

Abstract
The polycomb paralogs CBX2, CBX4, CBX6, CBX7, and CBX8 are epigenetic readers that rely on “aromatic cage” motifs to engage their partners’ methyllysine side chains. Each CBX carries out distinct functions, yet each includes a highly similar methyllysine-reading chromodomain as a key element. CBX7 is the only chromodomain that has yet been targeted by chemical inhibition. We report a small set of peptidomimetic agents in which a simple chemical modification switches the ligands from one with promiscuity across all polycomb paralogs to one that provides selective inhibition of CBX6. The structural basis for this selectivity, which involves occupancy of a small hydrophobic pocket adjacent to the aromatic cage, was confirmed through molecular dynamics simulations. Our results demonstrate the increases in affinity and selectivity generated by ligands that engage extended regions of chromodomain binding surfaces.
Keywords: Epigenetics, methyllysine reader proteins, Polycomb Group proteins, CBX6, peptidomimetics
Post-translational methylation exerts critical control over multiple gene expression pathways.1 Histone methylation is among the most prominent and diverse of post-translational modifications (PTMs) in which differences in the location and degree of methylation dictate the engagement of different partners and give varying downstream biological effects.2,3 The Drosophila protein polycomb protein reads trimethylation marks on histone tails and is the namesake of the polycomb group (PcG), a set of functionally diverse proteins that coordinate to modify gene expression at hundreds of loci.4 There are five human paralogs of polycomb, CBX2, CBX4, CBX6, CBX7, and CBX8, each with distinct functions in cellular differentiation during development, cancer progression, and stem cell maintenance.5−8 The canonical functions of CBX proteins involve participation in variations of polycomb repressive complex 1 (PRC1), within which they serve as readers of the mark histone 3, lysine 27 trimethylated (H3K27me3). In spite of functional differences, all polycomb paralogs rely on a common methyllysine reader module (chromodomain) with high structure and sequence similarity.9−11
Epigenetic reader proteins are growing as a class of potential drug targets. Inhibitors of acetyllysine-binding bromodomains (BRD) are well-known; clinical trials for diverse malignancies are underway, and their promise in control of inflammation, cancer, and viral infection is being actively explored.12−24 There are hundreds of methyl reader proteins, but progress in inhibiting methyllysine readers has been comparatively slow. The first examples were inhibitors of methyllysine-binding Malignant Brain Tumor (MBT) domains.25−29 Other recent reports of methyllysine reader protein inhibitors include agents targeting one tudor domain30 and two PHD fingers.31,32
In the family of chromatin organization modifier domains (chromodomains), the initial focus has been on CBX7, one of the five human polycomb paralogs. (The proteins CBX1, CBX3, and CBX5 are a more distantly related set that are heterochromatin protein 1 (HP1) paralogs). Our group reported peptidomimetic inhibitors developed from peptide leads,33 and the group of Zhou recently reported on small molecule inhibitors discovered through high-throughput screening.34 The initial focus on CBX7 is partly because it is strongly associated with many disease phenotypes, and partly also because it tends to give higher in vitro affinities for its native ligands than the other CBX proteins.9,10 This characteristic has also been identified in a computational analysis that predicted that CBX7 would have a relatively “druggable” binding site among methyl reader proteins.35
We sought to identify selective chemical or peptidic tools that would overcome this bias and target other members within the polycomb CBX family of epigenetic modifiers. The sequence and structural similarities within the human polycomb chromodomains are very high (Figure S1). Their highly diverse in vivo functions (above) are partially understood as arising from their divergent domain architecture outside of their methyl-reading chromodomains.11 However, there are no known sites for chemical binding outside of the chromodomains, so any efforts to create selective ligands must rely on being able to discriminate among the five highly similar CBX chromodomains.
The overall similarity of chromodomains makes their selective inhibition challenging. The aromatic cage pockets that bind to the histone’s Kme3 side chain are essentially indistinguishable among the five polycomb paralogs (Figure 1A,B). A small, hydrophobic adjacent pocket binds the conserved histone Ala side chains that uniformly occur (−2) to the trimethyllysine sites H3K9me3 and H3K27me3, the two sites known to be targeted in vitro by CBX proteins. The (−2) pocket is created and shielded from solvent by the closure of two residue side chains over the ligand in a motif called the hydrophobic clasp.9,33 The floor of the (−2) pocket is partially defined by a Val residue in CBX4 and CBX7 that is replaced by an Ala residue in CBX2, -6, and -8 (Figure 1C,D). It was recently shown that a Val/Ala exchange at this position can make CBX7 display CBX2-like binding affinities and functions, demonstrating the importance of these residues at the floor of the (−2) pocket in the CBX proteins’ intrinsic biological functions.8
Figure 1.
