

Abstract

Screening of the relatively new target class, the lysine and arginine methyltransferases (MTases), presents unique challenges in the identification and confirmation of active chemical matter. Examination of high throughput screening data generated using Scintillation Proximity Assay (SPA) format for a number of protein MTase targets reveals sensitivity to both the known pan assay interference compounds (PAINS) and also other scaffolds not currently precedented as assay interferers. We find that, in general, true actives show significant selectivity within the MTase family. With the exception of slight modifications of SAM-like compounds, scaffolds that are observed frequently in multiple MTase assays should be viewed with caution and should be carefully validated before following up.
Keywords: Methyltransferase, epigenetics, PAINS, promiscuity, HTS
The protein methyltransferase (MTase) gene family has emerged in the last several years as a target class of interest for drug development across multiple therapeutic areas. Chemical probes for target validation of both lysine and arginine MTases have been reported by the Structural Genomics Consortium (SGC),1 and pharmaceutical companies have entered the clinic with inhibitors of the lysine MTases, EZH2 and DOT1L, for treatment of hematologic cancers.2−5
To identify starting points, pharmaceutical companies have reached into their archives in high throughput screening (HTS) campaigns of compound sets ranging from 150,000 to 2.4 million.6−8 However, the triage of the screen output is complicated by several factors. First, the growing appreciation9 of the false positives due to promiscuous PAINS-type structures is highly relevant to this target class.10 Indeed, many molecules that have been reported as inhibitors of MTases have been revealed as PAINS or promiscuous compounds due to their functionality.11 Second, as a relatively new area, there are few well-characterized “privileged” chemotypes that can reliably translate activity from target to target. While analogues of the cofactor, S-adenosylmethionine (SAM), have been reported,12 the high polarity and resultant poor permeability of this adenosine chemotype may limit its utility in target validation using cell-based assays, although the DOT1L inhibitor, E-5676,5 clearly demonstrates that this can be overcome. For substrate competitive inhibition, the quinazolines associated with G9a13 and SETD814 and the methyllysine channel moiety found on PRMT inhibitors15,16 are notable exceptions. As a consequence, likely “true” actives are not easily recognized based solely on biochemical activity, but require additional confirmation in orthogonal binding assays and/or biochemical mechanism of action (MoA) studies.
Our own experiences with MTases led us to try to find rapid methods to eliminate false positives from the screens. In addition to removing compounds from consideration because they contain recognized PAINS chemotypes or impurities, we began to see structural motifs appear repeatedly in screens, which raised several questions: was this due to true chemical homology and features that caused the MTases to have particular affinity for them or was this an artifact associated with the method used for screening?
To answer these questions, we collated HTS data from eight protein MTase assays: EZH2 (w.t.), EZH2 (A677G), EZH2 (Y641N), PRMT5, PRMT6, SUV420H1, SMYD2, and SETD8, all of which were screened in Scintillation Proximity Assay (SPA) format. To better separate assay specific versus target specific chemotypes, we also included HTS data from five non-MTase assays using SPA format in this analysis (Table 1). Because these 13 assays were screened over a period of several years with different compound sets, very few compounds have been screened across all of them (Figure S1). Nonetheless, there is overlap by chemotypes if not exact structures, and the physicochemical properties of compounds screened in different assays are generally comparable as well (Table S1).
Table 1. Hit Rates of MTase and Non-MTase Assays.
| assayb | conc. (μM) | # screened | hit ratea |
|---|---|---|---|
| EZH2(w.t.) | 50 | 63,026 | 0.22% (0.84%) |
| EZH2(A677G) | 50 | 61,349 | 0.16% (0.70%) |
| EZH2(Y641N) | 50 | 54,948 | 0.25% (1.25%) |
| PRMT5 | 30 | 110,347 | 0.79% (2.46%) |
| PRMT6 | 50 | 9,977 | 0.61% (1.19%) |
| SETD8 | 10 | 158,178 | 0.96% (2.72%) |
| SUV420H1 | 30–50 | 16,923 | 0.64% (1.46%) |
| SMYD2 | 60 | 103,230 | 0.23% (0.97%) |
| NHR A | 25 | 66,925 | 0.81% (3.03%) |
| PDE A | 20 | 85,613 | 0.22% (0.93%) |
| PDE B | 20 | 49,688 | 1.47% (7.83%) |
| PDE C | 20 | 85,613 | 3.12% (13.17%) |
| PDE D | 20 | 66,429 | 1.40% (5.02%) |
Hits are defined as compounds with % inhibition more than 80% (50% in parentheses) in a single-point screening.
