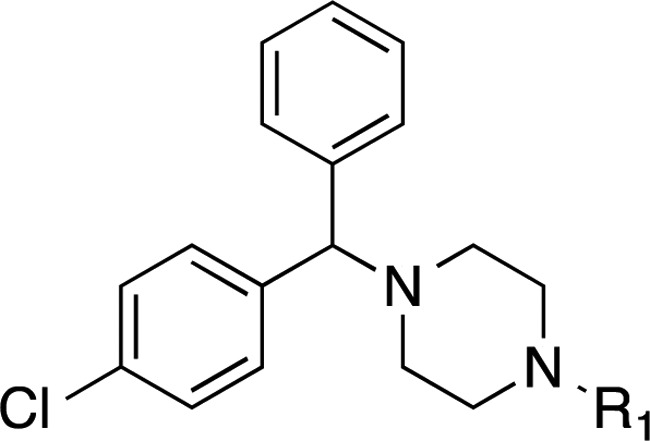
The synthesis of CCZ analogues is displayed in Schemes 1–3. Scheme 1A shows the synthesis of asymmetrical CCZ derivatives. Thus, following modified literature procedures, the corresponding aldehyde or ketone underwent reductive amination with commercially available chiral (R)-1 or (S)-1 and NaBH3CN in the presence of acetic acid (compounds 10–15)28 or p-toluenesulfonic acid (compound (S)-18) using an alcoholic solvent to afford the corresponding N-alkylated derivatives. Ti(OiPr)4 was required to carry out the reductive amination of the cyclopentyl ((S)-16) and cyclohexyl ((S)-17) derivatives.17 Acylation of (S)-1 with acetyl chloride afforded N-acyl derivative (S)-19. Deuterated derivatives (R)-20 and (S)-20 were synthesized from (R)-1 and (S)-1 with CD3I in the presence of aqueous NaOH. Likewise, trifluorinated derivatives (R)-21 and (S)-21 were prepared from the common chiral starting material and trifluoroethyl triflate in the presence of K2CO3.
Scheme 1. General Synthetic Route for Analogues Shown in Table 1 (A) with Alkyl Substituents and (B) with Oligoethylene Glycol Side Chain.

Scheme 3. General Synthetic Route for Analogues Shown in Table 3 (A) Compounds 44 and 45, and (B) Compound 48.

Scheme 1B displays the synthesis of racemic CCZ analogues with a solubilizing polyethylene glycol side chain. Starting from the commercially available hydroxyzine (Rac-5), introduction of the phthalimido moiety via standard Mitsunobu conditions afforded Rac-22. Subsequent hydrazine mediated deprotection afforded primary amine Rac-23. Elongation of the solubilizing side chain was obtained by alkylating Rac-5 to produce N-Boc derivative Rac-24, which was deprotected to the free amine Rac-25 and subsequently acylated to afford Rac-26 (Scheme 1B).
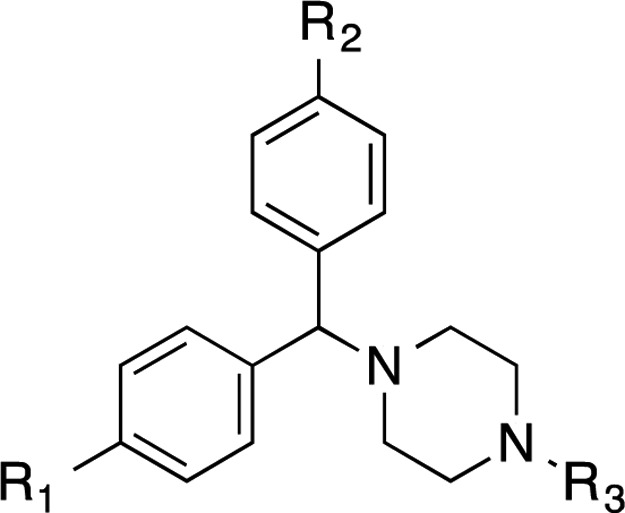
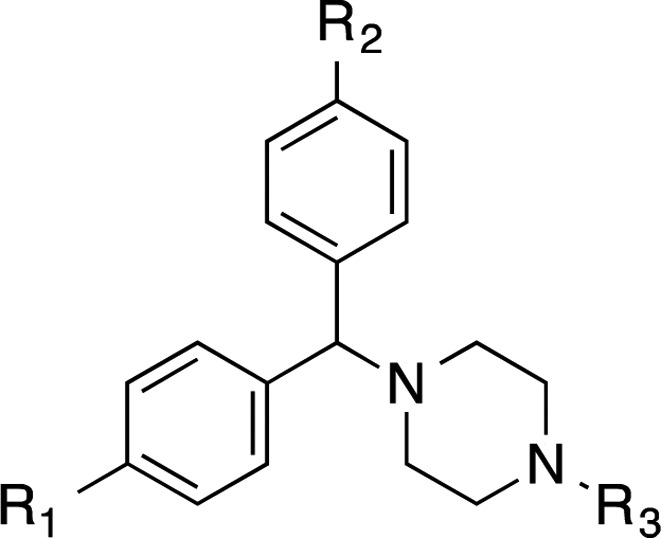
The synthesis of achiral dichlorcyclizine derivatives is displayed in Scheme 2A. Starting from dichlorobenzophenone 27, NaBH4 mediated reduction of the ketone, followed by chlorination with SOCl2 afforded chloride 28, which was used to alkylate a variety of cyclic amine derivatives following modified literature procedures to yield compounds 29–33 (Scheme 2A).29−31 Piperazine derivative 29 was further alkylated, following a modified literature procedure, to produce the diehtylene glycol-containing compound 34,32 which was further elongated to the pentaethylene glycol amine 35. To probe the halogen effect on the cyclizine scaffold, compounds 38 and 39 were prepared according to Scheme 2B, via reductive amination of acetaldehyde with the corresponding piperazines 36 and 37 in the presence of NaBH3CN and acetic acid in methanol.
Scheme 2. General Synthetic Route for Analogues Shown in Table 2 (A) with Two Chlorine Substituents and (B) with Bromide Substituents.

Cyclizine scaffold modification via exchange of the central tertiary carbon with the nitrogen analogue is displayed in Scheme 3A, where methyl piperidone underwent reductive amination with the corresponding aniline 40 and 41 to afford the intermediate amines 42 and 43, which subsequently underwent N-arylation under Buchwald conditions to afford amines 44 and 45. The last modification studied was the introduction of rigidity into the cyclizine scaffold. As displayed in Scheme 3B, this was accomplished by starting from the commercial bromofluorenone 46, which was reduced to the alcohol with NaBH4 and subsequently converted to the chloride 47 with CaCl2 and concentrated HCl. Chloride 47 was used to alkylate N-ethyl piperazine to afford the cyclizine rigid analogue 48 (Scheme 3B).
