

Abstract
Inhibition of the bromodomains of the BET family, of which BRD4 is a member, has been shown to decrease myc and interleukin (IL) 6 in vivo, markers that are of therapeutic relevance to cancer and inflammatory disease, respectively. Herein we report substituted benzo[b]isoxazolo[4,5-d]azepines and benzotriazolo[4,3-d][1,4]diazepines as fragment-derived novel inhibitors of the bromodomain of BRD4. Compounds from these series were potent and selective in cells, and subsequent optimization of microsomal stability yielded representatives that demonstrated dose- and time-dependent reduction of plasma IL-6 in mice.
Keywords: Bromodomain, IL-6 inhibition, BRD4, BET
The bromodomain and extra terminal (BET) family is made up of BRD2, BRD3, BRD4, and BRDT, each of which contains tandem bromodomains (BD-1 and BD-2) that recognize specific acetylated lysine residues in the N-terminal tails of histones.1 Members of the BET family modulate gene expression by recruiting transcriptional regulators to specific genomic locations.2 In 2009, the Mitsubishi Tanabe Pharma Corporation reported the first small molecule inhibitors of this family of chromatin adaptors,3 and publications followed on the use of BET inhibitors to demonstrate antitumor activity4 and modulate inflammation.5 Experiments applying genetic ablation or specific inhibitor treatment have established the role of BET family members in regulating genes crucial for normal cell growth.6−8
Subsequent studies9,10 have defined a mechanistic link between potency of BET inhibition and activity against hematologic malignancies and demonstrated that BET inhibition regulates the MYC oncogene, resulting in cell cycle arrest and apoptosis. In vivo BET bromodomain inhibition leads to efficacy in xenograft models representing multiple cancer types, and BET bromodomains are also implicated in nononcology indications, i.e., human immunodeficiency virus and human t-lymphotrophic viral gene expression, DNA damage response, and cardiac hypertrophy. Importantly, BET inhibitors have been shown to selectively regulate a subset of multiple inflammatory cytokines, including interleukins 6, 1β, 17, 21, and 23, and granulocyte macrophage colony-stimulating factor11,12 and have demonstrated efficacy in several in vivo models of inflammation, including endotoxic shock, experimental autoimmune encephalomyelitis, and arthritis.5,11
Because of the potential therapeutic application of BET inhibition, we initiated a drug discovery program and recently reported on a fragment-based effort that resulted in a series of isoxazolo[5,4-c]thieno[2,3-e]azepine BET inhibitors exemplified by CPI-3 (Scheme 1).13 Concurrent with that effort we were interested in exploring a different connectivity within the seven-membered ring that would provide a unique basis for modulation of the physicochemical properties and BET potency of the resultant compounds. Specifically, we hypothesized that changing the cyclic imine to a substituted aniline would retain the concave shape important for specific binding to the bromodomain while providing a different metabolic profile for the series. We were also interested in evaluating a modified linker in combination with the reported triazole binding element4,5 and in finding a replacement for the thiophene group of CPI-3 because of its potential metabolic instability. Here we describe the resulting benzo[b]isoxazolo[4,5-d]azepine and benzotriazolo[4,3-d][1,4]diazepine series, which were potent inhibitors of BET bromodomains in biochemical and cellular assays and which allowed for optimization of metabolic stability and demonstration of a pharmacodynamic effect in a mouse model of IL-6 suppression (Scheme 1).
Scheme 1. Overview of the Origin and Development of Different Azepine-Derived BET Inhibitors.

Our investigations began with the preparation of benzo[b]isoxazolo[4,5-d]azepine fragment 1 (Scheme 1), which exhibited a BRD4 BD-1 binding affinity in an AlphaLISA (AS) assay comparable to that of the 3-methyl-4-phenylisoxazol-5-amine fragment from which CPI-3 was derived (IC50 33 μM)13 and which provided several vectors to use to improve potency. Of them, N-arylation to form the 4-cyano compound 4 (Table 1) markedly improved biochemical potency and activity in a cellular readout of IL-6 suppression, a BET-related phenotype relevant to modulation of the immune response in vivo (see Supporting Information for details). Throughout our studies, BRD4 BD-1 was used as a surrogate for the entire BET family; tests of individual compounds against the bromodomains of BRD2, BRD3, and BRDT supported this assumption (vide infra). A variety of substitutions around the ring were explored; 4-CN-Ph was consistently the most potent substitution and is the only one shown here, for simplicity. Potency could be further improved with the addition of a heterocycle at the 8 position, as in compound 5 (Table 1, see numbering). Introduction of a methyl group on the azepine (Table 1, 6) maintained biochemical activity but improved translation of that potency into a cellular context, perhaps by affording greater conformational preorganization of the compound, which has been shown to improve cell permeability.14,15 A variety of substituted heterocycles were tolerated at the 8 position, as exemplified by compounds 7 and 8, but any groups attached to the azepine ring other than methyl were deleterious to activity (e.g., compound 9 in Table 1; see Table S7 for further examples). The limitation to methyl at the 4 position is in contrast to what we observed with the isoxazolo[5,4-c]thieno[2,3-e]azepine scaffold (cf. CPI-3)13 and with what others have observed with related scaffolds.4,5 Substitutions at other positions of the benzo ring or at the 1 position of the isoxazole diminished potency (data not shown).
