

Abstract

In this letter we report first nonpeptide inhibitors of hepatocyte growth factor (HGF) activation. These compounds inhibit the three proteases (matriptase, hepsin, and HGF activator) required for HGF maturation. We show that 6, 8a, 8b, and 8d block activation of fibroblast-derived pro-HGF, thus preventing fibroblast-induced scattering of DU145 prostate cancer cells. Compound 6 (SRI 31215) is very soluble (91 μM) and has excellent microsome stability (human t1/2 = 162 min; mouse t1/2 = 296 min). In mouse 6 has an in vivot1/2 = 5.8 h following IV administration. The high solubility of 6 and IV t1/2 make this compound a suitable prototype “triplex inhibitor” for the study of the inhibition of HGF activation in vivo.
Keywords: Serine protease, matriptase, hepsin, HGFA, iHGFa, triplex protease inhibitor, SRI 31215
Hepatocyte growth factor (HGF) binds to the receptor tyrosine kinase MET. It activates a signaling cascade that drives the growth and survival of cancer cells and supports their metastatic spread.1 HGF/MET signaling also promotes resistance to classic cytotoxic and targeted therapies, such as anti-EGFR therapy.1,2 Accordingly, constitutive activation of the HGF/MET signaling pathway results in tumor aggressiveness, resistance to therapy, and poor patient outcomes in many cancers. The current approach to preventing HGF/MET signaling is dominated by MET kinase inhibitors and biologics that target HGF or MET.1,3,4 Several of these agents are showing promise in the clinic as monotherapy or in combination with targeted therapies.4
HGF is secreted by tumor associated fibroblasts5 or in an autocrine fashion by some tumors6,7 as the inactive precursor pro-HGF. The trypsin-like serine proteases matriptase, hepsin, and HGF activator (HGFA) are the principal proteases for HGF activation.8−15 The endogenous inhibitors of HGF activation, HAI-1 and -2, inhibit these proteases,11,16,17 thereby controlling the production of active HGF. Reduced expression of the HAIs is a prognostic marker for poor patient outcomes18−23 and high plasma levels of HGF have been found in patients with advanced disease.
The proteolytic conversion of pro-HGF to active HGF is the rate-limiting step in HGF/MET signaling. Several lines of evidence have established that matriptase, hepsin, and HGFA are the most efficient pro-HGF activators. Positional scanning of a synthetic combinatorial peptide library revealed that pro-HGF is the preferred substrate for matriptase,8,9 HGFA, and hepsin,8,10 and they cleave pro-HGF to HGF 104-times more efficiently than urokinase plasminogen activator (uPA).11
To effectively control pro-HGF activation and HGF/MET signaling, it is our objective to develop “triplex inhibitors” of matriptase, hepsin, and HGFA that will mimic the activity of the HAIs. Although inhibitors that target either matriptase, hepsin, or HGFA are available,24 only recently a series of tetrapeptide ketothiazole have been described that are triplex protease inhibitors of HGF activation (iHGFa).25
Here we report the design and synthesis of the first nonpeptide tetrahydropyrimidin-2(1H)-one analogues as triplex protease inhibitors of HGF activation. In Figure 1A we illustrate the design of these inhibitors. The presumed binding mode is based upon (2-oxo-1,3-diazepan-1-yl)benzimidamide inhibitors of the pro-coagulation trypsin-like serine protease factor Xa.26,27 We developed a tetrahydropyrimidin-2(1H)-one core to facilitate the synthesis of analogues that can access the S3 subsite. This design makes use of three conserved structural features found in all trypsin-like serine proteases (Figure 1A): (1) a strong ionic interaction between the carboxylate side chain of Asp 189 in the S1 subsite and phenylamidine, (2) an H-bond interaction with Gly 216–NH– and the carbonyl of the urea core, and (3) a lipophilic interaction with Trp 215 in the S4 subsite.
Figure 1.