MTase assays are listed in bold text.
As illustrated by others,6 identifying a potent MTase inhibitor from diversity screening is extremely challenging perhaps because most compound collections are biased toward the targets worked on before, making it less likely that highly potent compounds will be found in an HTS. To overcome such issues, most HTS campaigns against protein MTases were conducted at high micromolar ligand concentration, while a relatively low inhibition cutoff was used for hit follow up.7,8 For example, Constellation Pharma screened at a concentration of 10–80 μM and followed up compounds with inhibition as low as 35%.6 GSK used 31–34% as primary hit criteria when screening at 10 μM.8 We too opted for this more inclusive strategy in our internal screens. While such efforts can prevent one from missing weaker actives; it pushes the limit of the screening technology and will inevitably bring in more false positives. Out of the eight MTase assays, three EZH2 assays and the SMYD2 assay showed relatively low hit rate, and while the hit rate of PRMT5, PRMT6, SETD8, and SUV420H1 screening is somewhat higher (Table 1), these rates are generally lower than for the PDE and NHR targets that were screened by the SPA format. In the 13 assays, 25,003 unique compounds are active (>50% inhibition) in at least one assay. Out of them, 285 compounds are active in at least four assays, which we defined as frequent hitters (Figure 1).
Figure 1.
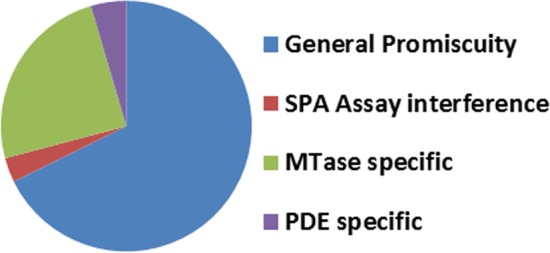
Classifications of frequent hitters.
The reason a compound becomes a frequent hitter may vary from privileged scaffold, assay/target interfering, or genuine promiscuous behavior. Here we leveraged historical data from assays spanning multiple therapeutic areas and formats to further investigate the 285 compounds. All of the 285 compounds have been tested in at least 45 different assays, and 66% of them have been tested more than 300 times (Table S2). Of these, 193 (68%) compounds displayed promiscuous behavior by hitting proteins within four or more gene families or by being active in more than 60 individual assays.17
Compounds displaying general promiscuity can be reactive compounds, aggregators, or contain impurities.10,17Table 2 lists three examples. Some general promiscuous compounds could be identified using rules such as PAINS. Compound 5721908 belongs to the PAINS class anil_di_alk_B.10 Compound 1913060, despite passing the PAINS filter, has an anthranilic acid moiety that exists in several existing PAINS rules. Further analysis around such scaffolds may serve as useful additions to the PAINS filter.
Table 2. Examples of Frequent Hitters and Their Classification.

