

Abstract

C646 inhibits the lysine acetyltransferases (KATs) p300 and CBP and represents the most potent and selective small molecule KAT inhibitor identified to date. To gain insights into the cellular activity of this epigenetic probe, we applied chemoproteomics to identify covalent targets of the C646 chemotype. Modeling and synthetic derivatization was used to develop a clickable analogue (C646-yne) that inhibits p300 similarly to the parent compound and enables enrichment of bound proteins. LC–MS/MS identified the major covalent targets of C646-yne as highly abundant cysteine-containing proteins, and follow-up studies found that C646 can inhibit tubulin polymerization in vitro. Finally, we provide evidence that thiol reactivity of C646 may limit its ability to antagonize acetylation in cells. These findings should enable a more precise interpretation of studies utilizing C646 as a chemical probe of KAT activity and suggest that an underappreciated liability of electrophile-containing inhibitors is a reduction in their cellular potency due to consumption by abundant protein and metabolite thiol sinks.
Keywords: Acetylation, acetyltransferase, epigenetics, probes, PAINS, pan-assay interference, reversible covalent inhibitor
Small molecule probes have provided valuable insights into the role of chromatin-modifying enzymes in disease.1 For example, the past five years have seen a transformation in our knowledge of how BET bromodomains impact cancer and other pathologies, primarily due to the development of tool compounds that can be used to probe the function of these proteins in cell and animal models.2,3 Lysine acetyltransferases (KATs) are an epigenetic enzyme family that has been comparatively recalcitrant to probe development. The most potent, useful, and broadly applied probe of KAT activity is the p300/CBP inhibitor C646 (Figure 1). C646 was identified by Cole and co-workers using a virtual ligand screening approach. These studies found that C646 inhibts p300 selectively in vitro over other KATs and metabolic acetyltransferase enzymes.4 Substantial evidence indicates C646 can also inhibit p300 activity in cells. For example, Andrews and co-workers recently showed that the biochemical effects of C646 on p300-catalyzed histone acetylation accurately predict the manner in which patterns of histone acetylation are affected by C646 in cells.5 C646 can be used to inhibit the p300/CBP-dependent histone modification H3K27Ac, which has been used in imaging studies to establish a causal role for histone acetylation in transcription in living cells.6 Furthermore, C646 can selectively induce cell death in leukemia cells containing the AML-ETO gene fusion, which encodes a transcription factor whose activity is dependent on p300/CBP KAT function.7,8
Figure 1.
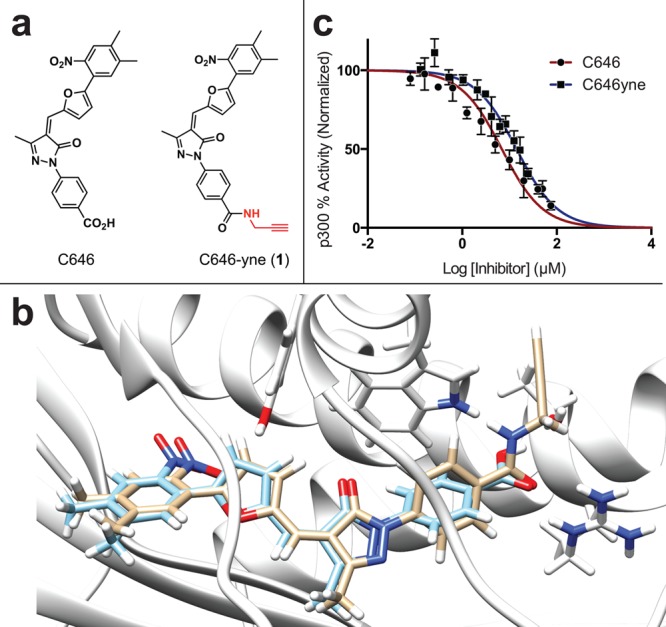
Design of a clickable C646 analogue. (a) Structure of C646 and C646-yne 1. (b) Docking of C646 and C646-yne to a structure of p300 (PDB: 3BIY) suggests the two molecules can adopt a similar conformation in the KAT active site. C646, blue; C646-yne 1, gold. (c) Relative in vitro inhibition of p300-catalyzed acetylation by C646 and clickable analogue C646-yne (1).