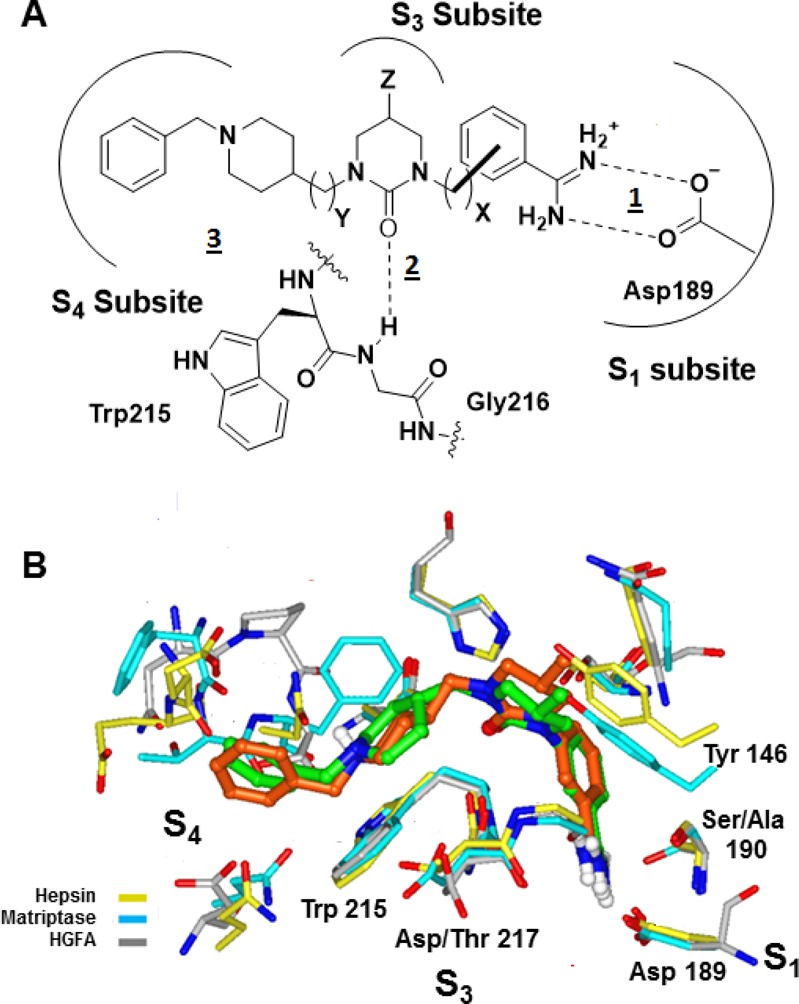
(A) Design hypothesis for the tetrahydropyrimidin-2(1H)-one series showing the main interactions. (B) Comparison of the binding pose for 6 (green) from the cocrystal structure in trypsin with the pose of 6 (orange) from the model in matriptase.
The focus of this study was to optimize the substitution pattern of phenylamidine with the S1 and N-benzylpiperidine with the S4 then explore the interaction of substituent Z with S3. In Table 1 we summarize the structure–activity relationship (SAR) with six proteases, the target enzymes, matriptase, hepsin, and HGFA, the prototype for the protease family, trypsin, and the antitargets, thrombin and factor Xa.
Table 1. Protease Panel Results as Ki [μM].

| substitutions | matriptase | hepsin | HGFA | trypsin | thrombin | factor Xa | ||
|---|---|---|---|---|---|---|---|---|
| 1 | meta | X = 0 | 0.83 | 3.4 | 9.8 | 0.78 | 35.9 | 0.24 |
| 2 | para | X = 0 | 48.9 | 61.2 | 46.5 | 35.4 | >100 | 25.5 |
| 3 | meta | X = 1 | 13.5 | 15.1 | >100 | 4.8 | >100 | 49.4 |
| 4 | para | X = 1 | 15.4 | 20.8 | 39.6 | 4.8 | 82.2 | 22.8 |
| 5 | 40.5 | 53.1 | 54.6 | 5.3 | >100 | 5.4 | ||
| 6 | Y = 1 (SRI 31215) | 0.53 | 0.54 | 0.48 | 0.43 | 11.6 | 0.38 | |
| 7 | Y = 2 | 0.51 | 2.12 | 1.11 | 2.77 | >100 | 0.40 | |
| 8a | Z = HO– | 0.28 | 0.21 | 0.88 | 0.39 | 21.9 | 0.85 | |
| 8b | Z = HOCH2– | 0.58 | 0.69 | 0.23 | 0.15 | 7.4 | 0.25 | |
| 8c | Z = H3CO– | 0.81 | 0.76 | 2.49 | 0.76 | >100 | 1.23 | |
| 8d | Z = H3COCH2– | 0.85 | 0.92 | 0.16 | 0.21 | 8.1 | 0.20 | |
| 8e | Z = PhCH2O– | 1.54 | 0.76 | 1.92 | 0.55 | 29.7 | 0.92 | |
| 8f | Z = PhCH2OCH2– | 0.85 | 0.56 | 0.43 | 0.10 | 2.13 | 0.18 | |
| 8g | Z = PhC(O)NHCH2– | 0.47 | 1.1 | 1.1 | 0.33 | > 100 | 0.75 | |
| 8h | Z = PhCH2C(O)NH– | 1.28 | 1.12 | 0.38 | 0.5 | 42.3 | 0.74 | |
| 8i | Z = PhCH2C(O)NHCH2– | 1.51 | 0.31 | 1.08 | 0.59 | >100 | 1.55 | |
| 8j | Z = H3CSO2NH– | 0.33 | 0.72 | 0.35 | 0.42 | >100 | 0.51 | |
| 8k | Z = PhSO2NH– | 0.52 | 0.67 | 0.52 | 0.34 | 17 | 0.61 | |
| 8l | Z = PhCH2SO2NH– | 0.35 | 0.53 | 0.44 | 0.31 | 13.4 | 0.66 | |