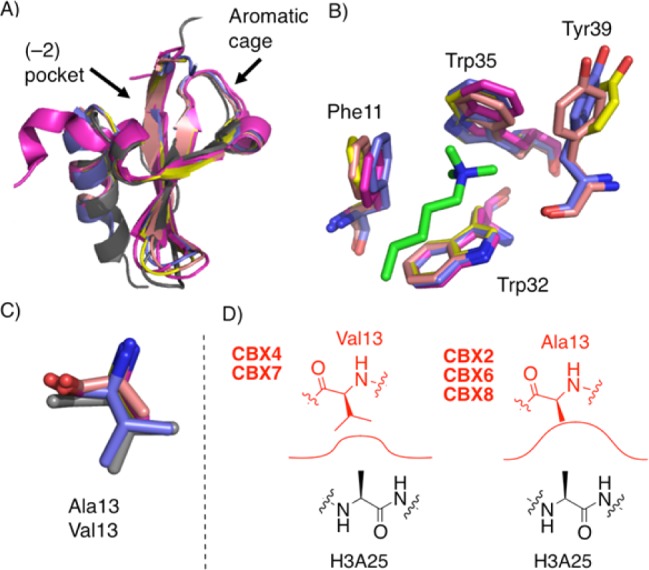
Overview of the chromodomains of human polycomb paralogs CBX2, CBX4, CBX6, CBX7, and CBX8. (A) Structural alignments show the overall similarity of CBX proteins (magenta = CBX2, pdb code 3H91; gray = CBX4, pdb code 3I8Z; yellow = CBX6, pdb code 3I90; purple = CBX7, pdb code 4MN3; salmon = CBX8, pdb code 3I91). (B) Overlay of the aromatic cages with the Kme3 native ligand in green. (CBX4 not shown, as the only available structure lacks bound Kme3 ligand.) (C) Overlay of the (−2) pocket floor Val/Ala residues. (D) Depiction of (−2) pocket size in each CBX protein with histone 3 alanine 25 as the native ligand. CBX7 numbering used throughout.
We report here that varying the ligand structure within the (−2) pocket has a large influence on CBX selectivity and in fact allows for the simple creation of potent CBX6-selective inhibitors.
We first established a panel of proteins for use in fluorescence polarization assays of ligand affinities and selectivities. Recombinant chromodomains for each polycomb paralog (CBX2, CBX4, CBX6, CBX7, and CBX8) were expressed and purified using minor modifications of reported protocols.9 We also prepared one member of the related HP1 family (CBX1; HP1β) as a representative from this more distantly related set of CBX proteins.
In spite of the canon that defines H3K9me3 and/or H3K27me3 as the targets of CBX proteins, H3K9me3 and H3K27me3 peptides have been shown not to bind measurably with multiple members of the CBX family.9,10 In order to ensure strong baseline affinity for our ligands, we started with a peptidic sequence (1) that we previously identified as a moderate-strength CBX7 binder (IC50 = 73 μM) and a chemically modified version (2) that has improved CBX7 affinity (IC50 = 1.7 μM) arising from a p-bromobenzamide group (Figure 2 and Figure S11).33 Competitive fluorescence polarization (FP) studies are limited in their ability to measure Kd values for potent ligands, especially for comparison across different proteins that have different intrinsic affinities for the FP probes.36 To overcome this limitation by using direct FP titrations of an entire panel of proteins into ligands (which gives reliable Kd values for all complexes), we modified 2 by adding a linking residue and fluorescent dye at the C-terminus. Compounds 3, 4, and 5 are such compounds that vary only in the identity of the side chain directed into the (−2) pocket.
Figure 2.

Peptide sequences Ac-FAYKme3S-NH2 (1) and pBr-FAYKme3S-NH2 (2) identified as CBX7 binders.
Figure 3 shows Kd values arising from direct FP titrations of all six chromodomains into all three ligands (see also Figures S13–15). The (−2) substitutions in 3–5 have dramatic effects on potencies and selectivity for CBX proteins. Compound 3, bearing the methyl substituent at the (−2) position, was potent and promiscuous. Its Kd values are, from strongest to weakest, CBX4/7 < CBX2/6 < CBX8. However, even the affinity for the weakest partner, at 1 μM, is >25-fold stronger than the affinities of any small molecule inhibitor for any chromodomain yet reported. Inhibitor 3 is moderately selective for all polycomb paralogs (0.1 to 1 μM) over the HP1 paralog CBX1 (5 μM).