Among the compounds that did not display a general promiscuous behavior from Lilly historical data, we found 79 (28%) compounds to be active in three or more MTase assays. Out of them, nine compounds are also active in at least one nonepigenetic assay. Such promiscuity could be attributed to nonspecific interference with the SPA assay format. Seventy compounds are only active in MTase assays, while either inactive or not tested in nonepigenetic assays and were analyzed in greater detail. For example, compound 1647171 was active in four MTase assays (EZH2(w.t.), EZH2(A677G), PRMT6, SUV420H1) and inactive in three non-MTase assays. This structural class could inhibit MTases in a nonspecific manner or could indeed contain a privileged chemotype to the targets. Finally, 13 compounds are only active in the PDE assays while inactive in the MTase assays. Compound 9857583 was active in all four PDE assays tested. It contains pyrazolo-pyrimidone, a privileged PDE scaffold that was further developed into a potent and selective PDE9 inhibitor.18
We also investigated whether PAINS substructures10 originally derived from Alphascreen assays were active in these protein MTase assays in SPA format. In our hands, compounds containing PAINS substructures indeed showed greater active rates in the screening than non-PAINS compounds. Across all 13 assays, compounds with PAINS substructures in general have a ∼2× higher hit rate (6.38%) than non-PAINS compounds (3.27%). This finding suggested at least certain PAINS substructures are promiscuous in an assay independent manner and not limited to the AlphaScreen technology originally used to define the substructures. Shown in Table S3 are the top 11 PAINS substructures that tested ≥30 times with a hit rate of ≥20%. Interestingly, certain PAINS rules like azo_A and hzone_phenol_B are also seen in a recent analysis using AstraZeneca internal HTS data.19
To further understand the impact of PAINS mechanisms in MTase assays, we tested ∼20,000 compounds from our diversity collection and 52 PAINS compounds in nine Cerep filter binding (FB) protein MTase assays. We then plotted the hit rate of the diversity library vs the hit rate of the PAINs compounds (Figure 2), with each MTase target represented as a dot. Similar to our in-house experience, Cerep MTase assays are also highly sensitive to PAINS compounds, with hit rates as high as 43%. This again illustrates the nuisance behavior of PAINS compounds beyond a specific assay format. We observed a very strong correlation between the hit rate in our diversity library and PAINS compounds, with a Spearman Rho of 0.86 (Figure 2), suggesting that testing PAINS compounds can serve as an efficient way to forecast MTase assay performance and potential assay interference in HTS. The reasons for this correlation are not fully clear. However, it could be that the PAINS compounds indeed find weakly ligandable pockets or possess recognition elements at rates that are correlated to the more diverse structures. Alternatively, while the screen set was designed to remove PAINS chemotypes, it is possible that additional PAINS structures, in particular those yet to be described in literature, remain in the screening deck. In fact, the authors of the original PAINS rules note that the rules were refined to precisely define the subgroups most responsible for assay interference with the highest possible enrichment value. Additional PAINS compounds will inevitably exist.20 A third explanation is that there are “PAIN-ful” MTase-specific chemotypes, and we sought to examine this possibility more closely.
Figure 2.
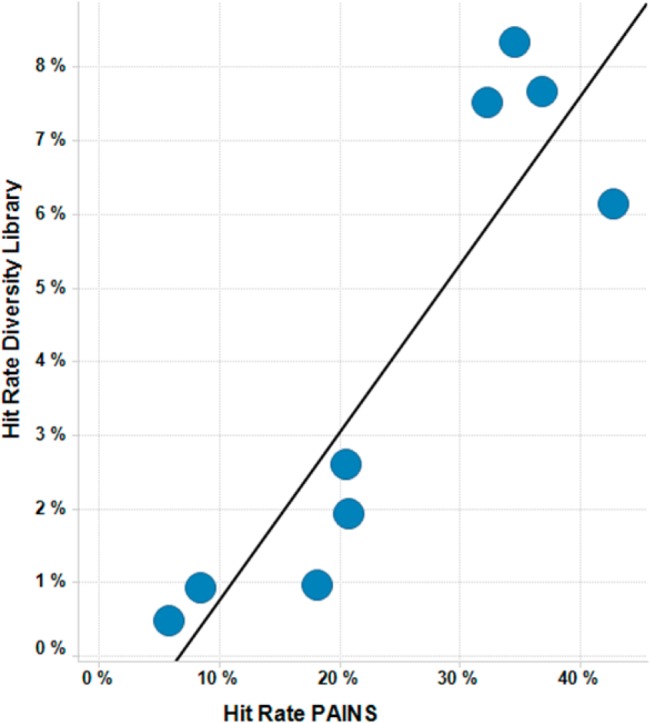
Hit rate correlation between diversity screening set and PAINS compounds in nine different MTase assays.
To compare to the selectivity profile of HTS screen actives, we collated a list of confirmed MTase inhibitors, which includes literature actives and a set of SAR compounds from internal medchem campaigns. These compounds were profiled in a panel of 11 MTase FB assays at Cerep. The heatmap in Figure 3 shows that with very few exceptions, compounds from target-specific projects tend to be highly selective. This includes a list of SAM-competitive analogues that contained the adenosyl moiety and were “SAM-like” yet were highly selective for the SAM pocket of their designed target.5,11,21−23 In addition, other SAM-competitive molecules of chemotypes such as the EZH2 inhibitors EPZ6438, GSK126, and UNC199924 (all containing a pyridone warhead) or tetramethylpiperidines reported by Constellation Pharma25 did not demonstrate notable cross-reactivity. Peptide competitive scaffolds such as the quinazoline UNC063813 also tended to a high level of selectivity. While it could be argued that the selectivity of these compounds was achieved through careful design, it is interesting to note that these selectivity patterns tend to be true of early SAR examples for which selectivity was not the primary focus. In contrast, the screen actives tended to hit against multiple targets, although the threshold for activity was raised to account for their low affinity relative to the focused compounds. This observation pointed to a need to dig deeper into whether the actives could indeed point to privileged chemotypes for these MTases, in a manner similar to the pan-gene family activity of some kinase inhibitors.26 Thus, we turned our attention to the compounds that appeared to be specific for MTases and not the PDE or NHR targets.