Table 1. BRD4 BD-1 Structure–Activity Relationships of Benzo[b]isoxazolo[4,5-d]azepines.

Binding was measured using an AlphaLISA assay with the isolated bromodomain. See Supporting Information for more details. N ≥ 3.
IL-6 EC50 = measurement of the suppression of IL-6 release from THP-1 cells following stimulation with lipopolysaccharide. N = 2.
clogP calculated using ChemDraw 14.
LM Clint = intrinsic clearance of compound when incubated with liver microsomes.
(m/r/d/h) = mouse/rat/dog/human.
PPB = plasma protein binding.
In the course of optimizing potency, we determined the in vitro metabolic stability of several compounds. As shown in Table 1, clearance in liver microsomes was generally high for the series across species, which we surmised may have been due to the high clogP values intrinsic to the class.14 Replacement of the isoxazole with a triazole binding element presented an opportunity to lower the clogP of the series; simple estimates of lipophilicity16 suggested that the more polar heterocycle would consistently lower clogP by one unit relative to the corresponding isoxazoles while maintaining the overall shape and binding mode of the initial series, allowing us to capitalize on similar structure–activity relationships (SARs).
To explore whether the triazole could be combined with the aniline azepine linker, we prepared the triazole fragment 2 (Scheme 1) and found that it, too, bound BRD4 with potency comparable to 1. Even with the change in heterocycle, much of the SAR from the benzo[b]isoxazolo[4,5-d]azepine series could be recapitulated; N-arylation improved potency (Table 2, 10), as did incorporation of certain heterocycles at the 8 position (Table 2, 11). In contrast with the benzo[b]isoxazolo[4,5-d]azepine series, 4-Cl-Ph was consistently the most potent N-aryl substituent for the benzotriazolo[4,3-d][1,4]diazepines, although, again, a variety of substitution patterns were tolerated. As before, subsequent methylation at the 4 position of the azepine ring (Table 2, 12) provided compounds highly active in both the biochemical and cellular assays. Also, azepine substitutions other than methyl resulted in a roughly 10-fold loss in potency, as exemplified by 14 (Table 2; further examples in Table S7). Separation of the individual enantiomers of elaborated compounds established that most of the binding activity resided within one enantiomer; compound 3 (98% ee) was greater than 50-fold more potent than 15 (>99% ee, Table 2). Gratifyingly, in vitro liver microsome intrinsic clearance in this series was generally lower than that in the corresponding isoxazoles (e.g., LM Clint of 6 vs 12 across species), even though incorporation of the chlorophenyl, which was optimal for potency, returned the clogP of most compounds to the same range as that of the isoxazole series. Finally, to determine the impact of alternate methyl substitution on the azepine ring, compound 16 was prepared. It demonstrated little change in potency relative to unsubstituted 10 even though both 4- and 5-methyl substituents would be expected to preorganize the azepine, suggesting that the position of the methyl azepine substituent was important beyond just its effect on the conformation of the azepine ring.
Table 2. BRD4 BD-1 Structure–Activity Relationships of Benzo[1,2,4][4,3-d]triazolo[1,4]diazepines.

Binding was measured using an AlphaLISA assay with the isolated bromodomain. See Supporting Information for more details. N ≥ 3.
IL-6 EC50 = measurement of the suppression of IL-6 release from THP-1 cells following stimulation with lipopolysaccharide. N = 2.
clogP calculated using ChemDraw 14.
LM Clint = intrinsic clearance of compound when incubated with liver microsomes.