Figure 3.
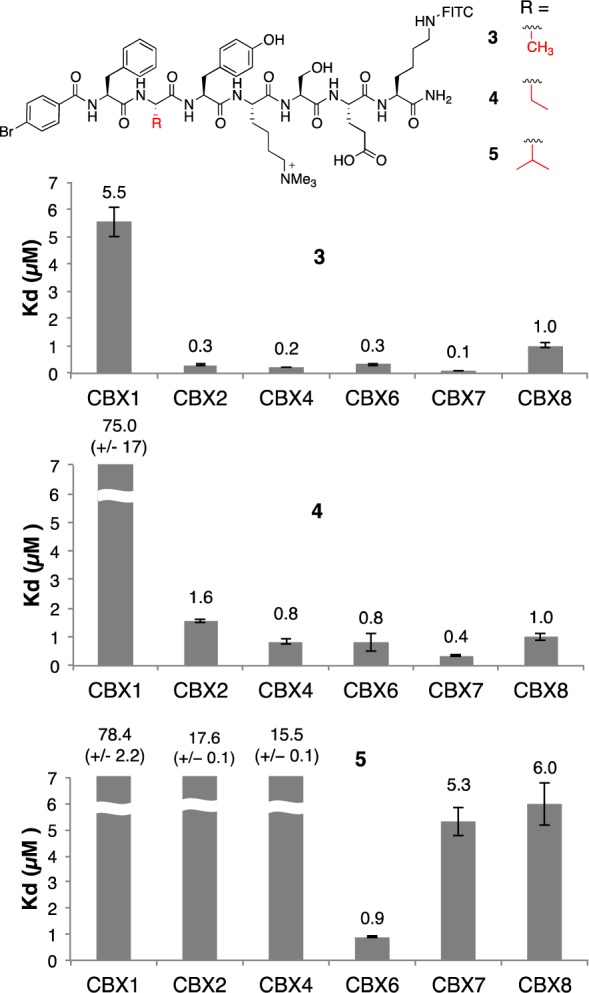
Series of CBX ligands with varying alkyl substitutions at the (−2) position and binding affinities determined by direct FP. Standard errors are shown as error bars or in parentheses for values that exceed axis limits.
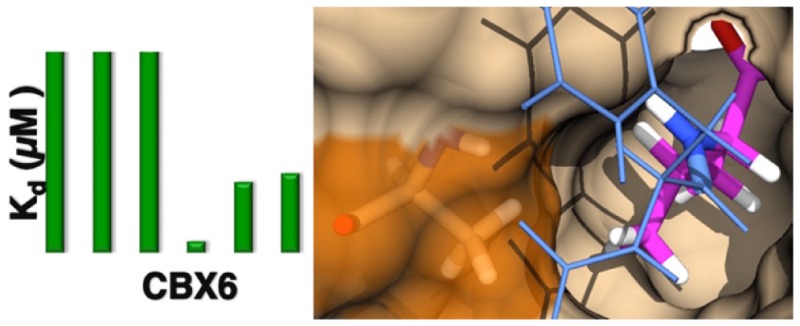
Addition of the ethyl substituent in 4 weakened binding to CBX1 (HP1β) by >10-fold, while generating smaller decreases in binding potency to CBX2/4/6/7 and no change in binding to CBX8. The isopropyl substitution in compound 5 decreased binding affinity of the peptide to all of CBX2/4/7/8, while not significantly changing binding to CBX6. Compound 5 is 90×, 20×, 18×, 6×, and 7× selective for CBX6 over CBX1/2/4/7/8, respectively. Analogues of compounds 3 and 5 lacking FITC labels were tested using a competitive FP assay (Figure S12). The IC50 values determined showed 7-fold selectivity for CBX6 over CBX7 for unlabeled 5, demonstrating that the FITC tag alone is not the source of CBX6 selectivity.
We further confirmed the affinities and selectivities of 3 and 5 by preparing 3-biotin and 5-biotin to enable orthogonal characterization of the complexes by surface plasmon resonance (SPR). The results echo the selectivity trends obtained by direct FP, while also generally agreeing with the absolute Kd values (Table 1 and Figures S16 and S17).