Tables 1–3 disclose the activity of synthesized compounds in our structural/chemical modification study centered around CCZ (Rac-2). The anti-HCV activity was reported in EC50 values (concentration of compound inhibiting 50% of viral levels in comparison with the DMSO control) using the HCV-Luc (HCV JFH-1 strain, genotype 2a) with insertion of the luciferase reporter gene) infection assay. The cytotoxicity was measured by CC50 values (concentration of compound exhibiting 50% cytotoxicity in comparison with the DMSO control) evaluated with an ATPlite assay in Huh7.5.1 cells, the same cell line used for measuring the antiviral activity. Thus, the activities of the hit compound, racemic CCZ (Rac-2), and its enantiomers ((R)-2 and (S)-2) were confirmed as having good selectivities (selective indices = 1132–1875, Table 1). Nor-CCZ (compound 1), a known in vivo metabolite of CCZ, showed comparable anti-HCV activity but with reduced selectivity (Table 1). It should be noted with interest nor-CCZ’s lack of antihistamine activity (Table 4).18−20
Table 1. Structural Modifications with Substituents on the Piperazine Ringa.

EC50 ± SEM (n ≥ 3) is from the HCV-Luc infection assay; CC50 ± SEM (n ≥ 3) is from the ATPlite cytotoxicity assay; selectivity index = CC50/ EC50.
Table 3. Structural Modifications within the Piperazine Corea.

EC50 ± SEM (n ≥ 3) is from the HCV-Luc infection assay; CC50 ± SEM (n ≥ 3) is from the ATPlite cytotoxicity assay; selectivity index = CC50/EC50.
Table 4. Anti-HCV, Metabolic, and Physical Properties Profile of Selected Analogues.

EC50 ± SEM (n ≥ 3) is from the HCV-Luc infection assay; CC50 ± SEM (n ≥ 3) is from the ATPlite cytotoxicity assay; selectivity index = CC50/EC50.
All compounds were tested at 10 μM, except that compounds (S)-1, Rac-23, and Rac-29 were tested at 3.2, 3.2, and 1 μM, respectively, to avoid potential cytotoxicity.
Assay description: the H1HR assay addresses the H1-histamine receptor (H1HR) inhibitory activity. The numeric value corresponds to the percentage of activation induced by histamine (250 nM) in the presence of 10 nM of compound; the HCVsc assay applies single-round infectious HCV to detect the inhibition of early stage prior to assembly in the virus replication cycle; the HCVpp assay uses an HCV pseudoparticle to determine HCVpp entry inhibition, while the murine leukemia virus pseudoparticle (MLVpp) and vesicular stomatitis virus G pseudoparticle (VSV-Gpp) were tested as controls to address viral specificity. In the HCV subgenomic replicon assay, the genotype 2a HCV replicon cell line was used to evaluate the inhibitory effect of HCV RNA replication.
In Table 1, we addressed the effect of two types of structural modifications: (1) chiral configurations and (2) side chains off the piperazine ring. The enantiomers of compounds 1, 2, 10, 11, 12, 13, 15, 20, and 21 showed comparable EC50 and CC50 values, suggesting that the chiral configuration of CCZ analogues does not have a major effect on the antiviral activity and selectivity. With compounds 10, 11, 12, 13, and 18, we investigated the effect of the length of the aliphatic chain on anti-HCV activity. When the number of carbons was less than 4, analogues showed comparable activity in the range of low double-digit nanomolar and comparable selectivity to CCZ (Rac-2), regardless of whether the chain was linear or branched. Chains with a carbon number of 4 led to decreased activity as shown for compounds 12 and 13. Surprisingly, compound 16, having a cyclopentyl ring exhibited improved activity (EC50 = 19 nM), while compound 17, having a cyclohexyl ring, showed an activity in the expected range (EC50 = 177 nM), comparable to that of compounds 12 and 13. Additionally, acetylation of the piperazine ring dramatically reduced the potency of analogue 19 to a double-digit micromolar value. Introduction of a bulky substituted benzyl group also reduced the anti-HCV activity (compound 14, EC50 = 456 nM). Deuterium, introduced in the side chain of compounds 20 and 15 with the aim of increasing their metabolic t1/2, did not impact their anti-HCV activity and indeed led to a slightly improved t1/2 value (2.9 h for (R)-15 in comparison with 2.2 h for (R)-10 in the rat microsomal stability assay). Moreover, the trifluoromethyl group in compound 21 led to a complete loss of activity, potentially indicating that the pKa value of the tertiary amine was important for anti-HCV activity.
The side effects of CCZ (Rac-2) include drowsiness, dizziness, and headache, attributed to its ability to cross the blood–brain barrier and its CNS penetration. For this reason, first generation antihistamine drugs were replaced by analogues like cetirizine, with reduced CNS-related side effects.21 In order to explore the possibility of reducing the CNS penetration of our molecules, we further expanded our synthetic efforts introducing oligoethylene glycol side chains. This structural modification would increase the molecular weight, solubility, total polar surface area, and number of free rotatable bonds, also potentially leading toward longer pharmacokinetic half-lives. By using ethylene glycol or polyethylene glycol as linker, we explore the effect of introducing a series of functional groups at the terminal position. Compound Rac-3 having a terminal carboxylic acid, completely lost the anti-HCV activity, potentially due to the lack of cell permeability. Moderate activities in the range of triple-digit nanomolar to single-digit micromolar were observed with carboxamide-substituted compounds Rac-4, Rac-22, Rac-24, and Rac-26 (Table 1). Compounds Rac-5, Rac-23 and Rac-25, having hydroxyl or amino groups at the chain terminal position, displayed high anti-HCV activity and moderate cytotoxicity. It was noteworthy that a 6-fold increased activity and retained selectivity were observed with compound Rac-23 (EC50 values below 8 nM with selective indices above 1000) (Table 1).
Table 2 shows the activity of analogues exploring different phenyl ring substituents. Thus, elimination of the para-chloro substituent reduced the activity from double-digit nanomolar (compound Rac-2, EC50= 44 nM) to single-digit micromolar (compound 6, EC50= 1.14 μM). Introduction of an additional para-cloro substituent in the other ring slightly increased the activity (compound 30, EC50= 17 nM). Following a similar trend, nonchiral dichloro compounds 29, 31, 34, and 35 also showed increased activity (1.3–8.7-fold) in comparison with that of the corresponding monochloro analogues Rac-1, (S)-10, Rac-5, and Rac-25. In general, the introduction of Br substituents led to comparable activity and selectivity to the corresponding Cl substituted compounds as shown in Rac-36, Rac-37, Rac-38, and Rac-39.
Table 2. Structural Modifications on the Phenyl Ringsa.

EC50 ± SEM (n ≥ 3) is from the HCV-Luc infection assay; CC50 ± SEM (n ≥ 3) is from the ATPlite cytotoxicity assay; selectivity index = CC50/ EC50.
Structural modifications to the piperazine core are displayed in Table 3, where compounds Rac-2, 29, 6, and Rac-38 were included for comparison. Compound Rac-7, having a one carbon extension of the piperazine ring, retained the activity but led to increased cytotoxicity. The replacement of the piperazine ring with other ring structures in compounds 32, 33, and 9 led to a dramatic loss of activity. In contrast, compounds 44, 45, and 8 retained the anti-HCV activity and selectivity.
Compound 48, which has a rigid cyclizine scaffold, showed an EC50 value that was more than 1 μM, indicating that the conformation of the rings was important for anti-HCV activity.