Figure 3.
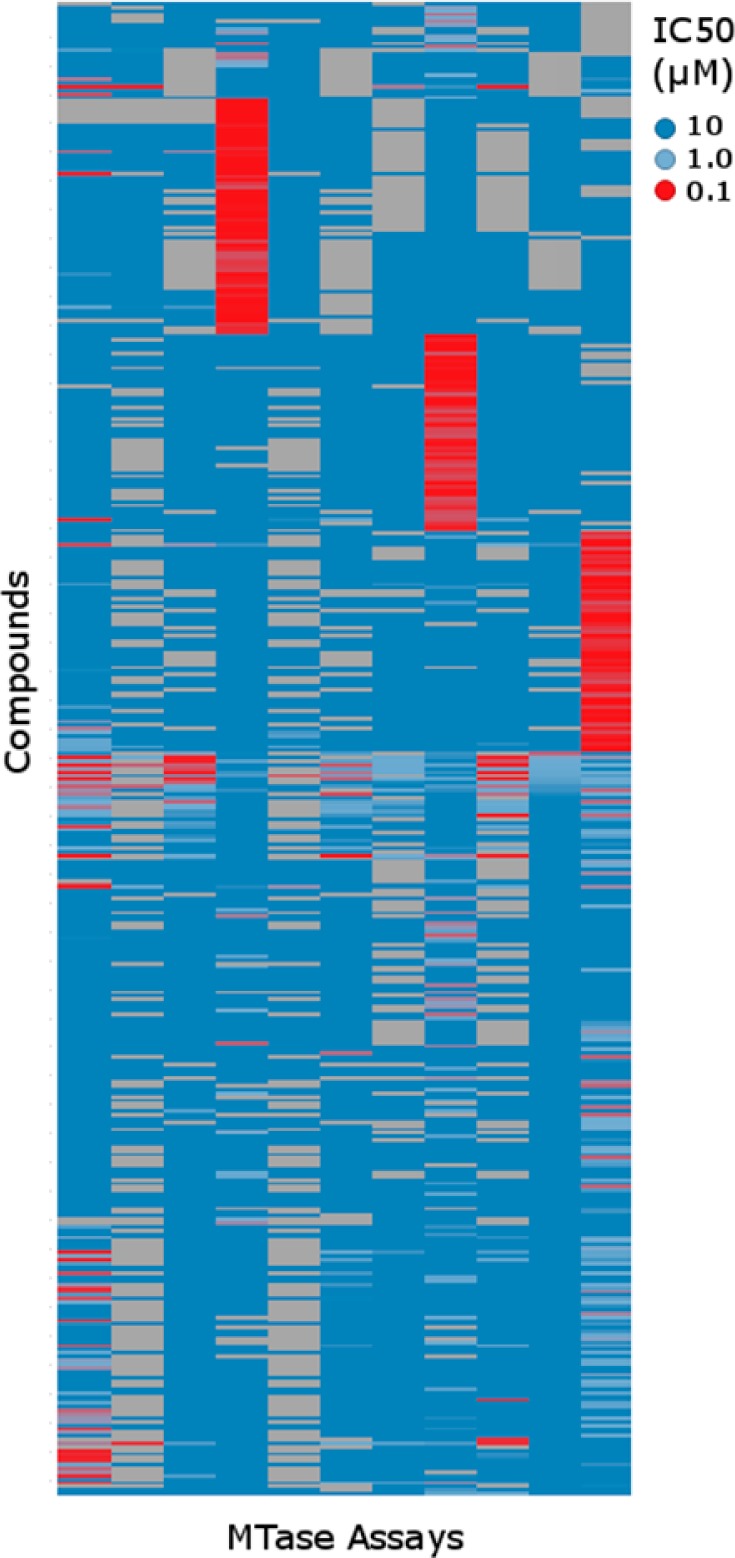
Selectivity profile of confirmed MTase inhibitors profiled in 11 Cerep MTase assays. These compounds included are literature actives and a set of project compounds with sub-μM activity. The heatmap is colored by compound IC50 in the corresponding assay. Empty values are shown in gray.