Table 1. Binding Affinities Determined by SPR.
| compd | protein | Kd (μM) |
|---|---|---|
| 3-biotin | CBX6 | 0.11 ± 0.01 |
| CBX7 | 0.16 ± 0.01 | |
| CBX7-V13A | 0.29 ± 0.05 | |
| 5-biotin | CBX6 | 0.91 ± 0.05 |
| CBX7 | 34 ± 8 | |
| CBX7-V13A | 24 ± 2 |
To probe the role of Val/Ala substitutions at the floor of the (−2) pocket (Figure 1C,D) in defining CBX protein selectivity, the CBX7-V13A mutant was prepared and tested by SPR (Table 1). The weak binding to compound 5 by this CBX6-like mutation of CBX7 shows that the V13A substitution alone is not sufficient to drive CBX6-like selective binding of 5. This is consistent with the observed low potency of 5 for binding CBX2 and CBX8, which also have the V13A substitution but differ in other residues.
We carried out energy minimizations (Moloc) and ligand docking (SeeSAR) for ligands 3 and 5 in order to gain more insight into this SAR. Both methods suggested that the large side chain on ligand 5 could be accommodated in identical modes and with identical energies in the (−2) pockets of CBX6 and CBX7, confounding the simple picture shown in Figure 1D. The overall picture is that the (−2) pocket is critical for the native binding preferences of CBX proteins, but that the way in which ligand 5 provides for CBX6 selectivity by occupying this pocket is too complicated to be understood using static X-ray structures.
To gain a dynamic view of the complexes, we carried out MD simulations of the complexes of CBX6 and CBX7 with each of 3 and 5. The trajectories show that the complex of CBX7 with ligand 5 (mismatched) undergoes opening of the hydrophobic clasp that envelopes the ligand. In contrast, the complex of CBX6 with 5 (matched) remains completely wrapped around the ligand throughout the simulation, as illustrated by clasp distance d1 in Figure 4 (see also Supporting Information for movies). Figure 4 includes other geometric parameters that quantify how CBX6 and CBX7 differ in their engagement of ligands. The ligand side chains always stay in the (−2) pockets, but the pocket shapes respond in different ways to different ligands. The overall “external pocket width” (d3 in Figure 4) is the same in all complexes, showing that the mouth of the pocket is (surprisingly) the same regardless of the Val13Ala swap at one edge of this measured distance. The “internal pocket width” (d2 in Figure 4) and hydrophobic clasp distance (d1) both show significant increases for CBX7-5 that are not observed for CBX6-5, showing increased strains that account for the observed experimental selectivity.
Figure 4.
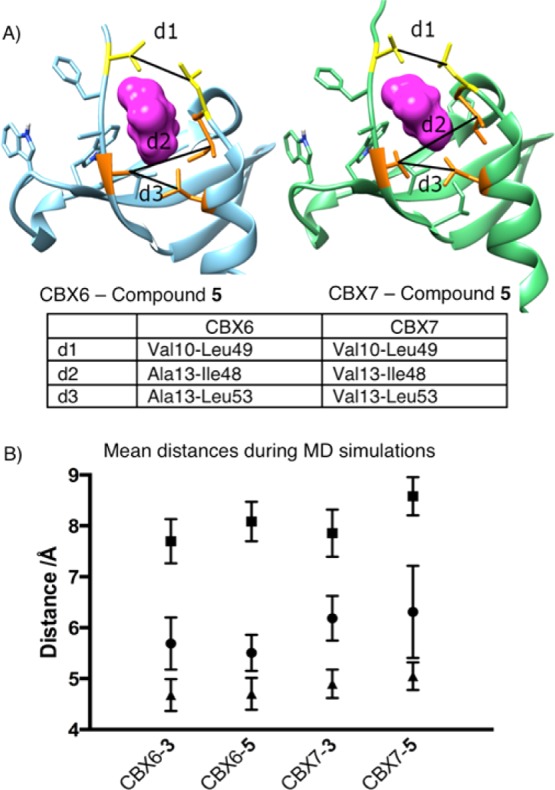
Molecular dynamic simulation results showing the change in distances within CBX6 and CBX7 when in complex with compounds 3 and 5. (A) Representative snapshots from MD trajectories, showing only those parts of the ligands that occupy the (−2) pocket as magenta surfaces. The distances d1, d2, and d3 define changes in pocket shape that can be compared between simulations (see text). (B) Mean values for distances d1 (circle), d2 (square), and d3 (triangle) show the changes induced in and around the (−2) pocket when the bulkier compound 5 is bound to CBX6 and CBX7. See also the movies of complete MD simulations (Supporting Information).