From our SAR studies, we selected a number of lead compounds based on their anti-HCV activity, selectivity, and structure diversity for an expanded characterization (Table 4). The cytotoxicity of these molecules was further evaluated in HepG2 cells and primary human hepatocytes. All compounds showed less than 1.8-fold difference in CC50 values in these two cell types in comparison with Huh7.5.1 cells (used for our anti-HCV activity assay), with the exception of compound 31, which was less cytotoxic in HepG2 cells than in Huh7.5.1 (approximately 3-fold higher CC50) (Table 4). The H1HR antagonistic activity of the chosen compounds were evaluated by measuring their capacity to block the β-arrestin internalization signal induced by histamine. The numeric value observed corresponds to the percentage of activation induced by histamine (250 nM) in the presence of 10 nM of compound, using CCZ (Rac-2) and (S)-1 as the positive and negative controls, respectively. Shown in Table 4, the results of CCZ (Rac-2) and (S)-1 were consistent with previously reported results.14 Lead compounds with R3 = H (compounds (S)-1 and 29) showed very low H1HR inhibition (approximately 10%). Meanwhile, when R3 = Me, Et, or the medium oligoethylene glycol chain, the H1HR inhibitory effects were generally lower than that of CCZ (Rac-2) (compounds Rac-23, 30, 31, and 34), and (S)-10 showed approximately 4-fold lower inhibition. Compound Rac-25 having a long oligoethylene glycol chain showed more than 3-fold lower inhibitory effect toward H1HR.
Additionally, HCV replication cycle assays were carried out to study the target stage of the CCZ analogues in the HCV replication cycle. The lead compounds exhibited potent inhibition in the HCV single-cycle assay, in which single-round infectious HCV (HCVsc) can infect hepatocytes but does not assemble into new virions (Table 4).13,14 The activity suggested that the CCZ analogues inhibited early steps of the HCV replication cycle prior to assembly. The analogues were tested in the HCV pseudoparticle (HCVpp) assay and the HCV subgenomic replicon assay,13 which detect whether the compounds target HCVpp entry and HCV replication, respectively. The HCVpp assay applies defective retroviral particles that harbor the HCV envelope glycoproteins to assess viral entry inhibition.13,22−24 No significant inhibitory effect was observed in the HCVpp (genotype 1a and 1b) assay with the lead compounds, except for Rac-23 possibly due to cytotoxicity (Table 4). To address viral specificity in the entry process, a vesicular stomatitis virus G pseudoparticle (VSV-Gpp) and a murine leukemia virus pseudoparticle (MLVpp) were tested as the control, in which no inhibitory effect was detected (Table 4). It is worth noting that the lack of HCVpp inhibition of a compound does not necessarily exclude virus entry as a mode of action. A compound could be targeting an entry step that is not otherwise captured by the HCVpp system. Multiple HCV entry inhibitors were reported without HCVpp inhibitory effect, including NPC1L1 antagonist ezetimibe and human apolipoprotein E peptides.25,26 In the genotype 2a HCV replicon cell line, all the lead compounds failed to reduce below 60% the replicon activity, as compared to the DMSO control, indicating that RNA replication was not the target of these analogues (Table 4).
Moreover, besides the in vitro physical properties of the chosen lead compounds, their in vitro metabolic properties were also evaluated using human, mouse, and rat microsomes. Compounds 29, 30, and 34 showed preferable human microsomal stability (t1/2 ≥ 30 min), while maintaining reasonable solubility.
In vivo pharmacokinetics and tissue distribution levels of compound 30 were measured in mice after a single dose of 10 mg/kg through the intraperitoneal (i.p.) route (Table S1). The half-life in the liver was 8.5 h, which was an improvement in comparison with the half-lives of (S)-CCZ [compound (S)-2] and (S)-nor-CCZ [compound (S)-1] (1.99 and 4.7 h, respectively) measured under the same conditions (Figure 2A and C).14 Preferential liver distribution was also observed, as shown by the liver/plasma AUClast ratio of 16 (Figure 2C). Within 24 h after a single dose of 10 mg/kg, the liver concentration of compound 30 (56.9–5.29 μM) was dramatically above its in vitro EC50 values (0.017 μM). To detect any potential hepatotoxicity effect, the alanine transaminase (ALT) levels27 in the mouse serum were measured (Figure 2B). When treated with compound 30, the ALT levels were around or below 80 U/L at most time points except when t = 2 and 4 h, which may suggest a potential transient mild liver toxicity. From these observations, we concluded that there was not a clear correlation between the ALT levels and liver concentration of the compounds. To study whether the mild elevations of ALT level were due to compounds or the vehicle, a control study was carried out dosing the vehicle only. The ALT level was elevated at multiple time points with the treatment of the vehicle only (Figure 2B). Overall, we concluded that no clear hepatotoxicity was detected with the treatment of compound 30 in this condition.
Figure 2.

In vivo pharmacokinetic profiles of the lead compounds. (A) Plasma and liver samples were collected at indicated time points after a single i.p. dosing of compound 30 at 10 mg/kg in a mouse model. Concentrations were measured using UPLC-MS/MS methods. (B) Alanine transaminase level of mouse serum samples collected from the pharmacokinetic study. Results from each mouse are shown with scatter plots, and error bars show the means ± SEM. (C) Pharmacokinetics parameters of compound 30 in comparison with previously reported results of compounds (S)-2 and (S)-1.
To evaluate the capacity of these compounds to potentially impact other viruses in the Flaviviridae family, their antiviral activity was tested against the dengue virus. Both lead compounds (S)-10 and 30 showed EC50 values in the DENV-RVPs assay that were approximately 1000-fold more than their EC50 values against HCV-Luc infection (Table 4), together with CC50 values consistent with the observation when cells were infected with HCV-Luc (Table S1). The selective indices were below 5 for both compounds against the dengue virus. Furthermore, the activity of (S)-10 was submitted for evaluation in the NIAID antiviral screen against 13 types of viruses: hepatitis B virus, HCV replicon, herpes simplex virus-1, human cytomegalovirus, vaccinia virus, dengue virus, influenza A (H1N1) virus, respiratory syncytial virus, SARS coronavirus, poliovirus 3, Rift Valley fever virus, Tacaribe virus, and Venezuelan equine encephalitis virus. Little or no antiviral activity was detected (selective index < 10 and/or EC50 ≥ 1 μM), suggesting that the antiviral effects and mechanism of these analogues were HCV-specific.
Discussion
Besides the advancement in developing efficacious treatments for chronic HCV infection, the cost and side effects of current approved methods point to the direction of developing alternative approaches to therapeutically intervene in the disease. In this sense, in our previous study, we disclosed the in vitro and in vivo anti-HCV properties of chlorcyclizine (CCZ, Rac-2), a first generation antihistamine compound approved for over-the-counter use.14 Its affordability and established clinical safety profile make CCZ (Rac-2) an attractive candidate for repurposing toward chronic HCV infection (https://clinicaltrials.gov/ct2/show/NCT02118012). Additionally, we decided to carry out structural modifications, in vitro and in vivo studies to further optimize this series with the aim of improving their anti-HCV profile. We focused the chemical modifications on four major structural motifs: chirality, side chains off the piperazine ring, substituents on the phenyl rings, and modifications on the piperazine core. As summarized in Figure 3, chirality had little effect on the antiviral activity and selectivity. Dual para-chloro substitution on the aromatic rings led to more potent nonchiral analogues. Furthermore, the introduction of an oligoethylene glycol off the piperazine ring nitrogen improved the activity without compromising the selectivity. Most of the structure modifications in the piperazine core were not tolerated, suggesting that its geometry and its pKa value were important for the anti-HCV activity of the series. Selected lead compounds were also analyzed for additional assessment of cytotoxicity in HepG2 hepatocytes and primary human hepatocytes. They showed profiles similar to those observed in Huh7.5.1 cells, suggesting that the cytotoxicity evaluation in Huh7.5.1 cells in parallel with the HCV infection assay provides a good estimation of the cytotoxicity in human hepatocytes. Overall, the lead compounds in Table 4 exhibited excellent antiviral activity and selectivity in host cells (SI = 8608–201). One of the goals of this medicinal chemistry study was to eliminate the antihistamine side effect of CCZ (Rac-2), which was achieved with lead compounds having R3 = H or long ethylene glycol chains (compounds 29 and Rac-25).