(m/r/d/h) = mouse/rat/dog/human.
PPB = plasma protein binding.
N = 1.
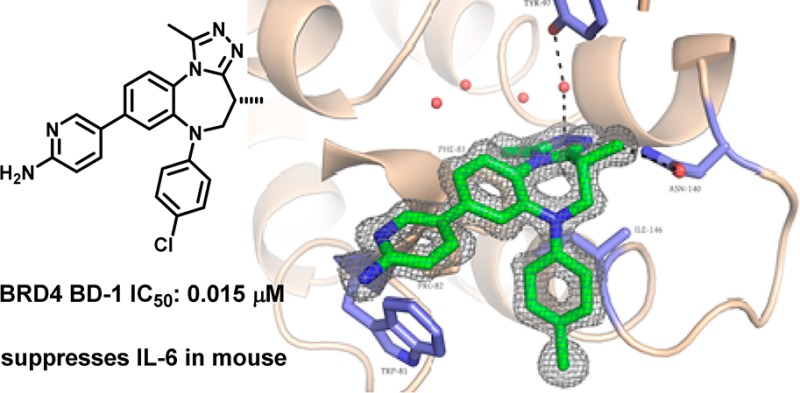
A co-crystal structure of 3 obtained in BRD4 BD-1 established the binding mode of these series (Figure 1a). Notably, it complemented the binding site similar to our reported isoxazolo[5,4-c]thieno[2,3-e]azepines, as exemplified by an overlay of the crystal structures of 3 (chosen for simplicity) and CPI-3; RMSD between the common heavy atoms of the two series was only 0.43 (Figure 1b). As intended, 3 adopted a cupped shape, filling the space between Asn140 and the WPF shelf (residues 81–83) and making lipophilic contacts around gatekeeper residue Ile146. The lone pair of the triazole nitrogen in the 3 position served as an H-bond acceptor in the crucial interaction with Asn140 (highly conserved among bromodomains), positioning the triazole methyl group in the same location as the N-acetyl of the natural acetylated lysine substrate and orienting the nitrogen in position 2 to engage in a through-water hydrogen bond with Tyr97. The 4-methyl azepine substituent provided a second, noncanonical H-bond donating interaction17,18 with the Asn and enforced the curvature of the azepine. The co-crystal structure allowed for the unambiguous assignment of the stereochemistry at that position (as did the co-crystal structure of the opposite enantiomer, 15, Figure S2). Interestingly, the proximity observed between the aminopyridine of 3 and Trp81 suggested that the increased potency of compounds substituted at that position may have been due to a productive edge to face interaction.
Figure 1.
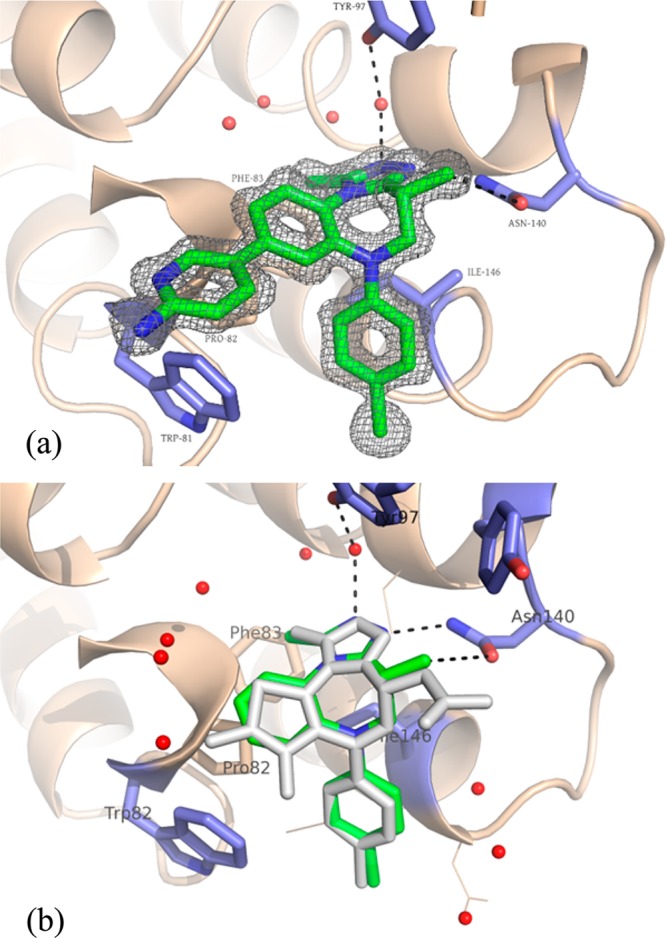
(A) A 1.4 Å cocrystal structure of 3 (green) bound to BRD4 BD-1. PDB code: 4Z1Q. Key H-bonding interactions are indicated. (B) An overlay of a co-crystal structure of 13 (green) with one of CPI-3 (gray) shows the similarity in binding modes between the two series (RMSD = 0.43 for common heavy atoms). PDB code: 4X2I.