The aromatic cage of CBX proteins are known to be important binding sites, but our results show how ligand affinity also depends on occupying other binding subsites. The histone tail ligands in native CBX–histone complexes occupy the beta groove and interdigitate between the existing two protein strands in order to make a short three-strand beta-sheet motif. The previously reported small molecule ligands for CBX7 engage only the region around the aromatic cage and have modest potencies (28–67 μM).34 The modified peptidic ligands we report here reach affinities as strong as 0.1 μM (for the complex CBX7-3) and are routinely sub-μM. We attribute this to their occupation of the beta groove, which includes the aforementioned (−2) pocket as well as a hydrophobic cleft that extends further from the Kme3 binding site. Comparison of 1 and 2 shows that occupying that cleft provides >40-fold enhanced potency.
The origins of selectivities for the small molecule vs the peptidic ligands are more subtle. The small-molecule ligand of Zhou shows impressive 3- to 22-fold selectivities for CBX7 over CBX2/4/6/8 even though it mainly binds the aromatic cage region.34 The aromatic cages of all CBXs are highly similar to each other in structure, so we infer that the aromatic cage of CBX7 has better preorganization and/or reduced solvent accessibility relative to its family members, rather than a large difference in protein–ligand interactions in the bound state. Promiscuous compound 3 shows stronger binding to CBX7 than any other CBX protein, suggesting that it is benefiting from similar effects.
Large groups in the (−2) pocket are able to overcome the inherent bias toward CBX7 binding. The particular CBX6 selectivity is not simply explained by the Val/Ala difference at position 13 of the chromodomains, as would have been predicted both by simple modeling and by the CBX2/7 results reported earlier this year. Kaustov et al. showed using a peptide array that CBX8 (but not CBX7) could accommodate a valine side chain in the (−2) position of a histone tail sequence,9 but their qualitative array-blotting result did not include CBX6 for comparison. Our solution phase data agree with this result to an extent, in that they show similar solution-phase affinities for valine-containing ligand 5 binding to CBX7 or CBX8 (ca. 5 μM each). However, the affinities for CBX6 are higher, providing selectivity for this polycomb paralog that would not have been anticipated based on prior studies of these proteins.
These results also uncover the previously unknown binding preferences of CBX6’s chromodomain. While it has been assumed to be a canonical polycomb reader of H3K9me3 and/or H3K27me3, the in vitro affinities of CBX6 for these marks are in fact unmeasurably weak.9,10 Each of these histone marks has an alanine residue occupying the (−2) pocket and, according to our results, would be poorly suited to bind CBX6. Our results suggest that CBX6 might be a reader of a different, as-yet undetermined trimethyllysine site.37 Identifying the unknown native target(s) of CBX6 would help prove that the SAR for CBX6 selectivity identified here also persists in a cellular context.
Chromodomain-containing proteins are increasingly suggested as targets for therapeutic intervention, and the functional biology of polycomb paralogs is an important frontier of epigenetics and stem cell biology. Studying the chemical biology and therapeutic potential of CBX proteins requires selective ligands that, until now, have not been available. Our results provide new inhibitors for CBX6, which has been the least studied of the human polycombs. They also inform the general design requirements of the next-generation of potent and selective small-molecule ligands for CBX proteins.
Acknowledgments
Plasmids were gifts of Cheryl Arrowsmith (Addgene plasmids 25245, 25158, 25237, 25296, 25241, and 62514). We thank Anna Hirsch and Milon Mondal for help with SeeSAR docking. F.H. and J.W. thank the Canada Research Chairs program. The computational component of this work was enabled in part by WestGrid (www.westgrid.ca) and Compute Canada/Calcul Canada (www.computecanada.ca).
Glossary
ABBREVIATIONS
- PTM
post-translational modifications
- CBX
chromobox
- HP1
heterochromatin protein
- PRC1
polycomb repressive complex 1
- BRD
bromodomain
- MBT
malignant brain tumor
- PHD
plant homeodomain
- FP
fluorescence polarization
- SPR
surface plasmon resonance
- MD
molecular dynamics
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00378.
Author Contributions
The manuscript was written by N.M. and J.M. with guidance from I.P., J.W., and F.H. N.M. carried out compound design and synthesis, with assistance of C.C., K.D., and C.S. M.G. carried out protein expression and binding studies with assistance of A.D. S.H. carried out SPR analyses. S.D. and T.Q. carried out CBX7 mutagenesis. J.M. carried out computational studies under supervision of I.P. All authors have given approval to the final version of the manuscript.
This research was supported by a CIHR fellowship and WestCoast Motorcycle Ride to Live award to N.M., Cancer Research Society Grant 19284, and Prostate Cancer Canada Movember Discovery Grant D2013–18,
The authors declare no competing financial interest.