| HAI-1 | 0.0032 | 0.0038 | 0.0034 | 0.0008 | >1.0 | >1.0 | ||
With compounds 1–4 we examined the substitution of the phenylamidine (X = 0 for 1 and 2) and benzylamidine (X = 1 for 3 and 4) for an ionic interaction with the Asp 189 carboxylate of the S1 subsite. Compound 1 is most active against matriptase (Ki = 0.83 μM), hepsin (Ki = 3.4 μM), and HGFA (Ki = 9.8 μM) compared to 2–4. Acyclic 5 illustrates the effect of constraining the urea with a six-membered ring; compared to 1, cyclization improves activity against matriptase (49-fold), hepsin (16-fold), and HGFA (6-fold). Compound 1 illustrates the challenge posed by off-target serine protease selectivity; while 1 is selective against thrombin (Ki = 25.9 μM), it is an inhibitor of factor Xa (Ki = 0.24 μM). Several factors contribute to this selectivity profile. This series originates from inhibitors of factor Xa26,27 and uses interactions common to serine proteases. In thrombin, access to the active site is restricted by a “60s insertion loop”, which forms a flap over the active site blocking access for most substrates.28 Compounds 6 and 7 show the effect of extending the N-benzylpiperidine further into the S4 subsite. For 6, the insertion of one methylene unit between the tetrahydropyrimidin-2(1H)-one and the N-benzylpiperidine leads to a more effective interaction with Trp 215 and improved potency over 1 for matriptase (Ki = 0.53 μM), hepsin (Ki = 0.54 μM), and HGFA (Ki = 0.48 μM). For 7, the insertion of an ethylene group gives a less effective inhibitor. Compounds 1–7 were synthesized by the methods outlined in Scheme 1.
Scheme 1. Synthesis of Compounds 1–7.

Reagents and conditions: (a) 1-benzylpiperidin-4-amine, DMF, r.t.; (b) 3-chloro-2-(chloromethyl)prop-1-ene, NaH, THF; (c) hydroxylamine hydrochloride, Et3N, EtOH; (d) Raney nickel, H2, 30–50 psi, r.t.; (e) for 6 (y = 1), tert-butyl 4-(aminomethyl)piperidine-1-carboxylate, DMF, r.t.; (f) for 7 (y = 2), tert-butyl 4-(2-aminoethyl)piperidine-1-carboxylate, DMF, r.t.; (g) TFA; (h) benzyl bromide, Et3N, DMF, r.t.