Recently there has been an increased interest in understanding the mechanisms and liabilities of pan-assay interference compounds (commonly referred to as PAINS).9−11 C646 is a unique molecule because, while it has proven effects on p300/CBP-mediated phenotypes in cells, it also has several chemical features that would render it a PAIN.12,13 These include a conjugated pyrazolone-furan, which is potentially reactive with cellular nucleophiles, as well as an aromatic nitro group, which could form reactive intermediates upon metabolic reduction. Initial structure–activity relationships found that the nitroaromatic group of C646 could be replaced by more metabolically benign functionalities with little loss in potency.4 In contrast, the electrophilic conjugated pyrazolone-furan of C646 was observed to be essential for p300 inhibition. This was hypothesized to be due to a requirement for planarity for the molecule to bind the p300 active-site, supported by modeling studies.3 Enzyme activity analyses suggest C646 does not covalently modify p300. However, whether C646 possesses covalent targets in a cellular context was not determined. This inspired us to develop a chemical proteomic approach to identify the covalent targets of the C646 pyrazolone-furan chemotype, in order to better understand the cellular activity of this KAT inhibitor and also gain insights into the major liabilities of electrophilic tool compounds in cells.
Since a structure of the p300-C646 complex has not yet been reported, we performed docking studies to facilitate the design of our chemical proteomic probe (Figure 1b). The C646 carboxylate represents the most straightforward route for derivatization of the parent molecule, and docking analyses suggested its modification would weaken p300–C646 interactions (deleting a hydrogen bond between the ligand and enzyme) but still enable the molecule to adapt its putative binding conformation in the KAT active site. This is also consistent with structure–activity analyses of C646 performed by Cole and co-workers.4 Therefore, we targeted the carboxylate of C646 for modification with a propargylamide moiety, providing a latent affinity handle to enable click chemistry-based enrichment and visualization (Figure 1).14 Initial studies found that little or no product was formed when attempting to directly couple C646 and propargylamine using traditional coupling reagents (EDC, PyBOP). However, in situ formation of the C646 mixed anhydride, using isobutylchloroformate/N-methylmorpholine, followed by addition of excess propargylamine, afforded the product (C646-yne, 1) in yields sufficient for further analysis (Scheme S1). With probe 1 in hand, we first compared the ability of C646 and its bioorthogonal analogue 1 to inhibit p300 activity in an electrophilic mobility shift assay.15 As expected from docking analysis, C646-yne inhibited p300, although more weakly than the parent compound (C646 IC50 = 6.8 μM, CI95 = 5.6–8.4 μM; C646-yne 1 IC50 = 14.1 μM, CI95 = 12.2–16.3 μM) (Figure 1c). This indicates C646-yne retains the key features of C646 required for KAT inhibition.
Next, we performed gel-based labeling experiments to assess whether C646-yne 1 possessed covalent targets in cellular settings (Figure 2). C646-yne (5–50 μM) was incubated with HEK-293 cell lysate for 16 h, followed by conjugation to a fluorescent azide using copper-catalyzed [3 + 2] cycloaddition.14 These reactions were quenched with loading buffer (without boiling, to avoid artifactual labeling) separated by SDS-PAGE and assessed for covalent labeling by fluorescence imaging. Incubation with C646-yne resulted in the dose-dependent fluorescent labeling of HeLa proteomes (Figure 2). Of note, the click chemistry step and SDS-PAGE procedure used for protein visualization in these experiments are strongly denaturing,16 and the labeling of proteins under these conditions is therefore indicative of irreversible covalent, rather than reversible covalent, labeling.17 Labeling was competed by preincubation of proteomes with iodoacetamide, suggesting it was cysteine directed (Figure S1). Also consistent with covalent cysteine labeling, upon prolonged (24 h) incubation we identified formation of a stable C646-cysteine adduct by LC–MS (Figure S2). Finally, C646-yne was found to strongly label bovine serum albumin (BSA), a cysteine-rich protein. These studies suggest the pyrazolone-furan of C646 is an electrophilic chemotype capable of irreversible protein reactivity.