Supplementary Material
References
- Berger S. L.; Kouzarides T.; Shiekhattar R.; Shilatifard A. An Operational Definition of Epigenetics. Genes Dev. 2009, 23, 781–783. 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. Translating the Histone Code. Science 2001, 293, 1074–1080. 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Fang J.; Bedford M. T.; Zhang Y.; Xu R. M. Recognition of Histone H3 Lysine-4 Methylation by the Double Tudor Domain of Jmjd2a. Science 2006, 312, 748–751. 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Gieni R. S.; Hendzel M. J. Polycomb Group Protein Gene Silencing, Non-Coding Rna, Stem Cells, and Cancer. Biochem. Cell Biol. 2009, 87, 711–746. 10.1139/O09-057. [DOI] [PubMed] [Google Scholar]
- O’Loghlen A.; Munoz-Cabello A. M.; Gaspar-Maia A.; Wu H. A.; Banito A.; Kunowska N.; Racek T.; Pemberton H. N.; Beolchi P.; Lavial F.; Masui O.; Vermeulen M.; Carroll T.; Graumann J.; Heard E.; Dillon N.; Azuara V.; Snijders A. P.; Peters G.; Bernstein E.; Gil J. Microrna Regulation of Cbx7Mediates a Switch of Polycomb Orthologs During Esc Differentiation. Cell Stem Cell 2012, 10, 33–46. 10.1016/j.stem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey L.; Aloia L.; Cozzuto L.; Benitah S. A.; Di Croce L. Rybp and Cbx7 Define Specific Biological Functions of Polycomb Complexes in Mouse Embryonic Stem Cells. Cell Rep. 2013, 3, 60–69. 10.1016/j.celrep.2012.11.026. [DOI] [PubMed] [Google Scholar]
- Klauke K.; Radulovic V.; Broekhuis M.; Weersing E.; Zwart E.; Olthof S.; Ritsema M.; Bruggeman S.; Wu X.; Helin K.; Bystrykh L.; de Haan G. Polycomb Cbx Family Members Mediate the Balance between Haematopoietic Stem Cell Self-Renewal and Differentiation. Nat. Cell Biol. 2013, 15, 353–362. 10.1038/ncb2701. [DOI] [PubMed] [Google Scholar]
- Tardat M.; Albert M.; Kunzmann R.; Liu Z.; Kaustov L.; Thierry R.; Duan S.; Brykczynska U.; Arrowsmith C. H.; Peters A. H. Cbx2 Targets Prc1 to Constitutive Heterochromatin in Mouse Zygotes in a Parent-of-Origin-Dependent Manner. Mol. Cell 2015, 58, 157. 10.1016/j.molcel.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Kaustov L.; Ouyang H.; Amaya M.; Lemak A.; Nady N.; Duan S.; Wasney G. A.; Li Z.; Vedadi M.; Schapira M.; Min J.; Arrowsmith C. H. Recognition and Specificity Determinants of the Human Cbx Chromodomains. J. Biol. Chem. 2011, 286, 521–529. 10.1074/jbc.M110.191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E.; Duncan E. M.; Masui O.; Gil J.; Heard E.; Allis C. D. Mouse Polycomb Proteins Bind Differentially to Methylated Histone H3 and Rna and Are Enriched in Facultative Heterochromatin. Mol. Cell. Biol. 2006, 26, 2560–2569. 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senthilkumar R.; Mishra R. K. Novel Motifs Distinguish Multiple Homologues of Polycomb in Vertebrates: Expansion and Diversification of the Epigenetic Toolkit. BMC Genomics 2009, 10, 549. 10.1186/1471-2164-10-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C. W. Fragment-Based Discovery of Bromodomain Inhibitors Part 2: Optimization of Phenylisoxazole Sulfonamides. J. Med. Chem. 2012, 55, 587–596. 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- Banerjee C.; Archin N.; Michaels D.; Belkina A. C.; Denis G. V.; Bradner J.; Sebastiani P.; Margolis D. M.; Montano M. Bet Bromodomain Inhibition as a Novel Strategy for Reactivation of Hiv-1. J. Leukocyte Biol. 2012, 92, 1147–1154. 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. W.; Dean A. W.; Woolven J. M.; Bamborough P. Fragment-Based Discovery of Bromodomain Inhibitors Part 1: Inhibitor Binding Modes and Implications for Lead Discovery. J. Med. Chem. 2012, 55, 576–586. 