To further explore that idea, we examined a number of resolved compounds with a greater variety of substituents at position 8. As shown in Table 3, heteroaromatic substituents such as pyridine (17), pyrolidinone (18), and N-linked triazole (19) yielded compounds with potent binding and cellular activity. A primary carboxamide (20) also improved cellular potency roughly 3-fold relative to unsubstituted 13 (Table 2). Interestingly, compounds unable to align a π system with the edge of Trp81, such as 21 and 22, lost potency relative to 13, consistent with the model of an edge to face interaction driving the binding affinity at the 8 position.
Table 3. BRD4 BD-1 Structure–Activity Relationship of 8-Substituted Benzotriazolo[4,3-d][1,4]diazepines.
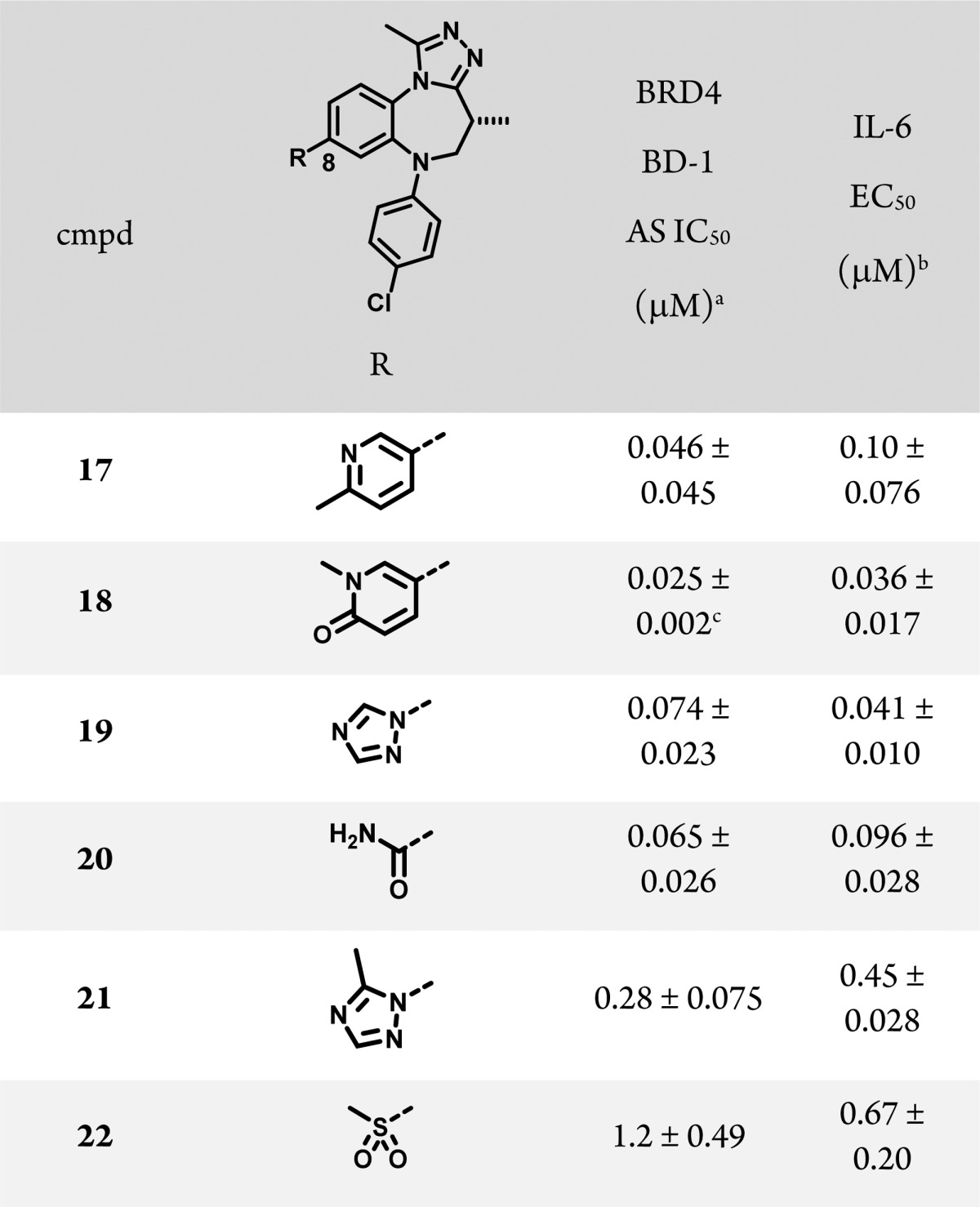
Binding was measured using an AlphaLISA assay with the isolated bromodomain; see Supporting Information for more details. N ≥ 3.