Compound 6, SRI 31215, inhibits the targets matriptase, hepsin, and HGFA with near equivalent potency. The off-target enzymes, trypsin and factor Xa, are inhibited as well; however, these enzymes are not known to be expressed in the DU145 cell line used here. Also, while the endogenous inhibitor HAI is an effective inhibitor of trypsin, its lack of activity against factor Xa indicates this enzyme is not a relevant activator of HGF. Therefore, 6 is a useful tool for studying the inhibition of HGF activation. We have compelling data demonstrating that 6 inhibits cleavage of pro-HGF to HGF, blocks fibroblast-induced activation of MET and its downstream effectors, prevents epithelial-mesenchymal transition, and overcomes the resistance to EGFR therapy in colon cancer cells.29
The cocrystal structure of 6 in complex with bovine pancreatic trypsin was obtained. From the trypsin structure (Figure 1B), the meta-phenylamidine of 6 engages the carboxylate side chain of Asp 189 through a bidentate ionic interaction with nitrogen to carboxylate distances of 2.8 and 2.9 Å. The carbonyl oxygen of the tetrahydropyrimidin-2(1H)-one is within H-bonding distance (3.3 Å) of the NH of Gly 216. The N-benzylpiperidine of 6 forms an edge-to-face interaction with Trp 215 of the S4 subsite. The 5-methyl group of the tetrahydropyrimidin-2(1H)-one projects toward the S3 subsite, providing a vector to exploit interactions with this pocket. From racemic 6, only the (S)-methyl enantiomer is found in this high resolution (1.3 Å) crystal structure. A binding model for 6 in matriptase (PDB code 2GV6, superposition with the α-carbon chain of trypsin gave a root-mean-square deviation (rmsd) = 0.79 Å) was developed using Glide software (Schrodinger). The benzamidine group was constrained in the S1 pocket and minimized. This pose was superimposed on the crystal structures of hepsin (1Z8G, rmsd = 0.93 Å) and HGFA (2WUC, rmsd = 0.90 Å). The model confirmed that the (S)-5-methyl projects toward the polar amino acids at position 217 in the S3 region of matriptase (amino acid, Asp 217), hepsin (amino acid, Thr 217), and HGFA (amino acid, Asp 217).
We prepared the 5-functionalized analogues (Table 1, 8a–8l, and analogues in SI Table 6) to study the effect substituents at Z directed to the S3 subsite have on off-target selectivity, especially factor Xa. We found that changes at Z yield some discernible trends; as with 1-7, they are poor inhibitors of thrombin and have variable inhibition activity against factor Xa. Like HAI, 8a–8l are inhibitors of trypsin. Compared to 6, compound 8a has improved inhibition for matriptase (Ki = 0.28 μM) and hepsin (Ki = 0.21 μM) but is less potent for HGFA (Ki = 0.88 μM); also, 8a is the first analogue to show a 3- to 4-fold separation between factor Xa inhibition activity and matriptase and hepsin.
Compound 8b has a triplex inhibition profile (matriptase Ki = 0.58 μM; hepsin Ki = 0.69 μM; HGFA Ki = 0.23 μM). Analogue 8d is an HGFA inhibitor (Ki = 0.16 μM), but is nonselective against factor Xa (Ki = 0.20 μM). Compound 8f is a triplex inhibitor (matriptase Ki = 0.85 μM; hepsin Ki = 0.56 μM; HGFA Ki = 0.43 μM) with potency comparable to 6. Amide 8h is an inhibitor of HGFA (Ki = 0.38 μM), while 8i inhibits hepsin (Ki = 0.31 μM) with 5-fold selectivity against matriptase and factor Xa, and 3-fold against HGFA. We have identified five analogues with an HAI-like triplex inhibitor profile (6, 8b, 8f, 8j, and 8l); two compounds with a selectivity profile favoring HGFA (8d and 8h) and a small molecule inhibitor of hepsin (8i). We confirmed that the 5-substituent is directed toward the S3 subsite by a cocrystal structure of 8f with trypsin (see SI Figure 1) .
Compounds 6, 8a, 8b, and 8d were studied in a cell model of HGF-driven scattering of DU145 prostate cancer cells (Figure 2), which express MET receptor and a high level of matriptase (data not shown). The source of pro-HGF was 18Co fibroblast conditioned media, which triggers scattering of DU145 cells. These analogues inhibit fibroblast-mediated cell scattering in a dose-dependent manner.
Figure 2.

Dose-dependent inhibition of fibroblast-mediated scattering of DU145 cells by 6, 8a, 8b, and 8d. The control image shows untreated DU145 cells with epithelial-like morphology. 18Co fibroblasts induce transition of DU145 cells to the mesenchymal state.
Compounds 6 and 8a–8f were examined in an in vitro ADME assay panel. These analogues have good solubility (47 to 91 μM) and LogD < 2 at pH 7.4 due to the basic phenylamidine functionality (pKa > 11). In human and mouse liver microsomes, the results vary with the 5-substituent (see SI Table 1). Compound 6 is stable in mouse and human microsomes and was examined in a mouse IV/PO pharmacokinetics model (Figure 3). Compound 6 has moderate clearance in the mouse (2283 mL/h, <70% of hepatic circulation) and a high volume of distribution with an in vivot1/2 of 5.8 h. Compound 6 is not orally bioavailable (F < 1%), due to poor membrane permeability (PAMPA – log Pe = 7.6).30
Figure 3.