Figure 2.
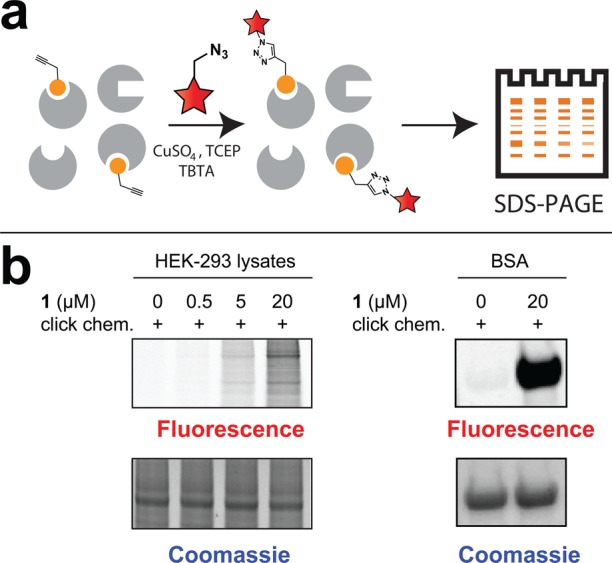
(a) Schematic for analyzing protein targets of C646-yne (1). (b) C646-yne labeling of HEK-293 cell proteome and BSA. Full gels for C646-yne labeling are provided in the Supporting Information, Figure S1.
To identify specific covalent protein targets of C646-yne, we next performed a similar experiment, again incubating cell lysates with probe 1 (20 μM), but this time using click chemistry to conjugate a biotin azide to labeled proteins (Figure 3). Probe-labeled samples were desalted, enriched on streptavidin beads, subjected to on-bead tryptic digest, and analyzed by LC–MS/MS.15 Consistent with the strong gel-based labeling of proteins between 40 and 60 kDa, we identified several proteins in this size range as strongly enriched. Notably, the five most enriched proteins were different chains of the abundant cytoskeletal protein tubulin. Other proteins that were highly enriched (>15 spectral counts, >4-fold enrichment relative to control) include elongation factor 1-alpha, the heat shock protein HSP60, and the glycolytic enzyme 3-phosphoglycerate dehydrogenase (Figure 3). Seven of the top ten proteins enriched have been identified as containing hyperreactive cysteines in proteomic studies.18 Whole proteome surveys indicate tubulins, heat shock proteins, and elongation factors are among the most abundant proteins in the cell.19 The three strongly enriched proteins that lack an identified hyperreactive cysteine (TUBA8, TUBB2A, TUBB4A) are all tubulin proteins, implying C646 reactivity may possess a degree of recognition toward this protein class. While we cannot rule out that lower abundance proteins outside the scope of our detection method are also modified, these studies suggest that the major targets of C646-yne reactivity are abundant cellular proteins containing reactive cysteine residues.
Figure 3.
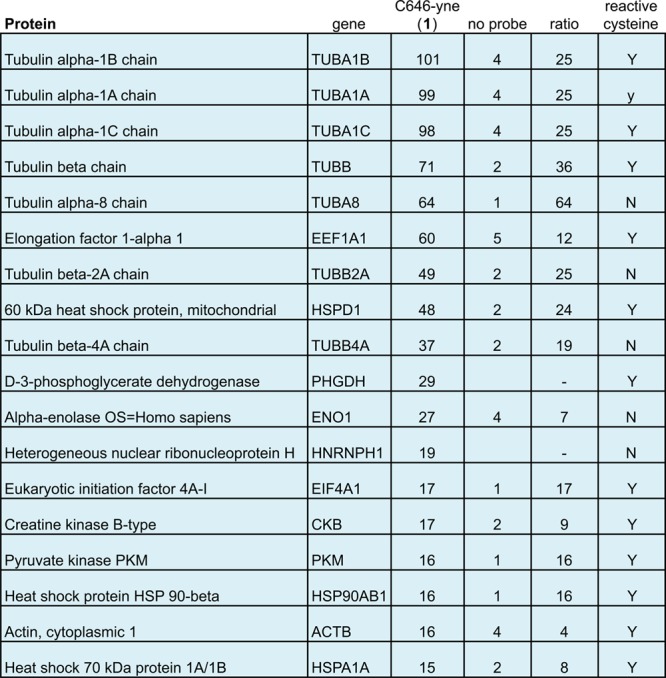
LC–MS/MS identified proteins significantly enriched by C646-yne 1 (>15 spectral counts, >4-fold ratio of probe-enriched relative to no probe control). Numbers correspond to spectral counts observed for each protein target. Reactive cysteine refers to proteins with annotated hyperreactive cysteine, as described in ref (18) of main text.