Figure 3.

SAR summary.
In the HCV replication cycle assays, the lead compounds showed inhibitory patterns similar to those of CCZ (Rac-2), displaying a dramatic inhibition in early stage HCV infection (in the HCVsc assay) but no inhibition on HCVpp entry or in the HCV replicon system. Although the exact mechanism of action of CCZ (Rac-2) is still under investigation, the observations here strongly suggest that the lead compounds likely follow the same mechanism of action as CCZ (Rac-2), only with improved activity and selectivity.
In vitro microsomal stability assays were carried out to evaluate the potential metabolic stability of the lead compounds. Among compounds 29, 30, and 34 with t1/2 > 30 min in human microsomes, compound 30 was selected for in vivo pharmacokinetic mouse studies because of its lower in vitro cytotoxicity. Compound 30 showed improved PK properties in comparison with that of (S)-CCZ ((S)-2), namely, a longer half-life, while retaining a high liver/plasma ratio.
Conclusions
Besides the progress in the treatment of chronic HCV infection, among other issues, the global affordability is still an important factor to consider within the current treatment options. Repurposing the over-the-count drug CCZ (Rac-2) may offer an affordable treatment for chronic HCV infection.14 Additionally, we presented a chemical/structural modification study, which resulted in optimized, nonchiral well-tolerated CCZ analogues with improved anti-HCV potency and pharmacokinetic properties that are able to provide good coverage in the liver at very reasonable doses. The lead compounds inhibited HCVsc infection without affecting HCVpp entry or HCV replication in the replicon assay, which is similar to the findings for CCZ (Rac-2), suggesting an unaltered mechanism of action. The lead compounds showing overall improved properties, will be selected for in vivo anti-HCV efficacy studies and potentially for further preclinical drug development efforts with the aim of moving additional compounds of this series toward anti-HCV human clinical trials.
Experimental Section
Chemistry
All air or moisture sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane, N,N-dimethylformamide (DMF), acetonitrile, methanol, and triethylamine were purchased from Sigma-Aldrich (St. Louis, MO). Preparative purification was performed on a Waters semipreparative HPLC system (Waters Corp., Milford, MA). The column used was a Phenomenex Luna C18 (5 μm, 30 × 75 mm; Phenomenex, Inc., Torrance, CA) at a flow rate of 45.0 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection at 220 nM. Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Method 1: A 7 min gradient of 4% to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 min run time at a flow rate of 1.0 mL/min. Method 2: A 3 min gradient of 4% to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 min run time at a flow rate of 1.0 mL/min. A Phenomenex Luna C18 column (3 μm, 3 × 75 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent diode array detector for both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers (Agilent Technologies, Santa Clara, CA). Chemical shifts are reported in ppm with undeuterated solvent (DMSO at 2.49 ppm) as internal standard for DMSO-d6 solutions. All of the analogues tested in the biological assays have a purity of greater than 95% based on both analytical methods.
High Resolution Mass
Spectrometry was recorded on an Agilent 6210 Time-of-Flight (TOF) LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (Version B.02). Enantiomerically pure compounds were purified to >99% purity using supercritical fluid chromatography (SFC) preparative systems at Lotus Separations, LLC (Princeton, NJ, USA). Compounds Rac-1, (S)-1, and (R)-1 were purchased from Albany Molecular Research (Albany, NY, USA). Compounds Rac-2, (S)-2, and (R)-2 were purchased from MP Biomedicals (Santa Ana, CA, USA). Compounds Rac-3 and 6 were purchased from Prestwick Chemical (France). Compound Rac-5 was purchased from TimTec (Newark, DE, USA). Compound Rac-7 was purchased from Biomol (Germany). Compounds 8 and 9 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Compounds 10,2812,2829,2930,3032,3133,31 and 34,32 were synthesized by modified literature procedures. Compounds Rac-36 and Rac-37 were purchased from Vitas-M Laboratory (The Netherlands).
(S)-1-((4-Chlorophenyl) (phenyl)methyl)-4-ethylpiperazine ((S)-10)
A solution of (S)-1 (50.0 mg, 0.174 mmol) in methanol (MeOH) (2.00 mL) was treated at room temperature with acetaldehyde (38.4 mg, 0.872 mmol), NaBH3CN (32.9 mg, 0.523 mmol), and acetic acid (30.0 mL, 0.523 mmol). The reaction mixture was stirred at room temperature overnight and then quenched with 1 N NaOH solution. The mixture was dried by blowing air, and the residue was redisolved in DMSO, filtered, and purified by preparative HPLC to afford (S)-10 (47.0 mg, 63%) as the TFA salt. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.22 (s, 1H), 7.50–7.29 (m, 8H), 7.29–7.19 (m, 1H), 4.54 (s, 1H), 3.42 (d, J = 12.23 Hz, 2H), 3.18–3.09 (m, 2H), 3.04 (q, J = 11.21 Hz, 2H), 2.84 (d, J = 13.01 Hz, 2H), 2.21 (q, J = 11.50 Hz, 2H), 1.18 (t, J = 7.27 Hz, 3H); LCMS RT (Method 1) = 4.566 min; RT (Method 2) = 3.035 min, m/z 315.1 [M + H+]; HRMS (ESI) m/z calcd for C19H24ClN2+ [M + H+] 315.1623; found, 315.1637.
Bis(4-chlorophenyl)methanol
A solution of 27 (3.00 g, 11.9 mmol) in MeOH (15.0 mL) was treated at 0 °C in portions with NaBH4 (0.678 g, 17.9 mmol). The reaction mixture was stirred at 0 °C for 15 min, allowed to warm to room temperature, and stirred for 2 h. The reaction was quenched with ice, diluted with H2O, and extracted with EtOAc. The organic layer was separated, dried over MgSO4, and concentrated to give the title compound as a white solid (3.00 g, 99%), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ ppm 7.31 (d, J = 8.8 Hz, 4H), 7.28 (d, J = 8.7 Hz, 4H), 5.78 (d, J = 3.2 Hz, 1H), 2.26 (d, J = 3.5 Hz, 1H). LCMS RT (Method 2) = 3.733 min, m/z 254.5 [M + H+].
4,4′-(Chloromethylene)bis(chlorobenzene) (28)
Bis(4-chlorophenyl)methanol (3.00 g, 11.8 mmol) was dissolved in CH2Cl2 (10.0 mL); to this was added 3–4 drops of DMF followed by thionyl chloride (2.60 mL, 35.6 mmol). The resulting reaction mixture was allowed to stir at room temperature for 45 min, after which TLC anlysis (20% EtOAc in Hex) showed completion. The reaction mixture was concentrated under reduced pressure to afford 28 (2.50 g, 78%) as a white solid, which was used without further purification. 1H NMR (400 MHz, CDCl3) δ ppm 7.42–7.27 (m, 8H), 6.06 (s, 1H). LCMS RT (Method 2) = 3.932 min, m/z 272.6 [M + H+].