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- Fish P. V.; Filippakopoulos P.; Bish G.; Brennan P. E.; Bunnage M. E.; Cook A. S.; Federov O.; Gerstenberger B. S.; Jones H.; Knapp S.; Marsden B.; Nocka K.; Owen D. R.; Philpott M.; Picaud S.; Primiano M. J.; Ralph M. J.; Sciammetta N.; Trzupek J. D. Identification of a Chemical Probe for Bromo and Extra C-Terminal Bromodomain Inhibition through Optimization of a Fragment-Derived Hit. J. Med. Chem. 2012, 55, 9831–9837. 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Cote A.; Nasveschuk C. G.; Taylor A. M.; Harmange J. C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Bangalore Srinivasamurthy N.; Birudukota S.; Albrecht B. K. Discovery, Design, and Optimization of Isoxazole Azepine Bet Inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Cote A.; Nasveschuk C. G.; Taylor A. M.; Harmange J. C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Bangalore Srinivasamurthy N.; Birudukota S.; Albrecht B. K. Discovery, Design, and Optimization of Isoxazole Azepine Bet Inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Gosmini R.; Toum J.; Clement C. A.; Barnathan M.; Brusq J. M.; Mordaunt J. E.; Grimes R. M.; Crowe M.; Pineau O.; Ajakane M.; Daugan A.; Jeffrey P.; Cutler L.; Haynes A. C.; Smithers N. N.; Chung C. W.; Bamborough P.; Uings I. J.; Lewis A.; Witherington J.; Parr N.; Prinjha R. K.; Nicodeme E. Discovery of Epigenetic Regulator I-Bet762: Lead Optimization to Afford a Clinical Candidate Inhibitor of the Bet Bromodomains. J. Med. Chem. 2013, 56, 7501–7515. 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Puissant A.; Frumm S. M.; Alexe G.; Bassil C. F.; Qi J.; Chanthery Y. H.; Nekritz E. A.; Zeid R.; Gustafson W. C.; Greninger P.; Garnett M. J.; McDermott U.; Benes C. H.; Kung A. L.; Weiss W. A.; Bradner J. E.; Stegmaier K. Targeting Mycn in Neuroblastoma by Bet Bromodomain Inhibition. Cancer Discovery 2013, 3, 308–323. 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani I. A.; Dommeti V. L.; Wang X.; Malik R.; Cieslik M.; Yang R.; Escara-Wilke J.; Wilder-Romans K.; Dhanireddy S.; Engelke C.; Iyer M. K.; Jing X.; Wu Y. M.; Cao X.; Qin Z. S.; Wang S.; Feng F. Y.; Chinnaiyan A. M. Therapeutic Targeting of Bet Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature 2014, 510, 278–282. 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. G.; Sanchez R.; Zhou M. M. Privileged Diazepine Compounds and Their Emergence as Bromodomain Inhibitors. Chem. Biol. 2014, 21, 573–583. 10.1016/j.chembiol.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective Inhibition of Bet Bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S.; Knapp S. Discovery of Bet Bromodomain Inhibitors and Their Role in Target Validation. MedChemComm 2014, 5, 288–296. 10.1039/c3md00291h. [DOI] [Google Scholar]
- Garnier J. M.; Sharp P. P.; Burns C. J. Bet Bromodomain Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2014, 24, 185–199. 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]
- Herold J. M.; Wigle T. J.; Norris J. L.; Lam R.; Korboukh V. K.; Gao C.; Ingerman L. A.; Kireev D. B.; Senisterra G.; Vedadi M.; Tripathy A.; Brown P. J.; Arrowsmith C. H.; Jin J.; Janzen W. P.; Frye S. V. Small-Molecule Ligands of Methyl-Lysine Binding Proteins. J. Med. Chem. 2011, 54, 2504–2511. 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerino M. A.; Zhong N.; Dong A.; Dickson B.; James L.; Baughman B.; Norris J.; Kireev D.; Janzen W. P.; Arrowsmith C.; Frye S. V. The Structure-Activity Relationships of L3mbtl3 Inhibitors: A Second Series of Potent Compounds Which Bind the L3mbtl3 Dimer. MedChemComm 2013, 4, 1501–1507. 10.1039/c3md00197k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. I.; Barsyte-Lovejoy D.; Zhong N.; Krichevsky L.; Korboukh V. K.; Herold J. M.; MacNevin C. J.; Norris J. L.; Sagum C. A.; Tempel W.; Marcon E.; Guo H. B.; Gao C.; Huang X. P.; Duan S. L.; Emili A.; Greenblatt J. F.; Kireev D. B.; Jin J.; Janzen W. P.; Brown P. J.; Bedford M. T.; Arrowsmith C. H.; Frye S. V. Discovery of a Chemical Probe for the L3mbtl3Methyllysine Reader Domain. Nat. Chem. Biol. 2013, 9, 184–191. 