IL-6 EC50 = measurement of the suppression of IL-6 release from THP-1 cells following stimulation with lipopolysaccharide. N ≥ 2.
N = 2.
On the basis of its favorable combination of cellular activity and ADME properties, compound 3 was chosen for in vivo pharmacokinetic profiling in rat. As shown in Table 4, 3 demonstrated IV clearance ten percent below liver blood flow, a reasonable half-life, and appreciable oral bioavailability. Importantly, this profile suggested that exposure of 3 would be sufficient to modulate IL-6 in an in vivo model.
Table 4. Pharmacokinetic and Selected in Vitro Parameters for 3 in Rat.
| parameter | compd 3 | |
|---|---|---|
| cell potency | IL-6 EC50 (μM) | 0.013 |
| binding efficiency | LEAN/LLEa | 0.26/3.1 |
| In vitro | LMrat Clintb (μL/min/mg) | 26 |
| PPBrat (%)c | 96 | |
| IVd (1 mg/kg) | Cl (L/h/kg) | 3.8 (90% LBFe) |
| Clunbound (L/h/kg) | 95 | |
| Vss (L/kg) | 2.2 | |
| t1/2 (h) | 0.67 | |
| POf (10 mg/kg) | Cmax (μM) | 0.29 |
| t1/2 (h) | 1.8 | |
| AUClast (μM·h) | 1.4 | |
| F (%) | 23 |
LEAN = pIC50/#heavy atoms; LLE = pIC50 – clogP.
LMrat Clint = intrinsic clearance of compound when incubated with rat liver microsomes.
PPBrat = rat plasma protein binding.
IV formulation: 1 mL of 1 mg/mL solution of 3 in 5:25:70 DMA/PEG400/20% SBECD. N = 3 rats.
LBF = liver blood flow.
PO formulation: 10 mL of 1 mg/mL suspension of 3 in 0.5% methylcellulose. N = 3 rats.
Before proceeding with experiments to gauge the BET activity of 3in vivo, we determined its binding affinity to the bromodomains of other members of the BET family. As mentioned above, BRD4 BD-1 proved representative of activity against other BET proteins; IC50s ranged from 0.003 to 0.017 μM (see Supporting Information for individual values). Because bromodomains are highly homologous across the class,19 we also assessed the activity of 3 against eight other non-BET bromodomains, as estimated by AlphaLISA or time-resolved fluorescence resonance energy transfer. With the exception of CREB binding protein (CBP) (AS IC50 = 2.2 μM), 3 demonstrated an IC50 greater than 15 μM on all of the bromodomains tested (see Supporting Information), suggesting that its activity in vivo would be limited to the BET family at the relevant concentrations.
Having established its BET activity, specificity, and potential for meaningful in vivo exposure, 3 was dosed PO in mouse at 15 mg/kg at various time points prior to LPS stimulation. Plasma IL-6 levels were measured 2 h later. As shown in Figure 2, IL-6 release was suppressed to baseline levels at higher plasma concentrations of 3 and returned toward vehicle levels over the time course of dosing, in conjunction with decreasing plasma concentrations of 3. Substantial suppression of the IL-6 response in that experiment was observed only when the predicted free plasma concentration of compound was above the cellular EC50,20 consistent with the free drug hypothesis.21 These results support the model of BET inhibition mediating a reduction in inflammatory response IL-6 levels and further recommend BET as a target for drug discovery.
Figure 2.