To determine whether covalent labeling by the pyrazolone-furan chemotype is capable of modulating protein function, we next assessed the effects of C646 on tubulin polymerization. Previous studies have shown that the reaction of tubulin with electrophiles can reduce the rate of microtubule formation.20 Accordingly, porcine tubulin was assayed for polymerization in the presence or absence of C646. We found C646 caused a dose-dependent decrease in the rate of microtubule formation, as well as a reduction in final polymer mass (Figure 4a). Consistent with a covalent modification mechanism, these effects required preincubation with C646 (Figure S3). Of note, the concentrations at which C646 biochemically inhibits tubulin polymerization are similar to concentrations that have been applied in cellular assays.4,6,8 These studies suggest the electrophilic reactivity of C646 can have functional effects on protein activity.
Figure 4.
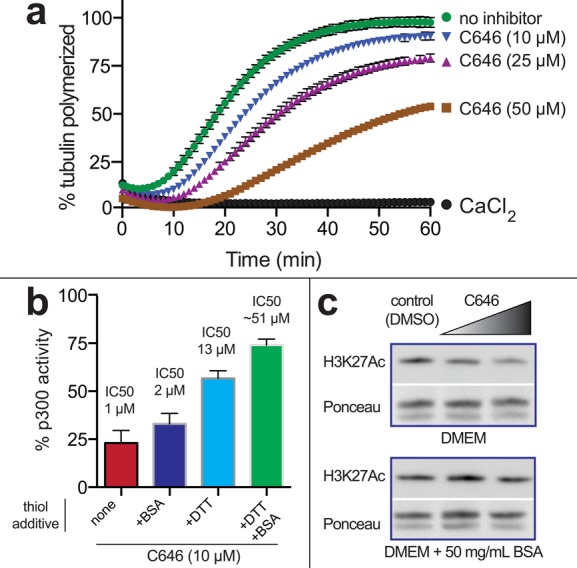
Biological consequences of C646 reactivity. (a) C646 can inhibit tubulin polymerization. Tubulin polymerization was monitored using a fluorescence assay. CaCl2 is a positive control that effectively inhibits tubulin polymerization. (b) In vitro inhibition of p300 by C646 is sensitive to metabolite and protein thiol-containing additives. Full dose–response curves are provided in Figure S4. (c) Cell-based inhibition of histone acetylation by C646 is sensitive to the presence of nucleophiles in cell growth media. Lane 1 (left), vehicle DMSO; lane 2 (middle), 20 μM C646; lane 3 (right), 40 μM C646.