1-(Bis(4-chlorophenyl)methyl)piperazine (29)
A solution of 28 (80.0 mg, 0.295 mmol) in THF (10.0 mL) was treated with piperazine (38.1 mg, 0.442 mmol) followed by K2CO3 (81.0 mg, 0.589 mmol). A catalytic amount of tetrabutylammonium iodide (10.9 mg, 0.029 mmol) was added to the mixture. The reaction mixture was refluxed for 8 h, after which LC/MS analysis showed completion. The reaction mixture was concentrated and redisolved in EtOAc. The organic layer was washed three times with saturated NaHCO3 solution, dried over MgSO4, filtered, and concentrated. The crude product was purified by preparative HPLC to give 29 (80.0 mg, 63%) as the TFA salt. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.50 (s, 2H), 7.43 (d, J = 8.7 Hz, 4H), 7.39 (d, J = 8.6 Hz, 4H), 4.56 (s, 1H), 3.11 (s, 4H), 2.46 (s, 4H). LCMS RT (Method 1) = 4.760 min, m/z 322.7 [M + H+]; HRMS (ESI) m/z calcd for C17H19Cl2N2+ [M + H+] 321.0920; found, 321.0930.
1-(Bis(4-chlorophenyl)methyl)-4-methylpiperazine (30)
To a stirred solution of 28 (0.800 g, 2.95 mmol) in THF (10.0 mL) was added K2CO3 (0.814 g, 5.89 mmol), 1-methylpiperazine (0.654 mL, 5.89 mmol), and catalytic potassium iodide (73.0 mg, 0.442 mmol). The reaction was heated to 100 °C for 48 h. The reaction mixture was partitioned between EtOAc and H2O, the layers separated, and the organic phase washed with brine, dried over MgSO4, filtered, and concentrated. The crude mixture was purified by flash column chromatography, silica gel with a gradient of 0–5% MeOH in CH2Cl2 to afford 30 (603 mg, 61%) as a free-base oil, which was then mixed in a 1:1 ratio with oxalic acid to form the oxalate salt as a white powder. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.41 (d, J = 8.6 Hz, 4H), 7.34 (d, J = 8.5 Hz, 4H), 4.33 (s, 1H), 2.32 (s, 4H), 2.27 (s, 4H), 2.14 (s, 3H). LCMS RT (Method 1) = 4.843 min, m/z 336.9 [M + H+]; HRMS (ESI) m/z calcd for C18H21Cl2N2+ [M + H+] 335.1076; found, 335.1086.
1-(Bis(4-chlorophenyl)methyl)-4-ethylpiperazine (31)
A solution of 28 (160 mg, 0.589 mmol) in THF (10.0 mL) was treated with 1-ethylpiperazine (101 mg, 0.884 mmol) followed by K2CO3 (163 mg, 1.18 mmol). A catalytic amount of tetrabutylammonium iodide (21.8 mg, 0.059 mmol) was added, and the resulting reaction mixture was heated to 100 °C for 48 h. The reaction mixture was partitioned between EtOAc and H2O, the layers separated, and the organic phase washed with brine, dried over MgSO4, filtered, and concentrated. The crude mixture was purified by flash column chromatography, silica gel with a gradient of 0–5% MeOH in CH2Cl2 to afford 31 (123 mg, 60%) as a free-base oil, which was then mixed in a 1:1 ratio with oxalic acid to form the oxalate salt. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.44 (d, J = 8.8 Hz, 4H), 7.40 (d, J = 8.8 Hz, 4H), 4.57 (s, 1H), 3.11–3.02 (m, 2H), 2.80 (s, 8H), 2.24 (s, 2H), 1.17 (t, J = 7.2 Hz, 3H). LCMS RT (Method 1) = 5.029 min, m/z 350.7 [M + H+]; HRMS (ESI) m/z calcd for C19H23Cl2N2+ [M + H+] 349.1233; found, 349.1239.
2-(2-(2-(4-((4-Chlorophenyl) (Phenyl)methyl)piperazin-1-yl)ethoxy)ethyl)isoindoline-1,3-dione (Rac-22)
Et3N (0.279 mL, 2.00 mmol) was added to a solution of Rac-5 (250 mg, 0.667 mmol) in THF (10.0 mL) at room temperature. The mixture was stirred for 15 min, then phthalimide (147 mg, 1.000 mmol) and triphenylphosphine (262 mg, 1.00 mmol) were added to the mixture followed by diisopropyl azodicarboxylate (0.130 mL, 0.667 mmol). The reaction mixture was stirred at room temperature for 4 h, after which LCMS analysis showed product formation. The reaction mixture was concentrated to dryness and residue purified by preparative HPLC to give Rac-22 (239 mg, 58%) as the TFA salt. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.42 (s, 1H), 7.72 (m, 4H), 7.46 (d, J = 8.4 Hz, 2H), 7.44–7.38 (m, 4H), 7.34 (t, J = 7.5 Hz, 2H), 7.25 (t, J = 7.4 Hz, 1H), 4.53 (s, 1H), 3.73 (d, J = 4.8 Hz, 4H), 3.58 (t, J = 5.2 Hz, 4H), 3.14 (d, J = 11.2 Hz, 2H), 3.04–2.97 (m, 2H), 2.82 (d, J = 12.8 Hz, 2H), 2.28 (m, 2H). LCMS RT (Method 1) = 5.205 min, m/z 505.7 [M + H+]; HRMS (ESI) m/z calcd for C29H31ClN3O3+ [M + H+] 504.2048; found, 504.2043.
2-(2-(4-((4-Chlorophenyl) (Phenyl)methyl)piperazin-1-yl)ethoxy)ethanamine (Rac-23)
Hydrazine (0.181 mL, 5.77 mmol) was added to a solution of Rac-22 (97.0 mg, 0.192 mmol) in EtOH (3.00 mL). The reaction mixture was stirred at 60 °C for 3 h, after which LCMS analysis showed completion. The reaction mixture was concentrated under reduced pressure and residue purified by preparative HPLC to give Rac-23 (58.0 mg, 63%) as the TFA salt. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.39 (s, 1H), 7.44 (d, J = 8.1 Hz, 2H), 7.41–7.38 (m, 4H), 7.35 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.3 Hz, 1H), 4.55 (s, 1H), 3.77 (d, J = 4.6 Hz, 2H), 3.55 (t, J = 5.0 Hz, 4H), 3.19 (d, J = 11.0 Hz, 2H), 3.09–2.95 (m, 2H), 2.80 (d, J = 11.5 Hz, 2H), 2.25 (m, 2H). LCMS RT (Method 1) = 3.959 min, m/z 374.7 [M + H+]; HRMS (ESI) m/z calcd for C21H29ClN3O+ [M + H+] 374.1994; found, 374.2002.
tert-Butyl (14-(4-((4-Chlorophenyl) (Phenyl)methyl)piperazin-1-yl)-3,6,9,12-tetraoxatetradecyl)carbamate (Rac-24)
A solution of Rac-5 (250 mg, 0.558 mmol) in DMF (5.00 mL) was treated with a 60% dispersion in mineral oil of NaH (89.0 mg, 2.23 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and room temperature for 30 min. To this mixture was added a solution of tert-butyl (2-(2-(2-bromoethoxy)ethoxy)ethyl)carbamate (174 mg, 0.558 mmol) in DMF (1.00 mL) and the resulting mixture allowed to stir overnight. The mixture was quenched with H2O and extracted with CH2Cl2. The organic layer was separated, dried over MgSO4, filtered, and concentrated. The crude residue was purified by preparative HPLC to give Rac-24 (220 mg, 55%) as the TFA salt. 1H NMR (400 MHz, CDCl3) δ ppm 7.45–7.37 (m, 4H), 7.37–7.18 (m, 5H), 4.44 (s, 1H), 3.86 (t, J = 4.4 Hz, 2H), 3.63–3.48 (m, 14H), 3.29 (s, 4H), 2.91 (s, 9H), 1.43 (s, 9H). 19F NMR (376 MHz, CDCl3) δ ppm −75.78. LCMS RT (Method 1) = 5.372 min, m/z 607.7 [M + H+]; HRMS (ESI) m/z calcd for C32H49ClN3O6+ [M + H+] 606.3304; found, 606.3307.