In vitro ADME data and mouse pharmacokinetic profile for compound 6.
In this letter we report the first nonpeptide inhibitors of HGF activation. These compounds mimic the endogenous inhibitors of HGF maturation, HAI-1 and -2. By blocking the three proteases, matriptase, hepsin, and HGFA, we can prevent the proteolysis of inactive pro-HGF to HGF and oncogenic HGF/MET signaling. In other work29 we show that 6 is an effective inhibitor of fibroblast-induced MET activation and downstream signaling by AKT, ERK, and STAT-3. Compound 6 inhibits fibroblast-mediated epithelial–mesenchymal transition and cell migration and overcomes HGF-dependent resistance to EGFR inhibitors in colon cancer cells.29 Compound 6 (SRI 31215) is currently being evaluated in in vivo models of cancer.
Acknowledgments
We thank John Gerdes and J. Robert Bostwick for their advice and useful discussions in the preparation of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00357.
Comprehensive protease panel data, preparative detail for 1–8v, in vitro ADME data, mouse PK study, cell scatter assay, and crystallography data for 6 and 8f (PDF)
Author Contributions
All authors contributed to this manuscript and have given approval to the final version.
This work was partially supported by Southern Research and an Alabama Innovation Fund grant (to R.A.G., L.K.). T.E.M. acknowledges support from the American Cancer Society, ACS-IRG-96-153-10.
The authors declare no competing financial interest.
Supplementary Material
References
- Comoglio P. M.; Giordano S.; Trusolino L. Drug Development of MET Inhibitors: Targeting Oncogene Addiction and Expedience. Nat. Rev. Drug Discovery 2008, 7, 504–16. 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- Wilson T. R.; Fridlyand J.; Yan Y.; Penuel E.; Burton L.; Chan E.; Peng J.; Lin E.; Wang Y.; Sosman J.; Ribas A.; Li J.; Moffat J.; Sutherlin D. P.; Koeppen H.; Merchant M.; Neve R.; Settleman J. Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors. Nature 2012, 487, 505–9. 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder J. P.; Vande Woude G. F.; Boerner S. A.; LoRusso P. M. Novel Therapeutic Inhibitors of the c-Met Signaling Pathway in Cancer. Clin. Cancer Res. 2009, 15, 2207–2214. 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- Sharma N.; Adjei A. A. In the Clinic: Ongoing Clinical Trials Evaluating c-MET-Inhibiting Drugs. Ther. Adv. Med. Oncol. 2011, 3, S37–50. 10.1177/1758834011423403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick N. A.; Neilson E. G.; Moses H. L. Stromal Fibroblasts in Cancer Initiation and Progression. Nature 2004, 432, 332–337. 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferracini R.; Di Renzo M. F.; Scotlandi K.; Baldini N.; Olivero M.; Lollini P.; Cremona O.; Campanacci M.; Comoglio P. M. The Met/HGF Receptor is Over-expressed in Human Osteosarcomas and is Activated by either a Paracrine or an Autocrine Circuit. Oncogene 1995, 10, 739–49. [PubMed] [Google Scholar]
- Seneviratne D.; Ma J.; Tan X.; Kwon Y. K.; Muhammad E.; Melhem M.; DeFrances M. C.; Zarnegar R. Genomic Instability Causes HGF Gene Activation in Colon Cancer Cells, Promoting their Resistance to Necroptosis. Gastroenterology 2015, 148, 181–191. 10.1053/j.gastro.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen K. A.; Qiu D.; Alves J.; Schumacher A. M.; Kilpatrick L. M.; Li J.; Harris J. L.; Ellis V. Pericellular Activation of Hepatocyte Growth Factor by the Transmembrane Serine Proteases Matriptase and Hepsin, but not by the Membrane-Associated Protease uPA. Biochem. J. 2010, 426, 219–228. 10.1042/BJ20091448. [DOI] [PubMed] [Google Scholar]