After eliminating PAINS and impure compounds whose contaminants potentially gave rise to the activity signal, a scaffold analysis demonstrated that there were indeed chemotypes that showed >50% activity across multiple MTases but in none of the five PDE and NHR targets, suggesting gene-family specific activity. Representative examples are shown in Table 3. While actives could be confirmed through biochemical MoA studies to provide a SAM or substrate competitive MoA, subsequent biophysical methods such as isothermal calorimetry (ITC) or hydrogen–deuterium exchange (HDX), or attempts at SAR expansion, either in parallel or in serial execution, generally resulted in an inability to demonstrate binding or SAR trajectory.
Table 3. Promiscuous MTase Scaffolds.

Several examples in the 4-aminoquinoline series (Table 3, row 2) were identified as SETD8 primary hits with IC50 values in the sub-micromolar to low micromolar range. This activity was confirmed in dose response from purified or resynthesized materials. Furthermore, several examples in this SAR were shown to be peptide competitive in substrate MoA studies using MS detection. However, this scaffold gave an inconsistent profile against an orthogonal set of biophysical tools and suggests that the SPA binding activity was a false SAR. For example, no heat could be detected in ITC experiments using either a truncated, SET-domain clone that was biochemically competent or using the same full length construct used in the biochemical screen. That is in contrast to similar experiments using either UNC037914 or peptide substrate H4K15–25 wherein high quality data sets were collected and accurate Kd values could be derived. We also attempted a similar series of experiments employing HDX and again observed no protection to exchange with a key member of this series. As with the ITC, the experimental method was validated with UNC0379 wherein protection was observed and mapped back to residues in the peptide pocket. Ultimately, a focused chemistry effort was initiated and resulted in a flat SAR against the biochemical assay and the same inconsistent activity profile in the biophysical assay systems.
Similarly, the quinoline scaffold was identified in screening for EZH2, with the most active example demonstrating an IC50 of 20 μM and a SAM-competitive MoA. However, additional examples drawn from our collection or specifically prepared as part of a hit assessment exercise showed no improvement in potency or LE, and the MoA was not retained. The scaffold was subsequently deprioritized as a likely false positive.
Thus, hit identification through HTS of MTases requires special considerations both to identify the initial hits and to confirm that they are acting through interactions that will allow for SAR/SPR optimization. Our experience with this data set suggests that despite sequence and active site homology in the gene family, there is limited cross-reactivity of ligands in the catalytic domains of these proteins. With the exception of slight modifications of SAM-like compounds, scaffolds that are observed frequently in multiple MTase assays should be viewed with caution. False positives can often be attributed to PAINS mechanisms, but may also reflect the highly charged nature of the surface of these proteins, whose native substrates include polar residues on histones wrapped by DNA. Therefore, weak electrostatic interactions result in modest affinity that does not translate into true SAR. The inability to optimize potency then leads one to conclude that, while the active was validated through retest or MoA studies, it is not bound to a truly druggable pocket and that minor changes in structure disrupt its weak interactions with the protein. Based on our experience, biophysical methods provide an efficient way to detect these compounds. However, it is worth noting that no single biophysical method is 100% reliable at identifying and triaging false positives. A screening hit is regarded as a viable starting point only if there are consistent evidence across multiple biochemical and biophysical methods. A scaffold analysis across multiple targets can help identify suspect structures, and common themes do emerge. We hope that sharing our learning from this challenging target class highlights the need for robust confirmation of actives with flow schemes that include confirmation of binding through biophysical or other orthogonal methods to avoid wasting further resources on likely false positives.
Acknowledgments
The authors thank Wayne Bocchinfuso, Min Xiao, Tonya Pohl, and Jerrold Bernard for their support of external collaborations. We also thank the SETD8 team and the quantitative biology laboratories, including John Schindler, Nathan Fite, Chris Bailey, Juan Felix Espinosa, Luis Pablo Calle, Feiyu Fred Zhang, and Alfonso Espada. The authors also thank Drs. Masoud Vedadi and Peter Brown and the SGC team for their collaboration on the MTase projects.