14-(4-((4-Chlorophenyl) (Phenyl)methyl)piperazin-1-yl)-3,6,9,12-tetraoxatetradecan-1-amine (Rac-25)
A solution of Rac-24 (217 mg, 0.358 mmol) in CH2Cl2 (10.0 mL) was treated with trifluoroacetic acid (5.00 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and room temperature for 30 min, after which LCMS analysis showed completion. The reaction mixture was concentrated, and the crude residue was purified by preparative HPLC to give Rac-25 (109 mg, 60%) as the TFA salt. 1H NMR (400 MHz, CDCl3) δ ppm 7.95 (s, 2H), 7.51–7.41 (m, 4H), 7.38–7.25 (m, 4H), 4.57 (s, 1H), 3.79 (dd, J = 11.2, 6.6 Hz, 4H), 3.70–3.49 (m, 9H), 3.58 (s, 7H), 3.36 (d, J = 4.8 Hz, 2H), 3.17 (s, 3H), 3.00 (s, 5H). 19F NMR (376 MHz, CDCl3) δ −75.78. LCMS RT (Method 1) = 3.916 min, m/z 507.2 [M + H+]; HRMS (ESI) m/z calcd for C27H41ClN3O4+ [M + H+] 506.2780; found, 506.2803.
Cells and Viruses
Human hepatoma cell line Huh7.5.1 and other HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life technologies, Grand Island, NY, USA) with 10% fetal bovine serum (FBS) (Life technologies, Grand Island, NY, USA) and antibiotics in 5% CO2 at 37 °C. HCV-Luc (HCV JFH-1 strain with insertion of the luciferase reporter gene) and pseudotyped viruses (HCVpp-1a, HCVpp-1b, and VSV-Gpp) were produced as reported before. HCV-Luc (HCV JFH-1 strain with insertion of the luciferase reporter gene), HCVsc (single-round infectious defective HCV particle), and pseudotyped viruses (HCVpp-1a, HCVpp-1b, and VSV-Gpp) were produced as reported before.13,22
HCV-Luc Infection and ATPlite Assays
Huh7.5.1 cells were plated in 96-well plates at 104 cells/well and incubated overnight. The cells were infected with HCV-Luc in the presence of increasing concentrations of the compound of interest. The viral level was measured 48 h after treatment using a Renilla luciferase assay system (Promega, Madison, WI, USA). ATP-based cell viability assay was carried out in parallel to evaluate the cytotoxicity with an ATPlite assay kit (PerkinElmer, Waltham, MA, USA). The concentration values that led to 50% viral inhibition and cytotoxicity (EC50 and CC50) were calculated using the nonlinear regression equation in GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA).
HCV Replication Cycle Assays
In the HCVsc assay, Huh7.5.1 cells were cultured in 96-well plates (104 cells/well) overnight before infection with HCVsc in the presence of compound treatments. After 48 h of incubation, the viral level was detected by a luciferase assay. In the HCV subgenomic replicon assay, HCV replicon (GT 2a) cells were plated into 96-well plates at 104 cells/well and incubated overnight. The cells were treated with tested compounds for 48 h, and luciferase activity was measured. In HCVpp assays, Huh7.5.1 cells were seeded in 96-well plates (104 cells/well) and cultured overnight. Then, the cells were infected with HCVpp GT 1a, 1b, VSV-Gpp, and MLVpp for 4 h in the presence of compound treatment. The cells were then washed and cultured for 48 h followed by a luciferase assay to detect the HCV entry. Positive controls (cyclosporin A at 10 μM and bafilomycin A1 at 10 nM) were tested in parallel.
H1HR Inhibition Assay
PathHunter β-Arrestin GPCR assay kit (DiscoveRx, Fremont, CA, USA) was used following the antagonist procedure. The PathHunter cells were plated in white 96-well plates with clear bottoms and cultured overnight. After incubation with the compound of interest for 3 h, agonist histamine at 0.25 μM was added, and the plates were incubated for an additional 2 h. The chemiluminescent signal was read after 60 min of incubation with detection agent at room temperature. Percent antihistamine activity was calculated based on the result from histamine-treated wells.
DENV-RVPs Assay and NIAID Screen
Huh7.5.1 cells were plated in 96-well plates at 104 cells/well and cultured overnight. Dengue RVPs (Integral Molecular, Philadelphia, PA, USA) were added to the cells with increasing concentrations of the compound of interest. Dengue RVP reproducibility was measured by luciferase signal 48 h after treatment. Lycorine HCl was tested as the positive control.33,34 The nonclinical and preclinical services program offered by the National Institute of Allergy and Infectious Diseases (NIAID) (http://www.niaid.nih.gov/labsandresources/resources/dmid/invitro/Pages/invitro.aspx) was used for the antiviral screen against the 13 viruses. The viruses include hepatitis B virus, HCV replicon, herpes simplex virus-1, human cytomegalovirus, vaccinia virus, dengue virus, influenza A (H1N1) virus, respiratory syncytial virus, SARS coronavirus, poliovirus 3, Rift Valley fever virus, Tacaribe virus, and Venezuelan equine encephalitis virus.
In Vitro and In Vivo Pharmacokinetics Properties
The in vitro microsomal stability was measured by incubation of compounds with human/mouse/rat liver microsomes at 37 °C in the presence of the cofactor, NADPH. The concentrations of compounds were measured by LC-MS/MS at 0, 5, 15, 30, and 45 min half-life (t1/2) was calculated as described before.35
The kinetic solubility of compounds was determined in phosphate buffer at pH 7.4, using μSOL Evolution from pION Inc. (www.pion-inc.com), with a fully automated system of sample preparation, sample analysis, and data processing. The effective permeability of compounds was determined via passive diffusion using the stirring double-sink PAMPA (Parallel Artificial Membrane Permeability Assay) method from pION Inc. (www.pion-inc.com) with a fully automated system of sample preparation, sample analysis, and data processing.