- Lee S. L.; Dickson R. B.; Lin C. Y. Activation of Hepatocyte Growth Factor and Urokinase/Plasminogen Activator by Matriptase, an Epithelial Membrane Serine Protease. J. Biol. Chem. 2000, 275, 36720–36725. 10.1074/jbc.M007802200. [DOI] [PubMed] [Google Scholar]
- Herter S.; Piper D. E.; Aaron W.; Gabriele T.; Cutler G.; Cao P.; Bhatt A. S.; Choe Y.; Craik C. S.; Walker N.; Meininger D.; Hoey T.; Austin R. J. Hepatocyte Growth Factor is a Preferred In Vitro Substrate for Human Hepsin, a Membrane-Anchored Serine Protease Implicated in Prostate and Ovarian Cancers. Biochem. J. 2005, 390, 125–136. 10.1042/BJ20041955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M.; Kataoka H. Mechanisms of Hepatocyte Growth Factor Activation in Cancer Tissues. Cancers 2014, 6, 1890–1904. 10.3390/cancers6041890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbs D.; Thiel S.; Stella M. C.; Sturzebecher A.; Schweinitz A.; Steinmetzer T.; Stürzebecher J.; Uhland K. In Vitro Inhibition of Matriptase Prevents Invasive Growth of Cell Lines of Prostate and Colon Carcinoma. Int. J. Oncol. 2005, 27, 1061–1070. 10.3892/ijo.27.4.1061. [DOI] [PubMed] [Google Scholar]
- Szabo R.; Rasmussen A. L.; Moyer A. B.; Kosa P.; Schafer J. M.; Molinolo A. A.; Gutkind J. S.; Bugge T. H. c-Met-Induced Epithelial Carcinogenesis is Initiated by the Serine Protease Matriptase. Oncogene 2011, 30, 2003–16. 10.1038/onc.2010.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H.; Hamasuna R.; Itoh H.; Kitamura N.; Koono M. Activation of Hepatocyte Growth Factor/Scatter Factor in Colorectal Carcinoma. Cancer Res. 2000, 60, 6148–59. [PubMed] [Google Scholar]
- Parr C.; Watkins G.; Mansel R. E.; Jiang W. G. The Hepatocyte Growth Factor Regulatory Factors in Human Breast Cancer. Clin. Cancer Res. 2004, 10, 202–211. 10.1158/1078-0432.CCR-0553-3. [DOI] [PubMed] [Google Scholar]
- Ye J.; Kawaguchi M.; Haruyama Y.; Kanemaru A.; Fukushima T.; Yamamoto K.; Lin C. Y.; Kataoka H. Loss of Hepatocyte Growth Factor Activator Inhibitor Type 1 Participates in Metastatic Spreading of Human Pancreatic Cancer Cells in a Mouse Orthotopic Transplantation Model. Cancer Sci. 2014, 105, 44–51. 10.1111/cas.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M.; Takeda N.; Hoshiko S.; Yorita K.; Baba T.; Sawaguchi A.; Nezu Y.; Yoshikawa T.; Fukushima T.; Kataoka H. Membrane-Bound Serine Protease Inhibitor HAI-1 is Required for Maintenance of Intestinal Epithelial Integrity. Am. J. Pathol. 2011, 179, 1815–1826. 10.1016/j.ajpath.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko S.; Kawaguchi M.; Fukushima T.; Haruyama Y.; Yorita K.; Tanaka H.; Seiki M.; Inatsu H.; Kitamura K.; Kataoka H. Hepatocyte Growth Factor Activator Inhibitor Type 1 is a Suppressor of Intestinal Tumorigenesis. Cancer Res. 2013, 73, 2659–2670. 10.1158/0008-5472.CAN-12-3337. [DOI] [PubMed] [Google Scholar]
- Oberst M. D.; Johnson M. D.; Dickson R. B.; Lin C. Y.; Singh B.; Stewart M.; Williams A.; al-Nafussi A.; Smyth J. F.; Gabra H.; Sellar G. C. Expression of the Serine Protease Matriptase and its Inhibitor HAI-1 in Epithelial Ovarian Cancer: Correlation with Clinical Outcome and Tumor Clinicopathological Parameters. Clin. Cancer Res. 2002, 8, 1101–1107. [PubMed] [Google Scholar]