Glossary
ABBREVIATIONS
- MTase
methyltransferase
- SPA
scintillation proximity assay
- PAINS
pan assay interference compounds
- SGC
Structural Genomics Consortium
- SAM
S-adenosylmethionine
- PDE
phosphodiesterase
- NHR
nuclear hormone receptor
- FB
filter binding
- ITC
isothermal calorimetry
- HDX
hydrogen–deuterium exchange
- MoA
mechanism of action
- LE
ligand efficiency
- SAR
structure−activity relationship
- SPR
structure−property relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00375.
Supplementary Tables S1–S3 and Figures S1–S2 (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare the following competing financial interest(s): C.G., B.J.M., J.M.S., L.R.V., and M.M.M. are employees and stockholders of Eli Lilly and Company.
Supplementary Material
References
- Kaniskan H. U.; Jin J. Chemical Probes of Histone Lysine Methyltransferases. ACS Chem. Biol. 2015, 10 (1), 40–50. 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aller G. S.; Pappalardi M. B.; Ott H. M.; Diaz E.; Brandt M.; Schwartz B. J.; Miller W. H.; Dhanak D.; McCabe M. T.; Verma S. K.; Creasy C. L.; Tummino P. J.; Kruger R. G. Long Residence Time Inhibition of EZH2 in Activated Polycomb Repressive Complex 2. ACS Chem. Biol. 2014, 9 (3), 622–629. 10.1021/cb4008748. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Scott M. P.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (19), 7922–7927. 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehling V. S.; Vaswani R. G.; Nasveschuk C. G.; Duplessis M.; Iyer P.; Balasubramanian S.; Zhao F.; Good A. C.; Campbell R.; Lee C.; Dakin L. A.; Cook A. S.; Gagnon A.; Harmange J.-C.; Audia J. E.; Cummings R. T.; Normant E.; Trojer P.; Albrecht B. K. Discovery, design, and synthesis of indole-based EZH2 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25 (17), 3644–3649. 10.1016/j.bmcl.2015.06.056. [DOI] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122 (6), 1017–1025. 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garapaty-Rao S.; Nasveschuk C.; Gagnon A.; Chan E. Y.; Sandy P.; Busby J.; Balasubramanian S.; Campbell R.; Zhao F.; Bergeron L.; Audia J. E.; Albrecht B. K.; Harmange J.-C.; Cummings R.; Trojer P. Identification of EZH2 and EZH1 Small Molecule Inhibitors with Selective Impact on Diffuse Large B Cell Lymphoma Cell Growth. Chem. Biol. 2013, 20, 1329–1339. 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Larsen N. A.; Howard T.; Pollard H.; Green I.; Grande C.; Cheung T.; Garcia-Arenas R.; Cowen S.; Wu J.; Godin R.; Chen H.; Keen N. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Diaz E.; Machutta C. A.; Chen S.; Jiang Y.; Nixon C.; Hofmann G.; Key D.; Sweitzer S.; Patel M.; Wu Z.; et al. Development and Validation of Reagents and Assays for EZH2 Peptide and Nucleosome High-Throughput Screens. J. Biomol. Screening 2012, 17 (10), 1279–1292. 10.1177/1087057112453765. [DOI] [PubMed] [Google Scholar]
- Baell J.; Walters M. A. Chemical Con-artists Foil Drug Discovery. Nature 2014, 513, 481–483. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V. M; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Müller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundström M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The Promise and Peril of Chemical Probes. Nat. Chem. Biol. 2015, 11 (8), 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung P.; Huang B.; Zehnder L.; Tatlock J.; Bingham P.; Krivacic C.; Gajiwala K.; Diehl W.; Yu X.; Maegley K. A. SAH Derived Potent and Selective EZH2 Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25 (7), 1532–1537. 10.1016/j.bmcl.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Liu F.; Chen X.; Allali-Hassani A.; Quinn A. M.; Wasney G. A.; Dong A.; Barsyte D.; Kozieradzki I.; Senisterra G.; Chau I.; Siarheyeva A.; Kireev D. B.; Jadhav A.; Herold J. M.; Frye S. V.; Arrowsmith C. H.; Brown P. J.; Simeonov A.; Vedadi M.; Jin J. Discovery of a 2,4-Diamino-7-aminoalkoxyquinazoline as a Potent and Selective Inhibitor of Histone Lysine Methyltransferase G9a. J. Med. Chem. 2009, 52 (24), 7950–7953. 