For in vivo pharmacokinetics, 27 male CD-1 mice (∼35 g) were obtained from Charles River Laboratories (Wilmington, MA). Mice were housed at the centralized animal facilities at the NIH (Bethesda, MD) with a 12 h light–dark cycle. The housing temperature and relative humidity were controlled at 22 °C and 55%, respectively. The animals had free access to water and food. All experimental procedures were approved by the Animal Care and Use Committee of the NIH. A dosing concentration of 2 mg/mL of the appropriate compound was freshly prepared in 10% PEG300 and 90% of 30% HP-β-CD in water. The pharmacokinetics was evaluated after single intraperitoneal administration at 10 mg/kg. The blood and liver samples were collected at predose, 0.083, 0.25, 0.5, 1, 2, 4, 7, and 24 h. Three samples (n = 3) were collected at each time point. The concentrations of the compound in the plasma and liver were determined by ultraperformance liquid chromatography–mass spectrometry analysis (UPLC-MS/MS). The pharmacokinetic parameters were calculated using the noncompartmental method (Model 200) of the pharmacokinetic software package Phoenix WinNonlin, version 6.2 (Certara, St. Louis, MO, USA). The area under the plasma and liver concentration versus time curve (AUC) was calculated using the linear trapezoidal method. Where warranted, the slope of the apparent terminal phase was estimated by log linear regression using at least 3 data points, and the terminal rate constant (λ) was derived from the slope. AUC0-∞ was estimated as the sum of the AUC0-t (where t is the time of the last measurable concentration) and Ct/λ. The apparent terminal half-life (t1/2) was calculated as 0.693/λ.
Acknowledgments
We thank S. Michael and M. Balcom for assistance with the robotic control in qHTS; P. Shinn, M. Itkin, and D. van Leer for help with compound management; and B. Zhang for technical assistance. We are grateful for the financial support from the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and Molecular Libraries. Primary human hepatocytes were provided by the NIH-funded Liver Tissue Procurement and Cell Distribution System (N01-DK-7-0004/HHSN26700700004C, principal investigator, D. Geller, University of Pittsburgh).
Glossary
Abbreviations Used
- HCV
hepatitis C virus
- IFN-α
interferon α
- RBV
ribavirin
- DAAs
direct-acting antivirals
- HTAs
host-targeting agents
- NPC
NCGC Pharmaceutical Collection
- qHTS
high-throughput screening
- CCZ
chlorcylizine HCl
- SAR
structure–activity relationship
- DMSO
dimethyl sulfoxide
- HCV-Luc
HCV JFH-1 strain with insertion of the luciferase reporter gene
- EC50
the concentration of compound that inhibited 50% of the virus level of DMSO
- CC50
the concentration of compound that exhibited 50% of the cytotoxicity of DMSO
- H1HR
H1-histamine receptor
- HCVsc
single-round infectious defective HCV particles
- HCVpp
HCV pseudoparticle
- ADME
absorption, distribution, metabolism, and excretion
- i.p.
intraperitoneal
- DMF
N,N-dimethylformamide
- LC/MS
liquid chromatography–mass spectrometry
- TOF
time-of-flight
- TFA
trifluoroacetic acid
- SFC
supercritical fluid chromatography
- THF
tetrahydrofuran
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- NIAID
National Institute of Allergy and Infectious Diseases
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00752.
Author Contributions
§ S.H. and J.X. contributed equally to this work.
The authors declare the following competing financial interest(s): T.J.L, M.F., S.H, X.H, Z.H, J.J.M., J.X., and W.Z are named as inventors on patent applications related to this work: U.S. Provisional Patent Appl. 62/011,462, “Heterocyclic compounds and methods of use thereof”.
Supplementary Material
References
- Scheel T. K. H.; Rice C. M. Understanding the Hepatitis C Virus Life Cycle Paves the Way for Highly Effective Therapies. Nat. Med. 2013, 19, 837–849. 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T. J.; Ghany M. G. Current and Future Therapies for Hepatitis C Virus Infection. N. Engl. J. Med. 2013, 368, 1907–1917. 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T. J. Current Progress in Development of Hepatitis C Virus Vaccines. Nat. Med. 2013, 19, 869–878. 10.1038/nm.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T. J.; Rehermann B.; Seeff L. B.; Hoofnagle J. H. Pathogenesis, Natural History, Treatment, and Prevention of Hepatitis C. Ann. Intern. Med. 2000, 132, 296–305. 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- Thomas D. L. Global Control of Hepatitis C: Where Challenge Meets Opportunity. Nat. Med. 2013, 19, 850–858. 10.1038/nm.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afdhal N.; Reddy K. R.; Nelson D. R.; Lawitz E.; Gordon S. C.; Schiff E.; Nahass R.; Ghalib R.; Gitlin N.; Herring R.; Lalezari J.; Younes Z. H.; Pockros P. J.; Di Bisceglie A. M.; Arora S.; Subramanian G. M.; Zhu Y.; Dvory-Sobol H.; Yang J. C.; Pang P. S.; Symonds W. T.; McHutchison J. G.; Muir A. J.; Sulkowski M.; Kwo P. Ledipasvir and Sofosbuvir for Previously Treated HCV Genotype 1 Infection. N. Engl. J. Med. 2014, 370, 1483–1493. 10.1056/NEJMoa1316366. [DOI] [PubMed] [Google Scholar]
- Afdhal N.; Zeuzem S.; Kwo P.; Chojkier M.; Gitlin N.; Puoti M.; Romero-Gomez M.; Zarski J. P.; Agarwal K.; Buggisch P.; Foster G. R.; Brau N.; Buti M.; Jacobson I. M.; Subramanian G. M.; Ding X.; Mo H.; Yang J. C.; Pang P. S.; Symonds W. T.; McHutchison J. G.; Muir A. J.; Mangia A.; Marcellin P. Ledipasvir and Sofosbuvir for Untreated HCV Genotype 1 Infection. N. Engl. J. Med. 2014, 370, 1889–1898. 10.1056/NEJMoa1402454. [DOI] [PubMed] [Google Scholar]
- Conteduca V.; Sansonno D.; Russi S.; Pavone F.; Dammacco F. Therapy of Chronic Hepatitis C Virus Infection in the Era of Direct-Acting and Host-Targeting Antiviral Agents. J. Infect. 2014, 68, 1–20. 10.1016/j.jinf.2013.08.019. [DOI] [PubMed] [Google Scholar]
- Sofia M. J.; Bao D.; Chang W.; Du J.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Wang P.; Zhang H. R.; Bansal S.; Espiritu C.; Keilman M.; Lam A. M.; Steuer H. M.; Niu C.; Otto M. J.; Furman P. A. Discovery of a β-D-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- Callaway E. Hepatitis C Drugs not Reaching Poor. Nature 2014, 508, 295–296. 10.1038/508295a. [DOI] [PubMed] [Google Scholar]
- Zeuzem S. Decade in Review - HCV: Hepatitis C Therapy - A Fast and Competitive Race. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 644–645. 10.1038/nrgastro.2014.164. [DOI] [PubMed] [Google Scholar]