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. I.; Korboukh V. K.; Krichevsky L.; Baughman B. M.; Herold J. M.; Norris J. L.; Jin J.; Kireev D. B.; Janzen W. P.; Arrowsmith C. H.; Frye S. V. Small-Molecule Ligands of Methyl-Lysine Binding Proteins: Optimization of Selectivity for L3mbtl3. J. Med. Chem. 2013, 56, 7358–7371. 10.1021/jm400919p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.; Herold J. M.; Kireev D.; Wigle T.; Norris J. L.; Frye S. Biophysical Probes Reveal a ″Compromise″ Nature of the Methyl-Lysine Binding Pocket in L3mbtl1. J. Am. Chem. Soc. 2011, 133, 5357–5362. 10.1021/ja110432e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetti M. T.; Baughman B. M.; Dickson B. M.; Mu Y.; Cui G.; Mader P.; Dong A.; Norris J. L.; Rothbart S. B.; Strahl B. D.; Brown P. J.; Janzen W. P.; Arrowsmith C. H.; Mer G.; McBride K. M.; James L. I.; Frye S. V. Identification of a Fragment-Like Small Molecule Ligand for the Methyl-Lysine Binding Protein, 53bp1. ACS Chem. Biol. 2015, 10, 1072. 10.1021/cb500956g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. C. R.; Rutherford T. J.; Birchall K.; Chugh J.; Fiedler M.; Bienz M. Competitive Binding of a Benzimidazole to the Histone-Binding Pocket of the Pygo Phd Finger. ACS Chem. Biol. 2014, 9, 2864–2874. 10.1021/cb500585s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. K.; Nath N.; Flemming R.; Feltenberger J. B.; Denu J. M. Identification and Characterization of Small Molecule Inhibitors of a Plant Homeodomain Finger. Biochemistry 2012, 51, 8293–8306. 10.1021/bi3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simhadri C.; Daze K. D.; Douglas S. F.; Quon T. T. H.; Dev A.; Gignac M. C.; Peng F. N.; Heller M.; Boulanger M. J.; Wulff J. E.; Hof F. Chromodomain Antagonists That Target the Polycomb-Group Methyllysine Reader Protein Chromobox Homo Log 7 (Cbx7). J. Med. Chem. 2014, 57, 2874–2883. 10.1021/jm401487x. [DOI] [PubMed] [Google Scholar]
- Ren C.; Morohashi K.; Plotnikov A. N.; Jakoncic J.; Smith S. G.; Li J.; Zeng L.; Rodriguez Y.; Stojanoff V.; Walsh M.; Zhou M. M. Small-Molecule Modulators of Methyl-Lysine Binding for the Cbx7 Chromodomain. Chem. Biol. 2015, 22, 161–168. 10.1016/j.chembiol.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago C.; Nguyen K.; Schapira M. Druggability of Methyl-Lysine Binding Sites. J. Comput.-Aided Mol. Des. 2011, 25, 1171–1178. 10.1007/s10822-011-9505-2. [DOI] [PubMed] [Google Scholar]
- Huang X. Fluorescence Polarization Competition Assay: The Range of Resolvable Inhibitor Potency Is Limited by the Affinity of the Fluorescent Ligand. J. Biomol. Screening 2003, 8, 34–38. 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]
- This is indirectly anticipated by a massive parallel computational docking effort that was backed up by microarray binding data.Li N.; Stein R. S. L.; He W.; Komives E.; Wang W. Identification of methyllysine peptides binding to chromobox protein homolog 6 chromodomain in the human proteome. Mol. Cell. Proteomics 2013, 12, 2750–2760. 10.1074/mcp.O112.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors identify 50 putative Kme3-containing sequences that should bind the CBX6 chromodomain. Two have Val and two have Ala at the (−2) position relative to Kme3 and are therefore potential candidates for binding CBX6. The other 46 candidates have larger and/or polar groups at the (−2) position that could not be accommodated by CBX6 or any other CBX chromodomain, based on extensive knowledge of X-ray structures and solution phase binding data for many CBX–peptide complexes. In the absence of solution phase or cellular binding studies, we are skeptical that these are true CBX6 chromodomain binders in vitro or in vivo.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.