Together with the substantial evidence from the literature, which suggests the effects of C646 on histone acetylation are KAT-dependent,4−6 our observations suggest a critical point: C646 is capable of both on-target inhibition of p300/CBP KAT activity, as well as off-target reactivity with tubulin and other abundant cellular proteins. This suggests that C646’s “on-target” anti-KAT activity may be limited by its “off-target” consumption by abundant cellular nucleophiles. To test this hypothesis, we first performed biochemical analyses of p300, assessing its inhibition by C646 in the presence of DTT. Indeed, we observed DTT reduced the inhibitory potency of C646 (Figure 4b). In addition, we found that extracellular nucleophiles, such as bovine serum albumin, could reduce the effects of C646 on histone acetylation in cellular models (Figure 4c). This suggests that a major limitation of C646 (and similar electrophile-containing tool compounds and PAINS) may be a diminishment of on-target activity due to consumption by intracellular and extracellular biological nucleophiles.21
Recent years have witnessed a major effort to raise awareness of PAINS in the scientific literature, so these molecules are not mistakenly applied as highly specific chemical probes.10,22 Paralleling this effort has been a growing reliance on phenotypic screens to identify inhibitors of challenging to target biological pathways.23 C646 is a molecule that lies at the intersection of these two paradigms, containing many chemical features of a PAIN compound, while also demonstrating the ability to phenocopy some effects of p300/CBP knockdown. Here we have applied a chemical approach to identify candidate covalent targets of this inhibitor, with the twin goals of better defining the mechanism by which it impacts acetylation, and to understand just how “PAIN-ful” the C646 chemotype is. Our results indicate that in complex mixtures the electrophilicity of C646 limits its KAT inhibitor activity and likely results in the modification of high abundance heat shock proteins, glycolytic enzymes, and tubulins. Investigating the effects of C646 on tubulin polymerization indicates that these covalent modifications may have functional effects on protein activity. Thus, the covalent targets of C646 identified here should be considered as potential influencers of C646-driven phenotypes, particularly when applying this compound at high concentrations and long time points. Of note, we did not observe substantial modification of low-abundance signaling proteins (i.e., kinases, nuclear proteins) whose inhibition would provide a straightforward “off-target” explanation for C646’s effects on histone acetylation.4 Although we cannot rule out that such targets were not detected due to limitations inherent to our method, our findings are also not inconsistent with the literature, which suggests that inhibition of KAT activity is a prominent mechanism by which C646 reduces acetylation in cells. This mechanistic ambiguity highlights the need to develop optimized probes of p300/CBP activity, an effort which, from a biochemical perspective, the discovery of C646 lends significant credence to.
A more subtle implication of our study is that PAINS molecules may be unexpected repositories of novel reactive chemotypes. Unlike other PAINS mechanisms (aggregation, redox cycling) covalent reactivity is a concept that has been validated in the development of chemical probes,24 as well as drugs.25 While it is important to note that in these cases reactivity was introduced late in development (rather than early as in the case of C646), an interesting question is whether some PAINS chemotypes possess reactivity that may be harnessed to useful effect. In these efforts, chemical reactivity profiling,11,26,27 as well as unbiased chemical proteomic methods28,29 may provide powerful approaches to help better define the scope and limitations of covalent chemotypes such as C646.
Acknowledgments
The authors thank Dr. Thorkell Andressen and Dr. Sudipto Das (Laboratory of Proteomics and Analytical Technology) for LC–MS/MS analyses. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC011488-02).
Glossary
ABBREVIATIONS
- KAT
lysine acetyltransferase
- p300
histone acetyltransferase p300
- CBP
Creb-binding protein
- PAINS
pan-assay interference compounds
- H3K27Ac
histone H3 lysine 27 acetylation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00385.
Supplemental figures, schemes, and methods (PDF)
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC011488-02).
The authors declare no competing financial interest.