- Puyang X.; Poulin D. L.; Mathy J. E.; Anderson L. J.; Ma S.; Fang Z.; Zhu S.; Lin K.; Fujimoto R.; Compton T.; Wiedmann B. Mechanism of Resistance of Hepatitis C Virus Replicons to Structurally Distinct Cyclophilin Inhibitors. Antimicrob. Agents Chemother. 2010, 54, 1981–1987. 10.1128/AAC.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.; Lan K. H.; He S.; Swaroop M.; Hu X.; Southall N.; Zheng W.; Liang T. J. Novel Cell-Based Hepatitis C Virus Infection Assay for Quantitative High-Throughput Screening of Anti-Hepatitis C Virus Compounds. Antimicrob. Agents Chemother. 2014, 58, 995–1004. 10.1128/AAC.02094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.; Lin B.; Chu V.; Hu Z.; Hu X.; Xiao J.; Wang A. Q.; Schweitzer C. J.; Li Q.; Imamura M.; Hiraga N.; Southall N.; Ferrer M.; Zheng W.; Chayama K.; Marugan J. J.; Liang T. J. Repurposing of the Antihistamine Chlorcyclizine and Related Compounds for Treatment of Hepatitis C Virus Infection. Sci. Transl. Med. 2015, 7, 282ra49. 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Southall N.; Wang Y.; Yasgar A.; Shinn P.; Jadhav A.; Nguyen D. T.; Austin C. P. The NCGC Pharmaceutical Collection: A Comprehensive Resource of Clinically Approved Drugs Enabling Repurposing and Chemical Genomics. Sci. Transl. Med. 2011, 3, 80ps16. 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoun-Emanuelli A. M.; Pecheur E. I.; Chen Z. Benzhydrylpiperazine Compounds Inhibit Cholesterol-Dependent Cellular Entry of Hepatitis C Virus. Antiviral Res. 2014, 109, 141–148. 10.1016/j.antiviral.2014.06.014. [DOI] [PubMed] [Google Scholar]
- Mattson R. J.; Pham K. M.; Leuck D. J.; Cowen K. A. An Improved Method for Reductive Alkylation of Amines using Titanium(IV) Isopropoxide and Sodium Cyanoborohybride. J. Org. Chem. 1990, 55, 2552–2554. 10.1021/jo00295a060. [DOI] [Google Scholar]
- Kuntzman R.; Phillips A.; Tsai I.; Klutch A.; Burns J. J. N-Oxide Formation: A New Route for Inactivation of the Antihistaminic Chlorcyclizine. J. Pharmacol. Exp. Ther. 1967, 155, 337–344. [PubMed] [Google Scholar]
- Gaertner H. J.; Breyer U.; Liomin G. Chronic Administration of Chlorcyclizine and Meclizine to Rats: Accumulation of a Metabolite Formed by Piperazine Ring Cleavage. J. Pharmacol. Exp. Ther. 1973, 185, 195–201. [PubMed] [Google Scholar]
- Dumasia M. C.; Grainger L.; Houghton E. Biotransformation of Cyclizine in Greyhounds. 1: Identification and Analysis of Cyclizine and Some Basic Metabolites in Canine Urine by Gas Chromatography-Mass Spectrometry. Xenobiotica 2002, 32, 795–807. 10.1080/00498250210144802. [DOI] [PubMed] [Google Scholar]
- Criado P. R.; Criado R. F.; Maruta C. W.; Machado Filho C. Histamine, Histamine Receptors and Antihistamines: New Concepts. An. Bras. Dermatol. 2010, 85, 195–210. 10.1590/S0365-05962010000200010. [DOI] [PubMed] [Google Scholar]
- Hsu M.; Zhang J.; Flint M.; Logvinoff C.; Cheng-Mayer C.; Rice C. M.; McKeating J. A. Hepatitis C Virus Glycoproteins Mediate pH-Dependent Cell Entry of Pseudotyped Retroviral Particles. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7271–7276. 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Choe S.; Walker R.; Di Marzio P.; Morgan D. O.; Landau N. R. Human Immunodeficiency Virus Type 1 Viral Protein R (Vpr) Arrests Cells in the G2 Phase of the Cell Cycle by Inhibiting p34cdc2 Activity. J. Virol. 1995, 69, 6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L. J.; Urlacher V.; Iwakuma T.; Cui Y.; Zucali J. Efficacy and Safety Analyses of a Recombinant Human Immunodeficiency Virus Type 1 Derived Vector System. Gene Ther. 1999, 6, 715–728. 10.1038/sj.gt.3300895. [DOI] [PubMed] [Google Scholar]
- Liu S.; McCormick K. D.; Zhao W.; Zhao T.; Fan D.; Wang T. Human Apolipoprotein E Peptides Inhibit Hepatitis C Virus Entry by Blocking Virus Binding. Hepatology 2012, 56, 484–491. 10.1002/hep.25665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainz B. Jr.; Barretto N.; Martin D. N.; Hiraga N.; Imamura M.; Hussain S.; Marsh K. A.; Yu X.; Chayama K.; Alrefai W. A.; Uprichard S. L. Identification of the Niemann-Pick C1-Like 1 Cholesterol Absorption Receptor as a New Hepatitis C Virus Entry Factor. Nat. Med. 2012, 18, 281–285. 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. D.; Morgan R. L.; Beckett G. A.; Falck-Ytter Y.; Holtzman D.; Teo C.-G.; Jewett A.; Baack B.; Rein D. B.; Patel N.; Alter M.; Yartel A.; Ward J. W.; Centers for Disease Control and Prevention . Recommendations for the Identification of Chronic Hepatitis C Virus Infection among Persons Born during 1945–1965. Recommendations and Reports: Morbidity and Mortality Weekly Report (MMWR); Centers for Disease Control and Prevention: Atlanta, GA, 2012; Vol. 61, pp 1–32. [PubMed]
- Wang L.; Jiang H.; Zhou Y.; Liu B.; Ji Z. Synthesis and Antiallergic Activities of Diphenylmethylpiperazine Derivatives. Zhongguo Yaowu Huaxue Zazhi 2002, 12, 125–129. [Google Scholar]
- Younes S.; Baziard-Mouysset G.; de Saqui-Sannes G.; Stigliani J. L.; Payard M.; Bonnafous R.; Tisne-Versailles J. Synthesis and Pharmacological Study of New Calcium Antagonists, Analogs of Cinnarizine and Flunarizine. Eur. J. Med. Chem. 1993, 28, 943–948. 10.1016/0223-5234(93)90049-K. [DOI] [Google Scholar]
- Baltzly R.; DuBreuil S.; Ide W. S.; Lorz E. Unsymmetrically Disubstituted Piperazines. III. N-Methyl-N′-benzhydrylpiperazines as Histamine Antagonists. J. Org. Chem. 1949, 14, 775–782. 10.1021/jo01157a009. [DOI] [Google Scholar]
- Phan T. B.; Nolte C.; Kobayashi S.; Ofial A. R.; Mayr H. Can One Predict Changes from S(N)1 to S(N)2 Mechanisms?. J. Am. Chem. Soc. 2009, 131, 11392–11401. 10.1021/ja903207b. [DOI] [PubMed] [Google Scholar]
- Morren H. G.; Denayer R.; Linz R.; Mathieu J.; Strubbe H.; Trolin S. New Derivatives of 1,4-Disubstituted Piperazine. Industrie Chimique Belge 1957, 22, 409–420. [Google Scholar]
- Wang P.; Li L. F.; Wang Q. Y.; Shang L. Q.; Shi P. Y.; Yin Z. Anti-Dengue-Virus Activity and Structure-Activity Relationship Studies of Lycorine Derivatives. ChemMedChem 2014, 9, 1522–1533. 10.1002/cmdc.201300505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing M.; Liu W.; Yuan Z.; Gu F.; Shi P. Y. A High-Throughput Assay using Dengue-1 Virus-Like Particles for Drug Discovery. Antiviral Res. 2010, 86, 163–171. 10.1016/j.antiviral.2010.02.313. [DOI] [PubMed] [Google Scholar]
- Obach R. S. Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of in vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.