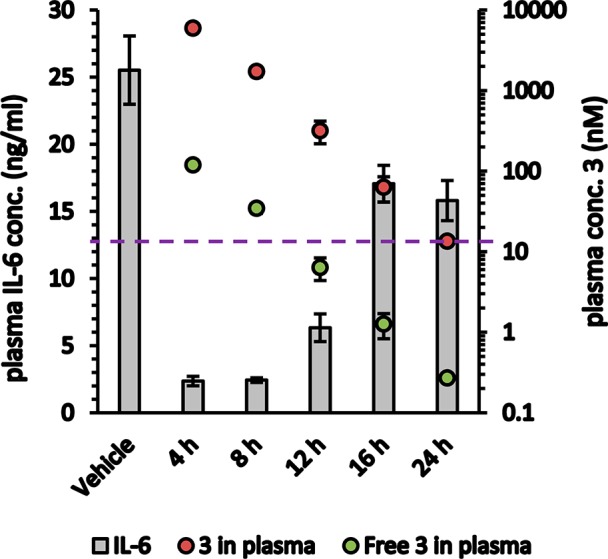
Suppression of IL-6 release in mouse correlates with plasma exposure of 3 following 15 mg/kg dosing PO. Free concentration of 3 calculated based on 98% PPBmouse. Dashed line indicates cellular IL-6 EC50.
In summary, we report the discovery of benzo[b]isoxazolo[4,5-d]azepines and benzotriazolo[4,3-d][1,4]diazepines as two new series of BET inhibitors that combine design elements from our previously reported isoxazolo[5,4-c]thieno[2,3-e]azepines with a unique azepine connection pattern. Both series afforded biochemically and cellularly potent compounds, some of which could be optimized for metabolic stability and applied in vivo to demonstrate dose-dependent modulation of IL-6 suppression via BET engagement. Further applications of these series will be reported in due course.
Acknowledgments
We are grateful to Ted Peters and Christina Lee in the Lead Discovery group for plating compounds used in these studies; PPD BioDuro for synthesizing some of the compounds presented here; and WuXi AppTec and Custom NMR Services for compound characterization.
Supporting Information Available
Further references describing the biological and pharmacological validation of BET as a drug discovery target; assay details; descriptions of the IL-6 PD assay and results; synthetic and characterization data for all compounds reported; cocrystal structures of 13 and 15; crystallographic methods and data; additional SAR data; BET family and other bromodomain potency data for 3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Editas Medicine, 300 Third Street, Cambridge, Massachusetts 02142, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Wu S. Y.; Chiang C. M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–45. [DOI] [PubMed] [Google Scholar]
- Dey A.; Chitsaz F.; Abbasi A.; Misteli T.; Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi M.; Ooike S.; Iwata K.; Hikawa H.; Sugahara K. Antitumor Compounds. WO 2009/084693A1, published Sept. 9, 2009.
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina A. C.; Denis G. V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant B. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. 3rd Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Nat. Acad. Sci. 2011, 108, 16669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Vakoc C. R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An extensive list of references describing specific biological and pharmacological studies is including in the Supporting Information.
- Mele D. A.; Salmeron A.; Ghosh S.; Huang H. R.; Bryant B. M.; Lora J. M. BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 2013, 210, 2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Y. M.; Kirkham P.; Barnes P. J.; Adcock I. M. Brd4 is essential for IL-1β-induced inflammation in human airway epithelial cells. PLoS One 2014, 9, e95051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Côté A.; Nasveschuk C. G.; Taylor A. M.; Harmange J. C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Bangalore Srinivasamurthy N.; Birudukota S.; Albrecht B. K. Discovery, design, and optimization of isoxazole azepine BET inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–23. [DOI] [PubMed] [Google Scholar]
- Bogdan A. R.; Davies N. L.; James K. Comparison of diffusion coefficients for matched pairs of macrocyclic and linear molecules over a drug-like molecular weight range. Org. Biomol. Chem. 2011, 9, 7727–33. [DOI] [PubMed] [Google Scholar]
- clogP values were generated using ChemBioDraw Ultra 14.0.
- Horowitz S.; Trievel R. C. Carbon-oxygen hydrogen bonding in biological structure and function. J. Biol. Chem. 2012, 287, 41576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The sequence homology between mouse and human BRD4 (100% for BD-1 and 97% for BD-2) and our previous observation that inhibition of mouse or human BRD4 has the same effect on myc expression support this analysis (see ref (7)).
- Smith D. A.; Di L.; Kerns E. H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discovery 2010, 9, 929–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.