- Zeng L.; Cao J.; Zhang X. Expression of Serine Protease SNC19/Matriptase and its Inhibitor Hepatocyte Growth Factor Activator Inhibitor Type 1 in Normal and Malignant Tissues of Gastrointestinal Tract. World J. Gastroenterol. 2005, 11, 6202–6207. 10.3748/wjg.v11.i39.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K.; Abarzua F.; Kodama J.; Hongo A.; Nasu Y.; Kumon H.; Hiramatsu Y. Expression of Hepatocyte Growth Factor Activator Inhibitors (HAI-1 and HAI-2) in Ovarian Cancer. Int. J. Oncol. 2009, 34, 345–353. 10.3892/ijo_00000157. [DOI] [PubMed] [Google Scholar]
- Hamasuna R.; Kataoka H.; Meng J. Y.; Wakisaka S.; Koono M. Reduced Expression of Hepatocyte Growth Factor Activator Inhibitor Type-2/Placental Bikunin (HAI-2/PB) in Human Glioblastomas: Implication for Anti-Invasive Role of HAI-2/PB in Glioblastoma Cells. Int. J. Cancer 2001, 93, 339–345. 10.1002/ijc.1349. [DOI] [PubMed] [Google Scholar]
- Morris M. R.; Gentle D.; Abdulrahman M.; Maina E. N.; Gupta K.; Banks R. E.; Wiesener M. S.; Kishida T.; Yao M.; Teh B.; Latif F.; Maher E. R. Tumor Suppressor Activity and Epigenetic Inactivation of Hepatocyte Growth Factor Activator Inhibitor Type 2/SPINT2 in Papillary and Clear Cell Renal Cell Carcinoma. Cancer Res. 2005, 65, 4598–06. [DOI] [PubMed] [Google Scholar]
- Franco F. M.; Jones D. E.; Harris P. K.; Han Z.; Wildman S. A.; Jarvis C. M.; Janetka J. W. Structure-based discovery of small molecule hepsin and HGFA protease inhibitors: Evaluation of potency and selectivity derived from distinct binding pockets. Bioorg. Med. Chem. 2015, 23, 2328–43. 10.1016/j.bmc.2015.03.072. [DOI] [PubMed] [Google Scholar]
- Han Z.; Harris P. K. W.; Jones D. E.; Chugani R.; Kim T.; Agarwal M.; Shen W.; Wildman S. A.; Janetka J. W. Inhibitors of HGFA, Matriptase and Hepsin Serine Proteases: a Nonkinase Strategy to Block Cell Signaling in Cancer. ACS Med. Chem. Lett. 2014, 5, 1219–1224. 10.1021/ml500254r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galemmo R. A. Jr.; Wells B. L.; Rossi K. A.; Alexander R. S.; Dominguez C.; Maduskuie T. P.; Stouten P. F.; Wright M. R.; Aungst B. J.; Wong P. C.; Knabb R. M.; Wexler R. R. The De Novo Design and Synthesis of Cyclic Urea Inhibitors of Factor Xa: Optimization of the S4 Ligand. Bioorg. Med. Chem. Lett. 2000, 10, 301–304. 10.1016/S0960-894X(99)00688-5. [DOI] [PubMed] [Google Scholar]
- Galemmo R. A. Jr.; Maduskuie T. P.; Dominguez C.; Rossi K. A.; Knabb R. M.; Wexler R. R.; Stouten P. F. The De Novo Design and Synthesis of Cyclic Urea Inhibitors of Factor Xa: Initial SAR Studies. Bioorg. Med. Chem. Lett. 1998, 8, 2705–2710. 10.1016/S0960-894X(98)00471-5. [DOI] [PubMed] [Google Scholar]
- Bode W.; Mayr I.; Baumann U.; Huber R.; Stone S. R.; Hofsteenge J. The Refined 1.9 Å Crystal Structure of Human α-Thrombin: Interaction with D-Phe-Pro-Arg Chloromethylketone and Significance of the Tyr-Pro-Pro-Tyr Insertion Segment. EMBO J. 1989, 8, 3467–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu B. Y.; Bansal N.; Venukadasula P. K. M.; Ross L. J.; Messick T. E.; Goel S.; Galemmo R. A. Jr.; Klampfer L.. Inhibition of HGF Activation, a Novel Approach to Block Oncogenic HGF/MET Signaling. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam P. Y.; Clark C. G.; Li R.; Pinto D. J.; Orwat M. J.; Galemmo R. A.; Fevig J. M.; Teleha C. A.; Alexander R. S.; Rossi K. A.; Wright M. R.; Bai S. A.; He K.; Luettgen J. M.; Wong P. C.; Knabb R. M.; Wexler R. R. Structure-Based Design of Novel Guanidine/Benzamidine Mimics: Potent and Orally Bioavailable Factor Xa Inhibitors as Novel Anticoagulants. J. Med. Chem. 2003, 46, 4405–4418. 10.1021/jm020578e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.