Supplementary Material
References
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11 (5), 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468 (7327), 1067–73. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468 (7327), 1119–23. 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers E. M.; Yan G.; Mukherjee C.; Orry A.; Wang L.; Holbert M. A.; Crump N. T.; Hazzalin C. A.; Liszczak G.; Yuan H.; Larocca C.; Saldanha S. A.; Abagyan R.; Sun Y.; Meyers D. J.; Marmorstein R.; Mahadevan L. C.; Alani R. M.; Cole P. A. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17 (5), 471–82. 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry R. A.; Kuo Y. M.; Bhattacharjee V.; Yen T. J.; Andrews A. J. Changing the selectivity of p300 by acetyl-CoA modulation of histone acetylation. ACS Chem. Biol. 2015, 10 (1), 146–56. 10.1021/cb500726b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasevich T. J.; Hayashi-Takanaka Y.; Sato Y.; Maehara K.; Ohkawa Y.; Sakata-Sogawa K.; Tokunaga M.; Nagase T.; Nozaki N.; McNally J. G.; Kimura H. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 2014, 516 (7530), 272–5. 10.1038/nature13714. [DOI] [PubMed] [Google Scholar]
- Wang L.; Gural A.; Sun X. J.; Zhao X.; Perna F.; Huang G.; Hatlen M. A.; Vu L.; Liu F.; Xu H.; Asai T.; Xu H.; Deblasio T.; Menendez S.; Voza F.; Jiang Y.; Cole P. A.; Zhang J.; Melnick A.; Roeder R. G.; Nimer S. D. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 2011, 333 (6043), 765–9. 10.1126/science.1201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X. N.; Lin J.; Ning Q. Y.; Gao L.; Yao Y. S.; Zhou J. H.; Li Y. H.; Wang L. L.; Yu L. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS One 2013, 8 (2), e55481. 10.1371/journal.pone.0055481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53 (7), 2719–40. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell J.; Walters M. A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513 (7519), 481–3. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Dahlin J. L.; Nissink J. W.; Strasser J. M.; Francis S.; Higgins L.; Zhou H.; Zhang Z.; Walters M. A. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 2015, 58 (5), 2091–113. 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2010, 2 (10), 1529–46. 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Ferrins L.; Falk H.; Nikolakopoulos G. PAINS: Relevance to Tool Compound Discovery and Fragment-Based Screening. Aust. J. Chem. 2013, 66 (12), 1483–1494. 10.1071/CH13551. [DOI] [Google Scholar]
- Speers A. E.; Cravatt B. F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004, 11 (4), 535–46. 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Montgomery D. C.; Sorum A. W.; Meier J. L. Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. J. Am. Chem. Soc. 2014, 136 (24), 8669–76. 10.1021/ja502372j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E.; Speers A. E.; Cravatt B. F. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)--a general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2007, 2 (6), 1414–25. 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- Serafimova I. M.; Pufall M. A.; Krishnan S.; Duda K.; Cohen M. S.; Maglathlin R. L.; McFarland J. M.; Miller R. M.; Frodin M.; Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8 (5), 471–6. 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B.; Bachovchin D. A.; Mowen K.; Baker D.; Cravatt B. F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468 (7325), 790–5. 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami A. M.; Hahne H.; Wu Z.; Auer F. J.; Meng C.; Wilhelm M.; Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013, 4 (3), 609–20. 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Stewart B. J.; Doorn J. A.; Petersen D. R. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 2007, 20 (8), 1111–9. 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang L.; Predina J.; Han R.; Beier U. H.; Wang L. C.; Kapoor V.; Bhatti T. R.; Akimova T.; Singhal S.; Brindle P. K.; Cole P. A.; Albelda S. M.; Hancock W. W. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19 (9), 1173–7. 10.1038/nm.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V.; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Muller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundstrom M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11 (8), 536–41. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J. G.; Rudolph J.; Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discovery 2014, 13 (8), 588–602. 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]
- Cohen M. S.; Zhang C.; Shokat K. M.; Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308 (5726), 1318–21. 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–17. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Weerapana E.; Simon G. M.; Cravatt B. F. Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol. 2008, 4 (7), 405–7. 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost C.; Nitsche C.; Scholz T.; Roux L.; Klein C. D. Promiscuity and selectivity in covalent enzyme inhibition: a systematic study of electrophilic fragments. J. Med. Chem. 2014, 57 (18), 7590–9. 10.1021/jm5006918. [DOI] [PubMed] [Google Scholar]
- Banerjee R.; Pace N. J.; Brown D. R.; Weerapana E. 1,3,5-Triazine as a modular scaffold for covalent inhibitors with streamlined target identification. J. Am. Chem. Soc. 2013, 135 (7), 2497–500. 10.1021/ja400427e. [DOI] [PubMed] [Google Scholar]
- Cisar J. S.; Cravatt B. F. Fully functionalized small-molecule probes for integrated phenotypic screening and target identification. J. Am. Chem. Soc. 2012, 134 (25), 10385–8. 10.1021/ja304213w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.