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A.; Yu W.; Li F.; Bleich R. M.; Herold J. M.; Butler K. V.; Norris J. L.; Korboukh V.; Tripathy A.; Janzen W. P.; Arrowsmith C. H.; Frye S. V.; Vedadi M.; Brown P. J.; Jin J. Discovery of a Selective, Substrate-Competitive Inhibitor of the Lysine Methyltransferase SETD8. J. Med. Chem. 2014, 57 (15), 6822–6833. 10.1021/jm500871s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Trainor G. L.; Lorenzi M. V.; Gottardis M.; Jayaraman L.; Purandare A. V. Benzo[d]imidazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 ( CARM1)-Hit to Lead studies. Bioorg. Med. Chem. Lett. 2009, 19 (17), 5063–5066. 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- Rioux N.; Mitchell L. H.; Tiller P.; Plant K.; Shaw J.; Frost K.; Ribich S.; Moyer M. P.; Copeland R. A.; Chesworth R.; Waters N. J. Structural and Kinetic Characterization of a Novel N-acetylated Aliphatic Amine Metabolite of the PRMT Inhibitor, EPZ011652. Drug Metab. Dispos. 2015, 43 (7), 936–943. 10.1124/dmd.115.064014. [DOI] [PubMed] [Google Scholar]
- Bruns R. F.; Watson I. A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- DeNinno M. P.; Andrews M.; Bell A. S.; Chen Y.; Eller-Zarbo C.; Eshelby N.; Etienne J. B.; Moore D. E.; Palmer M. J.; Visser M. S.; Yu L. J.; Zavadoski W. J.; Michael Gibbs E. The discovery of potent, selective, and orally bioavailable PDE9 inhibitors as potential hypoglycemic agents. Bioorg. Med. Chem. Lett. 2009, 19 (9), 2537–2541. 10.1016/j.bmcl.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Nissink J. W. M.; Blackburn S. Quantification of frequent-hitter behavior based on historical high-throughput screening data. Future Med. Chem. 2014, 6 (10), 1113–1126. 10.4155/fmc.14.72. [DOI] [PubMed] [Google Scholar]
- Baell J. B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2010, 2, 1529–1546. 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]
- Mendel D.; Antonysamy S.; Pelletier L.; Emtage J. S.; Gheyi T.; Nguyen A-Q. H.; Mader M.. High resolution structures of SMYD2 in complex with inhibitors that occupy the lysine substrate channel and SAM cofactor domain. Abstracts of Papers, 246th ACS National Meeting & Exposition, Indianapolis, IN, Sept 8–12, 2013; American Chemical Society: Washington, DC, 2013; MEDI 102. [Google Scholar]
- Mori S.; Iwase K.; Iwanami N.; Tanaka Y.; Kagechika H.; Hirano T. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorg. Med. Chem. 2010, 18 (23), 8158–8166. 10.1016/j.bmc.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Wahhab A.; Besterman J. M.; Delorme D.; Ljubomir I.; Llewellyn D.; Rahil J.; Saavedra O.; Deziel R.. Preparation of nucleoside analogs as inhibitors of DNA methyltransferase isoforms DNMT1 and DNMT3b2. US 20080132525, June 5, 2008.
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X.-P.; Kuznetsova E.; Rougie M.; Jiang A.; Pattenden S. G.; Norris J. L.; James L. I.; Roth B. L.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Hahn K. M.; Wang G. G.; Vedadi M.; Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8 (6), 1324–1334. 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasveschuk C. G.; Gagnon A.; Garapaty-Rao S.; Balasubramanian S.; Campbell R.; Lee C.; Zhao F.; Bergeron L.; Cummings R.; Trojer P.; Audia J. E.; Albrecht B. K.; Harmange J-C.P. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med. Chem. Lett. 2014, 5 (4), 378–383. 10.1021/ml400494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland J. J.; Gao C.; Cahya S.; Vieth M. What general conclusions can we draw from kinase profiling data sets?. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834 (7), 1425–1433. 10.1016/j.bbapap.2012.12.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.